
INTRODUCTION
Malaria is a deadly tropical disease caused by an infection of the protozoan Plasmodium, and it continues to be the most significant human parasitic illness in the world. It is a vector-borne illness. It spreads through the bites of female Anopheles mosquitoes infected with Plasmodium parasites [1]. Over the last century, despite the increased research efforts and control measures to drive down the malaria burden globally, eradication strategies and interventions have only been fairly successful. The parasite has co-evolved with new interventions, and eradication remains ongoing [2]. Plasmodium species have developed resistance to all known classes of antimalarial compounds and drugs which is one of the major challenges in the fight against malaria [3], in addition to inadequate research funding, healthcare professionals, facilities, research efforts in malaria-endemic regions, and limited knowledge about naturally acquired immunity to malaria [4,5].
Vaccination is an economical and successful solution to stop infectious diseases [6–10]. Advances have been made in blood-stage vaccine development, as they have reached clinical trials; however, they were unsuccessful in controlling human malaria infection on the field [11–13]. Antigen polymorphism, redundancy, parasite immune evasion, and low effectiveness of vaccine candidates have greatly hindered the rapid development of a licensed vaccine to neutralize malaria [14–18]. Furthermore, increasing insecticide resistance and asymptomatic infections have also been major setbacks [19,20], and cases of resurgence and increased malaria deaths have been reported in South American nations [21]. Vaccination has been identified as a critical therapeutic option for eliminating malaria; hence, it is a priority for sustained, substantial, and cost-effective control [22–24]. The only available licensed vaccine against malaria, RTS, S/AS01 shows low to modest efficacy in thwarting malaria caused by Plasmodium falciparum, which is enough to prevent clinical malaria but not enough for the global eradication of malaria [1]. Several other vaccines are currently under development, including the R21/Matrix-M (with 77% efficacy in preliminary trials), P. falciparum sporozoite, and the self-amplifying RNA vaccine. However, no vaccine with 100% efficacy remains available [25–27].
Post-genomic era vaccine development often involves preliminary computational analysis using bioinformatics tools to predict protective antigens rather than experimental studies with the pathogen, as in conventional vaccinology. Antigen characterization using in silico strategies and bioinformatics tools is crucial to the protein-based vaccine design and development [28]. Reverse vaccinology is named so because the vaccine discovery process employs computational methods to analyze genomic data instead of wet lab experimental studies with a pathogen, as with conventional vaccinology [29]. This technique has gained increasing global popularity and usage by research groups in the last 20 years for vetting the whole genome for vaccine antigens against several pathogens including Leishmania [30], Lassa fever [31], Dengue [32], Schistosomiasis [33], and P. falciparum [34], among others. This strategy is effective as it saves cost and time, and decrease the risk of failure compared to conventional vaccinology [35].
The World Health Organization set the goal to eradicate malaria in 1948, and efforts have been intensified to this end. Correspondingly, malaria-related fatalities progressively decreased, but this progress stalled due to the COVID-19 pandemic and attributed to the interference with malaria services and diagnostic practices [36]. In 2021, the number of malaria cases worldwide was recorded to be 241 million, with 627,000 related deaths [37]. In light of the available data and the current state of global malaria burden, this research aimed to develop a potent multi-epitope blood stage subunit vaccine candidate against P. falciparum by integrating epitopes from the most immunogenic regions of three merozoite surface proteins namely, liver-stage antigen 3 (LSA3-C), merozoite surface antigen 180 (MSA 180), and merozoite surface antigen 10 (MSA 10).
METHODOLOGY
A systematic flow chart was followed stepwise to identify immunodominant B- and T-cell epitopes and the design of a subunit vaccine via immunoinformatics techniques. A subsequent biophysical study was performed using integrated docking, immune simulation, and molecular docking. Finally, codon optimization was executed to ensure optimal expression in the microbial host (Fig. 1).
Amino acid sequence retrieval
To retrieve the primary sequence of liver stage antigen 3, MSA 180, and merozoite surface protein 10, the PlasmoDB [38] server was employed. However, only the most immunogenic regions of these proteins–(LSA3-C; V750-K1433), merozoite surface antigen 180 truncate-4 (MSA 180-T4; A805-P1093), and merozoite surface protein 10 region 1 (MSP10 R1; D29-N188) were further exploited for the vaccine construct (VC), following results from prior studies [39–41].
