
INTRODUCTION
Prosopis africana is a tree of great economic value used for various purposes by the rural dwellers in the Guinea Savanna of Nigeria (Agboola et al., 2004). It is known to contain alkaloids, flavonoids, tannins, and quinones and is useful as an analgesic, anthelminthic, antibiotic, antimalarial, antiemetic, and antiulcer agent (Henciya et al., 2017). The gum obtained from the plant’s seeds is a hemicellulose with desirable properties, making it useful as an excipient for pharmaceutical preparations (Olorunsola et al., 2014; Zarafi et al., 2012). The work of Selvi et al. (2010) showed that Prosopis gum (PRG) when used as a binder at concentrations of 8% and 10% produced tablets of relatively high crushing strength with drug release over a period of 5 hours.
Peptic ulcer disease is a break in the inner lining of the stomach, the duodenum, or sometimes the lower esophagus (Najm, 2011). It represents a widely prevalent gastrointestinal tract problem with annual incidence ranging from 0.10% to 0.19% (Sung et al., 2009). Duodenal ulcers account for about 80% of peptic ulcers, while gastric ulcers account for less than 20%. Only rarely do peptic ulcers occur outside the stomach and the duodenum (Gale, 2006). The disease is characterized by subsequent manifestation of mucosal damage in the gastric/duodenal epithelium, which develops as a result of the activity of pepsin and gastric acid.
Omeprazole is a substituted benzimidazole, i.e., 5-methoxy-2-[(4-methoxy-3,5-dimethylpyridin-2-yl)methylsulphonyl]-1H-benzimidazole. It is a potent inhibitor of gastric acid secretion by selectively inhibiting the gastric parietal cell proton pump (Young et al., 2001). It was the first proton pump inhibitor to be introduced to the market (Shin and Kim, 2013). This medicinal agent is widely used in the treatment of active duodenal ulcers, active benign gastric ulcers, gastroesophageal reflux disease, erosive esophagitis, Zollinger–Ellison syndrome, and other pathological hypersecretory conditions (Sweetman et al., 2005). It has a greater antisecretory activity than the H2 antagonists and is now more widely recommended (El-Sayed et al., 2007).
When omeprazole comes in contact with an acidic medium, there is a significant degradation of the drug and the bioavailability is consequently reduced. Hence, various oral formulations such as enteric-coated tablets and capsules (Thomson et al., 1997) have been developed to prevent drug release in the gastric region so as to optimize the oral bioavailability of the drug (Choi et al., 2000). With these, the absorption of a substantial amount of the drug takes place in the small intestine, and this is usually completed within 3–6 hours. Extended release and delayed release of proton pump inhibitors provide longer effective plasma concentrations of this class of drugs (Shin and Kim, 2013). It is, therefore, very beneficial to target the intestine for the delivery of omeprazole.
The duodenum and jejunum possess the greatest surface areas due to the highest concentration of villi and microvilli in these regions. Targeting the duodenum for drug delivery has benefits such as high surface area and short transit time which is less variable than those of the colon. Targeted drug delivery to the duodenum is most easily accomplished by circumventing drug release in the stomach by introducing pH-dependent solubility characteristics to the dosage form in the form of applying an outer enteric polymeric film coat or by dispersing the drug in a matrix with suitable swelling and solubility characteristics. It is, however, more economical to formulate a matrix tablet.
Since PRG used as a binder at concentrations of 8 and 10% produced tablets with drug release over a period of 5 hours (Selvi et al., 2010), formulating omeprazole tablets with relatively high and controlled concentrations of the gum could ensure delaying drug release and targeting the duodenum. In addition, our recent studies (Olorunsola et al., 2021) showed that a polymer could inhibit the gastric degradation of omeprazole. The aim of this research work, therefore, is to evaluate PRG as a matrix former for targeting the duodenum for the delivery of omeprazole.
MATERIALS AND METHODS
Materials
The materials used include omeprazole powder (Thode and Scobel, Hamburg, Germany), hydroxypropyl methylcellulose (E. Merk. Darmstadt, Germany), lactose (Riedel-de Haën AG, Seelze, Hannover), magnesium stearate (BDH Poole, England), talc (BDH Poole England), sodium hydroxide (FSA, Loughborough, England), hydrochloric acid (Qualikems, India), xylene (BDH Poole, England), ethanol (Thode and Scobel, Hamburg, Germany), and potassium dihydrogen phosphate (Guangdong Chemical Reagent Centre, China). PRG was obtained from P. africana seeds purchased from Karu Market, Abuja, and authenticated by the taxonomist in the Herbarium Unit of the Department of Biological Sciences, University of Abuja, Abuja, Nigeria. The gum was extracted as described earlier in the previous work of Olorunsola et al. (2014).
Omeprazole-prosopis gum compatibility study using Fourier transform infrared (FTIR) spectroscopy
Samples of PRG, omeprazole powder, and a simple Mixture of PRG and omeprazole were separately prepared in potassium bromide disks in a hydrostatic press at 6–8 Ton pressure. The FTIR spectra of the prepared samples were recorded at a scanning range of 500–5,000 cm−1 using a spectrophotometer (model 8400S, Shimadzu Corporation, Kyoto, Japan).
Formulation of omeprazole granules
Wet granulation was selected for the production of the tablets because it is known to produce stronger tablets than direct compression. One hundred tablets per batch were formulated based on the tablet formula in Table 1. The mixtures of omeprazole and lactose were mixed thoroughly before granulation with the addition of the binder [PRG or hydroxypropyl methylcellulose (HPMC)] previously dispersed in 4 ml of 95% ethanol. The wet mass was screened through a 2 mm sieve, and the formed wet granules were air-dried for 6 hours. The granules were further sieved using a 0.5 mm stainless steel sieve.
Characterization of the granules
Bulk density
The bulk volume of a 10 g sample of granules was determined by pouring into a graduated cylinder. The bulk density was calculated as the ratio of the weight to the volume occupied by the granules.
True density
The true density of the granules was determined using the liquid displacement method as described in the work of Olorunsola et al. (2021). An empty 25 ml pycnometer was weighed, and the weight was noted as W. It was filled with xylene and then reweighed, the new weight being taken as W1. The difference between W1 and W was calculated as the weight of xylene (W2). A 0.5 g sample of the granules (W3) was transferred into the pycnometer, and the excess liquid was wiped off. The final weight of the system was noted as W4. The true density Dt (g/cm3) of the granules was calculated as follows:
where W2/25 is equivalent to the density of xylene.
Granule porosity
The porosity of each batch of granules was determined using the following equation:
Compaction
The granules were mixed with the required amount of magnesium stearate and talc (as specified in Table 1). The mixture was compressed at a compaction pressure of 30 KN using a single-punch (12.5 mm diameter) automatic tableting machine (Model D63150, ERWEKA, Germany).
Quality Assessment of Tablets
Tensile strength
Five tablets were selected randomly from each batch, and their individual diameter and thickness were determined using a micrometer screw gauge. Each tablet was placed diametrically between the jaws of the Monsanto hardness tester, and the force needed to just crush the tablet was noted. The tensile strength was calculated using the following equation (Odeku et al., 2005):
where T is the tensile strength, F is the crushing force, d is the diameter, and t is the thickness of the tablet.
Friability
Friability was determined using a plastic chamber that revolves at a speed of 25 rpm, dropping the tablets at a distance of 6??” in each revolution. A sample of 10 preweighed tablets from each batch was dusted and placed in a Roche friability tester (Kumar Mfg. Ltd., India) and allowed to rotate for 4 minutes. The tablets were then removed, dusted, and weighed again. The friability of the tablet was calculated as percentage weight loss.
Tablet porosity
Five tablets were selected randomly from each batch, and their individual diameter and thickness were determined using a micrometer screw gauge. The tablet porosity was calculated using the following equation:
where m is the tablet’s weight, et is the particle density of the granules, r is the tablet’s radius, and h is the thickness.