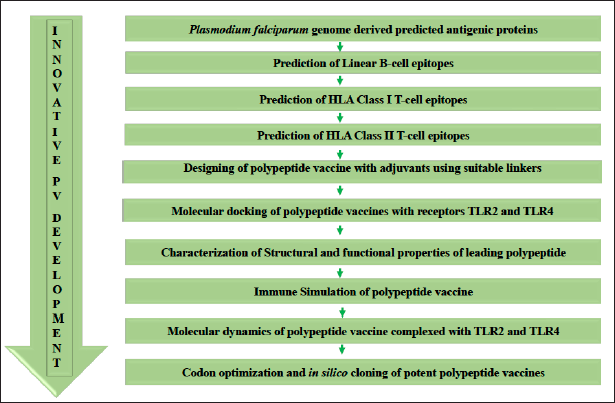 | Figure 1. Methodology flow chart showing the step-by-step procedures used. [Click here to view] |
Forecast of B-cell epitopes
Using default parameters for both, two servers were used to forecast the B-cell epitopes, including ABCpred [42] and BepiPred 2.0 [43]. Matching epitopes forecasted by both servers were selected. Furthermore, finalized epitopes were vetted for antigenicity (VaxiJen 2.0 server) [44], allergenicity (AllergenFP v.1.0) server [45], toxicity (ToxinPred server) [46], and conservancy (Protein Blast - NCBI). For conservancy, the epitopes were screened against Homo sapiens proteome (taxid: 9606), with nonhomology indicated by e-value scores ≥0.05 [47].
Forecast of CD4+ and CD8+ T-cell epitopes
The NetMHCIIpan 4.0 server [48] was utilized to identify helper T lymphocyte (HTL) epitopes with a threshold of 1% (strong binding) and 5% (weak binding). Twelve (12) major histocompatibility complex (MHC) supertypes available on the NetCTL 1.2 server were screened to predict CD8+ epitopes, with default parameters [49,50]. Also, the predicted epitopes were screened for antigenicity (VaxiJen 2.0 server) and immunogenicity (IEDB server), set at a threshold of 0.4 and default parameters respectively.
Designing of polypeptide subunit vaccine
The finalized B- and T-cell epitopes were integrated using several linkers, including the EAAAK (L1), AAY (L2), GPGPG (L3), and KK (L4) linkers. The adjuvant (ADV) RS09 was included at the N-terminals of the VC and is joined to the CD8+ epitopes by the EAAAK linker. The CD8+ epitopes were intralinked through the AAY linker and interlinked with the CD4+ epitopes by the GPGPG linker, which also intralinks the CD4+ epitopes. KK linkers interlink the CD4+ and B-cell epitopes and intralink the B-cell epitopes. The final VC comprises a His-tag (HHHHHH) at the C-terminal and is arranged ADV-CTL epitopes-HTL epitopes-B-cell epitopes–6X His tag.
Secondary and tertiary structure prediction, evaluation, and validation
Psipred [51] and RaptorX [52] servers were employed to forecast the secondary organization of the multi-epitope construct. Robetta [53], which creates three-dimensional (3D) models by employing ab initio and comparative modeling simulations, was used to predict the constructs’ tertiary structure [54]. Pymol 2.5 visualization software was used to envision the protein’s 3D organization. Subsequent refinement to improve the 3D model was evaluated using the GalaxyWEB [55] server.
The ProSA-web server [56] and Saves v.6.0 [57] server were employed to ascertain the quality of the 3D models. The Z-scores of each protein solved experimentally using data from X-ray and nuclear magnetic resonance sources are presented in a plot by ProSA. It determines whether the z-score of the 3D model of the query protein falls within the normal range for scores for native proteins of comparable size. The Ramachandran plot examines the model’s phi/psi distribution and C-beta deviation to determine the backbone (C-alpha) geometry [58].
Conformational B-cell epitopes
The VCs tertiary structure was put in the Ellipro server, which forecasts and visualizes discontinuous B-cell epitopes [59].
Structural and functional characterization of vaccine construct
The ExPASy-ProtParam tool [60] was used to estimate the physicochemical parameters of the VC. Furthermore, the antigenic and allergenic properties were predicted.