Drug release studies
Dissolution test
In vitro drug release studies of the six prepared formulations of the tablets were conducted for a period of 60 minutes using a six-station USP basket-type apparatus (UNIMACH RCZ-6C3, China). A tablet from each batch was placed in a wire mesh basket suspended in a 900 ml pH 1.2 dissolution medium maintained at the temperature of 37 ±1ºC. The basket was rotated at a speed of 50 rpm, and the experiment was allowed for 60 minutes. Aliquots (10 ml) were withdrawn from each batch at 20 minutes intervals and filtered through Whatman filter paper No. 2. An equal volume of fresh dissolution medium was replaced to maintain the volume of the dissolution medium.
At time 60 minutes, the pH of the medium was raised to 5.5 using 1 N sodium hydroxide. Sample withdrawal with replacement was continued at 30 minute intervals over a period of 4 hhours. The absorbance of the filtered samples was determined at 302 nm using a spectrophotometer (Labomed Inc., USA). A graph of percentage drug release in the medium was plotted against time.
Determination of the release models
The drug release data were fitted into different models, zero order, first order, and Higuchi, to determine the kinetics of drug release and fitted into a Korsmeyer–Peppas model in order to determine the mechanism of drug release (Bangale et al., 2019; Varshosaz et al., 2006). For the zero-order kinetic model, cumulative% drug released was plotted against time, for the first-order kinetic model, log cumulative% drug remaining was plotted against time, for Higuchi’s model, cumulative% drug released was plotted against the square root of time, while for the Korsmeyer–Peppas model, log cumulative% drug released was plotted against log time.
Data analysis
Data obtained from the characterization of the granules and tablets were expressed as mean ± standard deviation. The statistical analysis was done using the one-way analysis of variance, followed by the Tukey–Kramer multiple comparison test using GraphPad Instat3 software. p-value < 0.05 was taken as being significant. Regression coefficients and diffusion exponent were determined from the various release models to establish the release characteristics of the formulations.
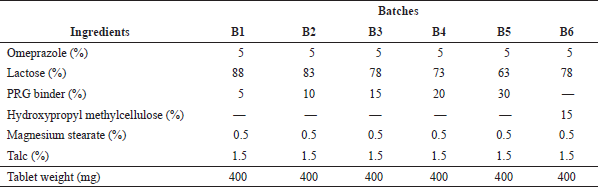 | Table 1. Tablet formula. [Click here to view] |
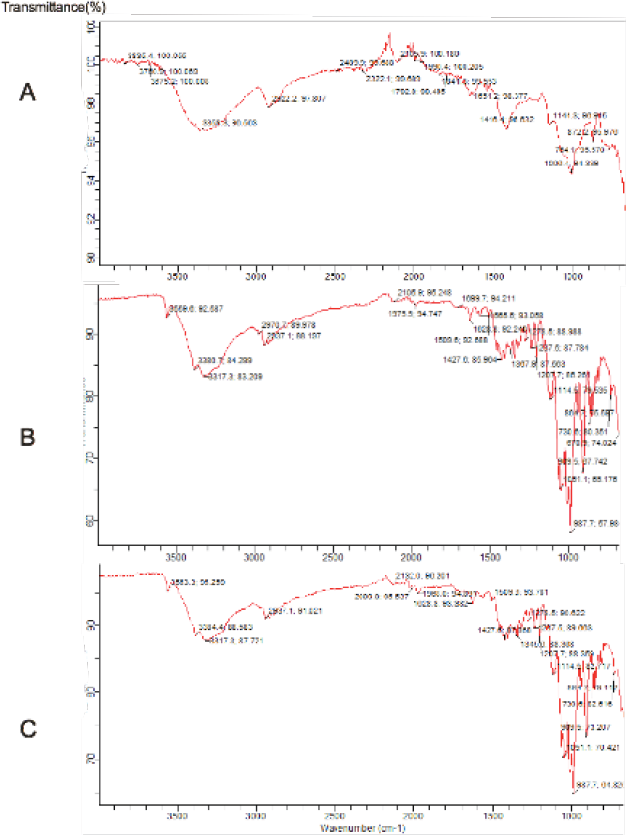 | Figure 1. Fourier transform infrared spectra of (a) PRG (b) Omeprazole and (c) PRG + Omeprazole. [Click here to view] |
RESULTS AND DISCUSSION
FTIR spectra
The FTIR spectra of PRG, omeprazole and PRG+ omeprazole are shown in Figure 1. In the spectrum of PRG, the peak at 1,006.4 cm−1 could be attributed to the bending vibration of C-H in the cyclic hydrocarbon; the peak at 3,358.3 cm−1 could be attributed to the O-H bending vibration, while that at 1,416.4 cm−1 could be assigned to the C-O bending vibration (Coutts, 2008).