Molecular docking and immune simulation of vaccine construct
Toll-like receptors (TLR)-2 and -4 were selected as effective immunological targets for provoking the innate immune system. The ClusPro 2.0.php server was employed in accessing the interaction between the TLRs (receptor) and VC (ligand) [61,62]. The C-IMMSIM web server, which uses machine learning techniques and a position-specific scoring matrix, was utilized to determine the immune profile of the VC for 3 injections administered 4 weeks apart [63,64]. Default parameters were set with 1, 84, and 170 specified time steps [65].
Molecular dynamics (MD) simulation of vaccine construct-TLR complexes
The iMODS server [66] predicts the MD, which uses the normal mode analysis (NMA) in internal coordinates to delineate the collective protein motions.
Codon optimization and in silico cloning of vaccine construct
To forecast a viable option for the expression and isolation of the VC, the JCat server [67] was used to improve the construct’s nucleotide sequence to commonly utilized codons of the Escherichia coli K 12 strain to enable optimal protein production by the expression host. Also, in silico cloning requires the SnapGene software to incorporate restriction sites (EcoRI and BamHI), after which the genetic sequence is integrated into the vector pET-28a(+).
RESULTS AND DISCUSSION
Proteins that encrust the extracellular surface of merozoite and are released from its specialized secretory apical organelles are considered potential vaccine candidates for the erythrocytic stage malaria [68]. Robust experimental evidence supports the importance of these surface proteins to host cell invasion, parasite growth, and survival and as potential preventive measures against P. falciparum malaria in humans [11,40,69–75]. In this study, three experimentally validated vaccine candidates against falciparum malaria were selected for further downstream immunoinformatics analysis.
The LSA3-C, MSA 180-T4, and MSP10 R1 are regions of blood-stage P. falciparum proteins that possess characteristics which have been experimentally validated to be malaria vaccine candidates, and this makes them attractive targets for developing a sub-unit vaccine [39–41]. Liver-stage antigen 3 (PF3D7_0220000) is a ~175kDa protein expressed in liver and blood stages on sporozoites surface, infected hepatocytes, and blood-stage merozoites, and several research groups have reported it to be a viable pre-erythrocytic and blood stage vaccine target [39,76–78]. Furthermore, LSA3 was reported to be highly conserved following genomic sequence analysis of 20 P. falciparum clinical isolates from diverse geographical regions and localized in the parasitophorous vacuole during the ring-stage following merozoite ingress [78,79]. Previous studies on the full-length and discrete regions of LSA3-C have reported its viability as a pre-erythrocytic and blood-stage vaccine candidate in addition to being the most immunologic antigen segment [39,78,79].
Plasmodium falciparum merozoite surface antigen 180 (PfMSA180) is a 170kDa protein that is essential and conserved in all Plasmodium sp.; it is expressed on the periphery of merozoites and has been implicated in merozoite ingress and egress during the asexual blood stage of the parasite’s lifecycle [41,80]. PfMSA180 (PF3D7_1014100) has been reported to be critical for parasite invasion of the erythrocyte, and antibodies against the C-terminal region of PfMSA180 [MSA 180 Tr-4 (A805-P1093)] abrogated merozoite invasion in vitro and conferred protective immunity against malaria [41]. Furthermore, the PfMSA180 (PfMSA180-Tr4) C-terminal region was reported to be highly conserved across isolates, interacts with the red blood cell (RBC) surface protein-CD47, and stimulates antibodies that abrogate parasite invasion; hence, Nagaoka et al. [41] proposed MSA 180 Tr-4 as a potential vaccine candidate. PfMSP10 (PF3D7_0620400) is an 80 kDa protein that directly interacts with PfGAMA, which is key for erythrocyte invasion. The PfMSP10 R1 (D29-N188) region was hypothesized to be the interacting region [40]. Also, bioinformatics genome-wide screening has predicted PfMSP10 to be a putative vaccine candidate [81].
Prediction of linear B-cell epitopes
B-cells comprise one of the two main types of cells in the adaptive immune system. Their epitopes are antigenic components, which trigger the synthesis of antibodies [82]. To overcome the challenge of prediction inaccuracy, we employed two servers for better B-cell epitope mapping. The results were compared, and consistent epitopes with both servers were selected. Also, nonantigenic, allergenic, toxic, and conserved epitopes in humans were discriminated against. The top three epitopes from each vaccine candidate that passed the screening (antigenicity, allergenicity, toxicity, and conservancy) for inclusion in the VC were selected (Table 1) from the epitopes with scores ≥ 0.52 (Table S1).