The prominent peaks of omeprazole at 987.7 and 1,051.1 cm−1 could be assigned to the C-C stretching vibration. The absorption peaks at 3,317.3 and 3,380.7 cm−1 could be assigned to the O-H bending and N-H stretching vibrations (Coutts, 2008), showing omeprazole as a benzimidazole.
The FTIR spectra of omeprazole and PRG + omeprazole are almost similar. All the major peaks observed in the FTIR spectrum of pure omeprazole were also seen in the FTIR spectrum of PRG + omeprazole. However, there were slight changes in the location of these peaks.
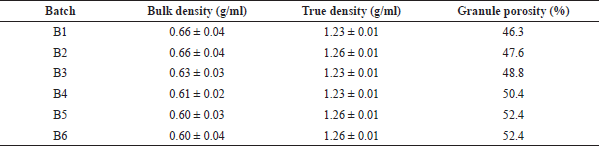 | Table 2. Micromeritic properties of the omeprazole granules. [Click here to view] |
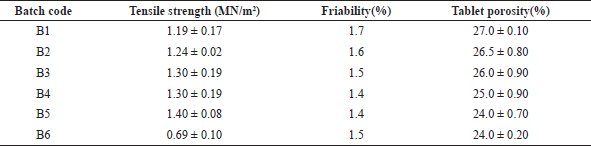 | Table 3. Physical properties of tablets. [Click here to view] |
Characteristics of the granules
The results of the micromeritic properties of the granules which were studied are presented in Table 2. The bulk density ranged from 0.60 to 0.66 g/ml, and the values were not significantly different. In the same manner, the true density of the granules ranged from 1.23 to 1.26 g/ml showing no significant difference. The porosity of the granules, however, increased with the increase in the concentration of the binder.
There was a significant difference between the bulk density and the true density of the granules. Since the bulk density takes into consideration the bulk volume while the true density only considers the volume occupied by the particles, the granules are generally characterized by high porosity as shown in Table 2. The increase in the porosity with the increase in the concentration of PRG can be attributed to the low density of this polymer. This observation is also consistent with the formation of larger granules as the concentration of the polymer increases leading to larger voids in between larger granules (Stanifort, 2002).
Physicomechanical properties of the tablets
Some physical and mechanical properties of the tablets are shown in Table 3. The tensile strengths of tablets containing PRG increased with the increase in the concentration of the gum used as a binder, and their values are significantly higher than those of the tablets containing hydroxypropyl methylcellulose. The friability of the formulations was in the range of 1.4% and 1.7%, and the porosity decreased with the increase in the concentration of PRG.
Higher concentrations of the polymer ensured higher tensile strength of the tablets. This is in agreement with the previously published works (Odeku et al., 2005; Olorunsola et al., 2017). Since a high tensile strength is required for a tablet to withstand the mechanical shocks during tablet manufacturing, packaging, and transportation, higher concentrations of the polymer will ensure better physical stability of the tablets. Tablet hardness affects parameters such as disintegration and dissolution of tablets (Quereshi et al., 2014). Since the purpose of the formulation is to design a drug product with drug release outside the stomach, a moderately high tensile strength will not have a negative impact on drug delivery.
There was an observed decrease in tablet friability as the concentration of the binder was increased which of course was due to the increasing binding strength of PRG. The friability values of all the formulations are outside the acceptable limit of less than or equal to 1%. Friability is a measure of the resistance of a tablet formulation to abrasion. It thus implies that even though the tablets have sufficient strength to withstand mechanical shock, the resistance to abrasion is not adequate. Since the tablets failed the friability test, it is necessary to modify the tablet composition and/or modify the compression parameters for the formulation to be considered for scale-up. The modification can be in the form of incorporation of an additional binder such as polyvinylpyrrolidone of high molecular weight or compaction at a higher pressure above the 30 KN used in the study.