CD4+ T-cell epitopes prediction
MHC II encrusts the periphery of antigen-presenting cells (APCs). These MHCs exhibit nonself-peptides to helper T-cells, which coordinates other immune responses [83]. Hence, it is essential to predict epitopes with a higher likelihood of being displayed by the MHC-II molecule. Strong interaction between the HTL epitope and HLA-DR is critical for epitope immunogenicity, and excellent HTL epitope candidates are expected to interact optimally with numerous HLA alleles [84,85]. To this end, LSA3-C, MSA-Tr4, and MSP10 R1 were subjected to CD4+ epitope prediction, and only the epitopes that can bind to three or more HLA-DR alleles were agreed upon [86]. The top three epitope sequences for each are included as shown in Tables 2 and S2.
CD8+ T-cell epitopes prediction
APCs and infected RBCs cells display peptides of pathogen origin on MHC I molecules expressed on their surface to cytotoxic T-cells, facilitating clearance and immunological memory [87]. LSA3-C, MSA-Tr4, and MSP10 R1 were screened for their MHC-1 epitopes using all 12 MHC-I supertypes available on the NetCTL1.2 server. Table 3 shows the finalized epitopes after the proteins were subjected to further screening parameters, including; antigenicity, allergenicity, immunogenicity, and conservancy predictions.
Polypeptide subunit vaccine design
The finalized B- and T-cell epitopes were integrated using suitable linkers alongside two ADVs. ADVs present a classical approach to targeted delivery of subunit vaccines and maximizing protective immunity, as protein vaccine candidates are poorly immunogenic [88]. They bridge the antigens with the APCs by projecting visibility of the otherwise non or weakly immunogenic antigen to the immune cells to trigger a better immune response [89]. RS09, a synthetic TLR4 agonist comprising seven amino acid residues [89], was incorporated as ADVs at the N-terminal of the VC [90,91]. The EAAAK linker increases the consistency of the overall structure and limits protein components from joining through effective detachment; hence, it was used to join the ADV with the polypeptide vaccine [92]. The AAY linker improves the vaccine’s ability to elicit an immunological response [93]. The need to impinge the development of junctional epitopes and improve epitope presentation necessitates the GPGPG linkers [94]. The KK linker was used to connect linear B-cell epitopes so that their self-regulating immunogenic responses may be preserved [95]. The VC follows the sequence “ADV-CD8+ epitope-CD4+ epitopes-B-cell epitopes- 6X His tag,” joined by appropriate linkers to form a 437 amino acid-long peptide as illustrated in Figure 2.
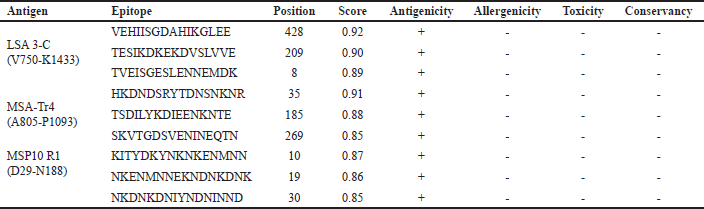 | Table 1. Selected linear B-cell epitopes with their antigenicity, allergenicity, toxicity, and conservancy. [Click here to view] |
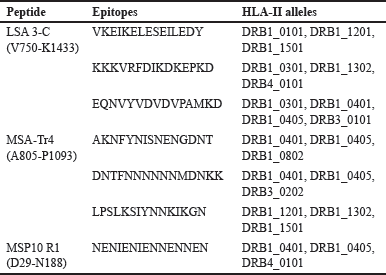 | Table 2. Details of selected CD4+ T-cell epitopes predicted by NetMHCIIpan 4.0. [Click here to view] |
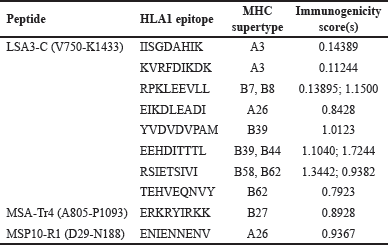 | Table 3. Selected CD8+ T-cell epitopes with MHC supertypes and immunogenicity score. [Click here to view] |
Secondary and tertiary structure prediction, evaluation, and validation