There was approximately 50% decrease in the porosity as the granules were compressed to form tablets for all the batches. This is due to change in the volume occupied by the granules as they were compressed. Furthermore, tablets containing higher amounts of the binder were characterized by lower porosity because of better compaction.
In vitro dissolution
The dissolution profiles of the tablets are shown in Figure 2. Batch 1 which contains 5% PRG as binder was found to be the fastest to dissolve, with about 42% of the drug released in the simulated gastric fluid (pH 1.2) medium within an hour. This was followed by Batch 2 containing 10% PRG from which the amount of drug release within 1 hour was 34%. Batch 3 (containing 15% PRG) and Batch 6 (control) had an approximately equal amount of drug release of about 30% within 1 hour. The gastric residence time ranges from 5 minutes to 2 hours with the average residence time being 1 hour, while the small intestine transit time ranges from 3 to 4 hours (Ashford, 2018). This is the reason why the dissolution at pH 1.2 was allowed to take place for 1 hour, while that at pH 5.5 was allowed to take place for 4 hours. The rates of drug release at pH 1.2 for Batches 1 and 2 were fast, thereby leaving just about 58% and 66% of the drug, respectively, for release at pH 5.5; thus, the two formulations will not allow the release of a substantial amount of omeprazole at the targeted site (the duodenum).
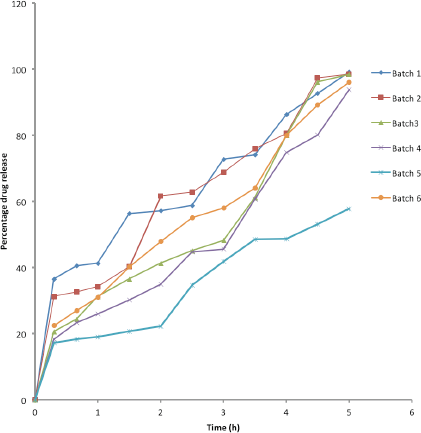 | Figure 2. Percentage drug release versus time (Batch 1 = 5% PRG, Batch 2 = 10% PRG, Batch 3 = 15% PRG, Batch 4 = 20% PRG, Batch 5 = 30% PRG, and Batch 6 = 15% HPMC). [Click here to view] |
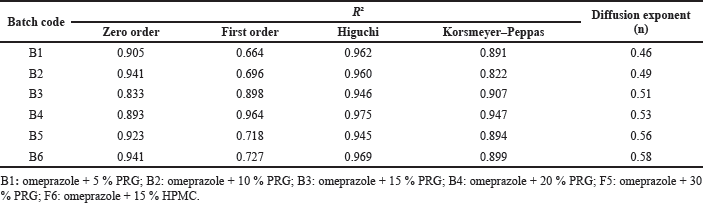 | Table 4. Regression coefficients (R2) and diffusion exponents (n) of the various batches. [Click here to view] |
On the other hand, B4 and B5 had greater levels of duodenum-targeted delivery properties as only 24% and 18% of the drug were released in the pH 1.2 dissolution medium at 1 hour with 76% and 82% of the drug reserved, respectively, for release in the pH 5.5 dissolution medium. The results showed that the amount of the drug that could reach the target site increased with the increase in the concentration of the binder.
There was a limitation for B5 because the drug was not completely released within 5 hours as at time 5 hours only 60% of the drug had been released. Conversely, there was complete drug release from B4 at time 5 hours. Hence, there was 76% drug release in the pH 5.5 dissolution medium for B4 (which contains 20% PRG) at time 5 hours which corresponds to the time the formulation would be leaving the duodenum. PRG, a hemicellulose, is solubilized at a higher pH above the gastric condition (Olorunsola et al., 2015). This attribute contributes to the duodenum-targeting ability of this polymer which swells at the gastric pH.