The VCs comprise 26% α-helix, 7% β-strand, and 66% coil, as predicted by Psipred, and RaptorX in plots representing the structural moieties (Fig. 3). Also, 76% was forecasted to be exterior, 8% mildly exterior, and 14% to be buried in relation to the availability of amino acid residues. The α-helix and β-turn have internal localization within the protein and are less likely to act as epitopes; instead, they maintain the structure of the protein due to the high chemical bond energy they contain. Random coils are exposed on the protein surface, have fewer rigid regions of the protein, and may be probable epitopes [96]. Over half the overall structure of the construct comprises random coils, hinting at the greater potential of the protein to trigger immune responses. The 3D structure was predicted by the Robetta server, of which five models were generated, and model 1 was selected as it had higher comparative model quality following subjection to the Saves v.6.0 server for the ERRAT and Ramachandran plot analysis. GalaxyRefine server was used to refine and improve the quality of the selected model (Fig. 4A). Structural validation involves the identification of probable faults in the forecast 3D structure [97]. The model had a Z-score of -7.37, predicted by ProSA-web (Fig. 4B). Also, to determine the nonbonded atom-atom interactions, the ERRAT server was utilized [57], which revealed an overall quality factor of 94.72, suggesting a high quality. The Ramachandran plot enables the visualization of energetically favorable and unfavorable dihedral angles psi (ψ) and phi (?) of amino acids, which delineates the stereochemical quality of the 3D structure [98]. The Ramachandran plot analysis revealed that 93.8% of the initial model was in the favored regions, 5.2% in the allowed regions, and 0.8% in the disallowed regions. However, the refinement process improved the protein quality to 95.3% favoured, 3.4% allowed, and 0.5% disallowed regions, representing a very good model (Fig. 4C and D).
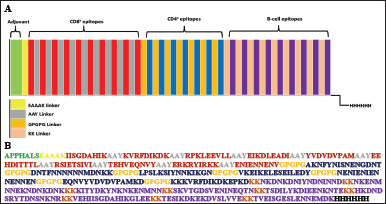 | Figure 2. (A) Schematic diagram of polypeptide vaccine. It comprises several segments: an ADV, RS09 (TLR4 agonists), 10 CD8+ epitopes, 7 CD4+ epitopes, 9 B-cell epitopes, and a His tag. These segments are integrated by EAAK, AAY, GPGPG, and kk linkers. (B) Sequence of polypeptide vaccine. [Click here to view] |
Conformational B-cell epitopes
B-cell epitopes usually form discontinuous epitopes resulting from the spatial proximity of a bunch of amino acid residues on the tertiary structure of the antigen [99]. Ellipro forecasted 4 epitopes with a maximum score of 0.758 and a minimum score of 0.512, and the number of epitope residues ranged from 3 to 81 (Table S3). Figure 5 reflects the structure of the epitopes in relation to the VC.
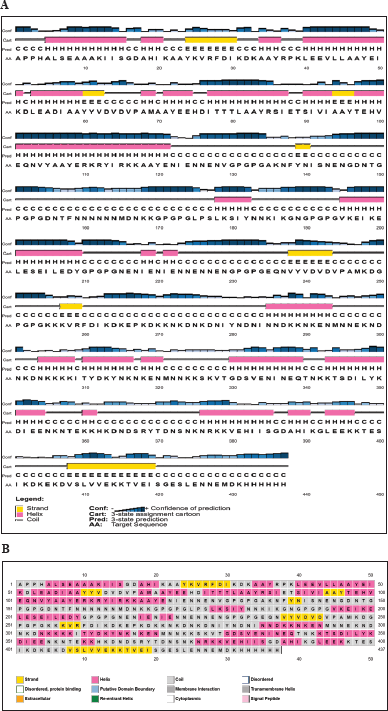 | Figure 3. Schematic diagram of the secondary structure of VC. (A) Predicted secondary structure (H: α-helix, E: β-strand, C: coil) by PSIPRED. The bar chart represents the percentage of confidence. (B) RaptorX generated the chimeric protein’s secondary structure, and the results show a structure made up of 26% α-helix, 7% β-strand and 66% coil. [Click here to view] |
Structural and functional characterization of VC
The physicochemical properties prediction revealed that the protein has a molecular weight and theoretical pI of 49.2 kDa and 5.99, respectively. The half-life was predicted to be 4.4 hours (mammalian reticulocytes, in vitro), >20 hours (yeast, in vivo), and >10 hours (Escherichia coli, in vivo). The instability index and GRAVY were forecasted to be 39.36 and −1.294, respectively. The score from the instability index suggested that the protein is stable. The GRAVY score represented that the protein is hydrophilic, which is a desired quality for a vaccine as it indicates the ability to trigger an elevated humoral immune response [100]. The aliphatic index was predicted to be 61.40, reflecting thermostability (Table S2).
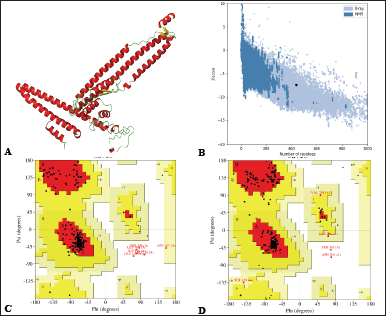 | Figure 4. (A) 3D structure cartoon representation (Red, green, and yellow represent α-helix, coil, and β-sheet). (B) ProSA map of the sub-unit vaccine with Z-score of −7.37. (C) Ramachandran plot before protein refinement with 93.8% in the favored region, 5.2% allowed region, 0.8% in the disallowed region. (D) Ramachandran plot after protein refinement with 95.3% favored region, 3.4% allowed region, and 0.5% disallowed region. [Click here to view] |
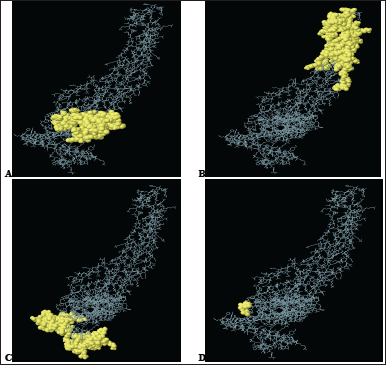 | Figure 5. 3-D representation of the four conformational B-cell epitopes. (A–D) A yellow surface represents the epitopes. [Click here to view] |
Molecular docking of vaccine construct with toll-like receptors
The contact between ligand and receptor molecules is embroiled in molecular docking to generate a stable ligand-receptor product [101]. The interaction between antibodies and their targeted antigens is critical to the humoral immune response to aid pathogen elimination. The VC-TLR2 complex has a total of 81 members in its cluster, while VC-TLR4 has 72 members in its cluster. The lowest energy score and binding affinity of −945.1 and −919.8 kcal/mol were obtained for the VC-TLR2 and VC-TLR4 complexes, respectively (Table 4). These complexes were stable as shown in Figure S1.
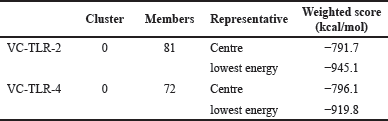 | Table 4. Molecular docking score between VC and TLR-2 and -4. [Click here to view] |
Immune simulation of vaccine construct
The immune simulation was performed to determine the natural immune response to the VC. The immune simulator C-ImmSim tool was employed to simulate the natural responses formed by the immune system. Figure 6 indicated that the antigenic recognition and desired corresponding immune responses would manifest. There was a marked increase in IgM and IgG production and the expansion of Helper T-cells due to memory development following primary immunization. There was also a depletion in antigen concentration, reflecting a rise in memory B-cell production (Fig. 6A–D). Figure 6F shows an elevation in the concentration of cytotoxic T-cells following vaccine administration. Similarly, there is an increase in the proliferation of cytokines, including IFN-γ, TGF- β, IL-10, and IL-12 (Fig. 6I), indicating that the vaccine is capable of provoking the immune response to act against malaria.
MD simulation of vaccine construct-TLR complexes
MD simulation is useful for analyzing the stability of protein-ligand complex by incorporating Newton’s laws of motion [102]. It was done by enumerating protein dynamics to their normal modes using the iMODS server [103]. NMA is commonly used to evaluate the collective functional motions of docked protein-protein complexes [66]. The 3D interaction between VC-TLR2 and VC-TLR4 complexes is presented, with the arrows representing the direction of amino acids (Fig. 7A and B). The peaks on the deformability graph delineate deformability, as higher peaks depict higher deformability. From the result, the deformability plots report the stability of the complexes with individual amino acid residues having a lesser likelihood of deforming (Fig. 7C and D). The B-factor graph comparatively evaluates the NMA and the protein data bank field of the docked complexes (Fig. 7E and F). The eigenvalue corresponds with motion stiffness; a lower value is congruent with higher deformability and vice versa. The predicted high values of 9.12572e-07 and 1.117255e-06 for VC-TLR2 and VC-TLR4, respectively, reflect the less deformability and high stability of both complexes (Fig. 7G and H). The variance corresponding to each normal mode is inversely proportional to the eigenvalue (Fig. 7I and J) [104]. The covariance matrix reflects if the pairs of residues’ motions were correlated (red), uncorrelated (white), or anti-correlated (blue) (Fig. 7K and L). The elastic network graph reflects atom pairs joined by springs, which are correspondingly colored by the degree of stiffness. The lighter grey area corresponds to less rigid springs and vice versa (Fig. 7M and N).