Release kinetics and mechanism of release of omeprazole from the tablets
The results of the in vitro release kinetics and mechanism of release of omeprazole tablets are presented in Table 4. The regression values for the zero-order, first-order, and Higuchi models for Batch 1 containing 5% of PRG were 0.905, 0.664, and 0.962, respectively. From the result, the Higuchi model showed the highest linearity with a regression value of 0.962; thus, the kinetics of the drug release followed the Higuchi model.
Similarly, the kinetics of drug release for the other five batches (B2, B3, B4, B5, and B6) followed the Higuchi model as the highest regression values were obtained in the Higuchi model for all the batches. Thus, drug release is proportional to the square root of time, indicating that the drug release is diffusion controlled.
The drug release data fitted into the Korsmeyer–Peppas model showed diffusion exponent (n) values of 0.46, 0.49, 0.51, 0.53, 0.56, and 0.58 for B1, B2, B3, B4, B5, and B6, respectively. The diffusion exponent derived from the Korsmeyer–Peppas plot is used for predicting the release mechanism of the formulations. It is often interpreted as Fickian transport (case I) for n ≤ 0.45, non-Fickian or anomalous transport for 0.45 < n < 0.89, case II transport (which often characterizes zero-order kinetics) for n = 0.89, and super case II for n > 0.89 (Bangale et al., 2019; Varshosaz et al., 2006).
All the batches are characterized by non-Fickian transport as their diffusion exponents are within the range 0.45 < n < 0.89. Also, there was an observed increase in the value of the diffusion exponent (n) as the polymer concentration increased, showing that the release mechanism shifted in the direction of anomalous transport.
CONCLUSION
Increase in concentration of PRG is characterized by improvement in the mechanical strength of the tablet. The work has shown that the gum, if used as a binder at concentrations of 20–30%, is suitable for targeting the duodenum for the delivery of omeprazole. The drug release from the formulations follows the Higuchi model, and the transport is non-Fickian, an indication of a combination of diffusion and erosion mechanisms. From the in vitro studies, the formulation is promising and could be considered for in vivo studies in a suitable animal model to assess the in vivo performance and bioavailability.
AUTHORS’ CONTRIBUTIONS
The conception of the work and its design and the drafting of the manuscript were carried out by Emmanuel O. Olorunsola, while data collection and analysis were done by Precious C. Olisakwe. Mfonobong F. Alozie and Musiliu O. Adedokun were involved in critical revision of the manuscript for the scientific content. All the authors gave final approval of the version of the manuscript being published.
CONFLICTS OF INTEREST
There are no conflicts of interest regarding the publication of this paper.
ACKNOWLEDGMENTS
This work was supported by the Nigerian Tertiary Education Trust Fund through the 2019 National Research Grant (Reference No. TETFund/DR&D/CE/NRF/STI/37/VOL1).
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included within this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation
REFERENCES
Agboola DA. Prosopis africana (Mimosaceae): stem, roots, and seeds in the economy of the savannah areas of Nigeria. Econ Bot, 2004; 58(suppl):34–42. CrossRef
Ashford M. Gastrointestinal tract – physiology and absorption. In: Aulton ME, Taylor KMG (eds.). Aulton’s pharmaceutics—the design and manufacture of medicines, Elsevier, China, pp 300–18, 2018.
Bangale GS, Shinde GV, Rajesh KS. Formulation and optimization by 32 factorial design for colon targeting. Global J Pharm Pharm Sci, 2019; 7(1):1–15.
Choi HG, Jung JH, Yung CS, Rhee CD, Lee MK, Han JH. Formulation and in- vivo evaluation of omeprazole buccal adhesive tablet. J Contr Rel, 2000; 68:405–12. CrossRef
Coutts RT. Infrared spectroscopy. In: Chatten LG (ed.). Pharmaceutical chemistry—instrumental techniques, CBS Publishers and Distributors PVT Ltd, New Delhi, India, pp 59–125, 2008.
El-Sayed A, Boraie NA., Ismail FA., El-Khordagui LK, Khalil SA. Assessment of pharmaceutical quality of omeprazole capsule brands marketed in Eygpt. Eastern Med Health J, 2007; 13(6):1427–37. CrossRef
Gale T. Ulcer (Digestive). Encyclopedia of medicine, 3rd edition. Emerald Group Publishing, Bingley, England, 2006.
Henciya S, Seturaman P, James AR, Tsai Y-H, Nikam R, Wu Y-C, Dahms H-U, Chang FR. Biopharmaceutical potentials of Prosopis app. (Mimosaceae, Leguminosa). J Food Drug Anal, 2017; 25(1):187–96. CrossRef
Najm WI. Peptic ulcer disease. Primary Care Clinics Office Pract, 2011; 38:383–94. CrossRef
Odeku OA, Awe OO, Popoola B, Odeniyi MA, Itiola OA. Compression and mechanical properties of tablet formulations containing corn, sweet potato and cocoyam starches as binders. Pharm Technol, 2005; 29:82–90.
Olorunsola EO, Bhatia PG, Tytler BA, Adikwu MU. Some chemical characteristics of novel hemicellulosic gums from seeds of Afzelia africana and Prosopis africana. West Afr J Pharm, 2014; 25(2):31–40.
Olorunsola EO, Bhatia PG, Tytler BA, Adikwu MU. Physicosurface properties of afzelia and prosopis hemicellulosic gums: potential surface active agents. IOSR J Pharm Biol Sci, 2015; 10(4 version 3):1–7.
Olorunsola EO, Adedokun MO, Bhatia PG, Tytler BA, Adikwu MU. Optimization of artesunate delivery by formulation in a delayed release prosopis hemicellulose matrix. J Appl Pharm Sci, 2017; 7(05):142–6.
Olorunsola EO, Udoh IE, Majekodunmi SO, Odiong IJ, Ebong UO. Inhibition of gastric degradation of omeprazole using a pH-sensitive polymer as a binder in tablet formulation. Int J Appl Pharm, 2021; 13(5):331–5. CrossRef
Quereshi J, Ijaz H, Sethi A, Zaman M, Bashir I, Hanif M, Danish Z, Azis M. Formulation and in vitro evaluation of sustained release matrix tablets of metoprolol tartrate using synthetic and natural polymers. Lat Am J Pharm, 2014; 33:1533–9.
Selvi RS, Gopalakrishanan S, Ramajayam M, Soman R. Evaluation of mucilage of Prosopis juliflora as tablet binder. Int J Pharm Pharm Sci, 2010; 2(3):157–60.
Shin JM, Kim N. Pharmacokinetics and pharmacodynamics of the proton pump inhibitors. J Neurogatroenterol Motil, 2013; 19(1);25–35. CrossRef
Stanifort J. Powder flow. In: Aulton ME (ed.). Pharmaceutics—the science of dosage form design. Churchill Livingstone, London, UK, pp 197–209, 2002.
Sung JJY, Kuipers ES, El-Serag, HB. Systematic review: The global incidence and prevalence of peptic ulcer disease. Alim Pharmacol Therap, 2009; 29(9): 938–46. CrossRef
Sweetman SC (ed.). Martindale—the complete drug reference. Pharmaceutical Press, London, UK, 2005.
Thomson AB, Kirdeikis P, Lastiwka R, Rohss K, Sindair P, Olofsson B. Pharmacokinetics and pharmacodynamics during treatment with omeprazole 20 mg enteric coated tablet and 20 mg capsule in asymptomatic duodenal ulcer patients. Int J Gastroenterol, 1997; 11:657–60. CrossRef
Varshosaz J, Tavakoli N, Kheirolahi. Use of hydrophilic natural gums in formulation of sustained-release matrix tablets of tramadol hydrochloride. AAPS PharmSciTech, 2006; 7(1) Article 24. CrossRef
Young CS, Jung JH, Rhee JD, Kook KC, Choi HG. Physicochemical characterization and evaluation of buccal adhesive tablets containing omeprazole. Drug Dev Ind Pharm, 2001; 27:447–55. CrossRef
Zarafi AB, Ayuba IS. Fungicidal activity of Prosopis africana and Anogeissus leiocarpus aqueous extracts against leaf blight of Jatropha curcas L. Arch Phytopathol, 2012; 45:413–22. CrossRef