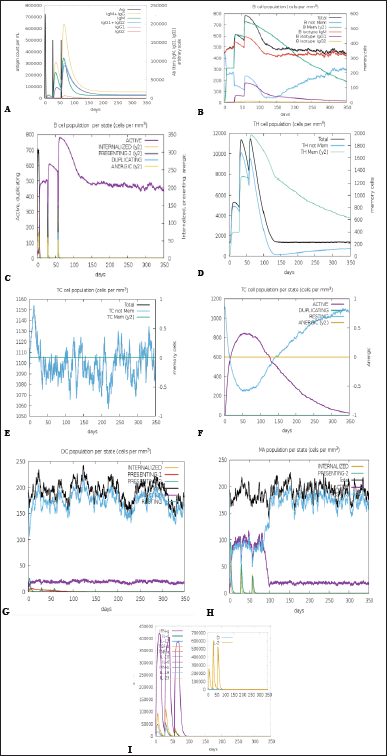 | Figure 6. Characterisation of the immune profile of the construct. (A) The antibody production denotes a rise in immune response following the vaccine shot. Antibody subtypes (IgM, IgG1, and IgG2) are depicted as colored peaks. (B) The active B-cell population is observed with the vaccine shots. (C) B-lymphocyte population per entity-state (D) CD4+ T-helper lymphocytes count sub-divided per entity-state (E) The generation of cytotoxic-T cells. (F) CD8+ T-cytotoxic lymphocytes count per entity-state (G) Dendritic cells for MHC class I and II. Shows the total number of active, resting, internalized, and presenting antigen (H) Macrophages. Total count, internalized, presenting on MHC class-II, active and resting macrophages (I) Concentration of cytokines and interleukins. [Click here to view] |
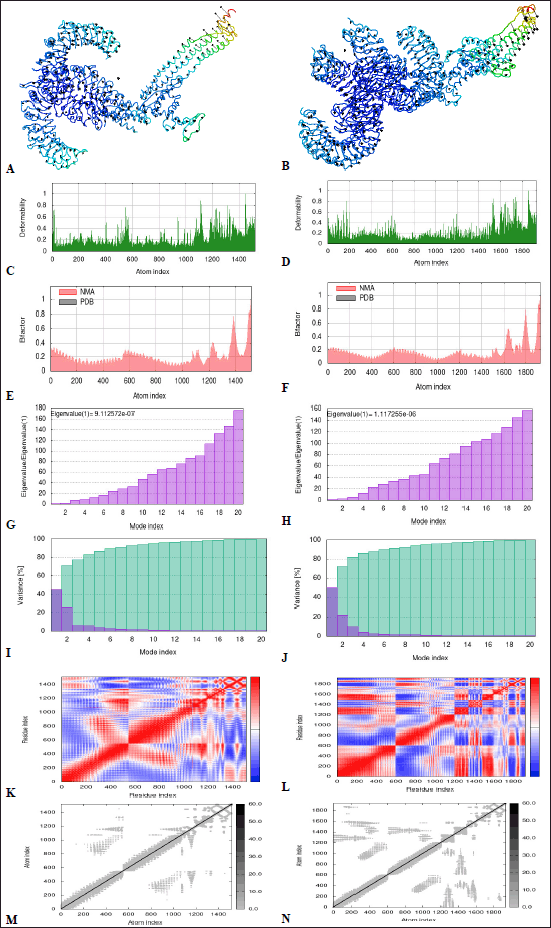 | Figure 7. (A, B) NMA mobility, (C, D) deformability, (E, F) B factor, (G, H) Eigenvalue, (I, J) variance map, (K, L) Covariance map and (M, N) Elastic network graph of docked complexes VC-TLR2 and VC-TLR4, respectively. For the covariance map, red: correlated, white: uncorrelated and blue: anti-correlated motions, while for the elastic network graph of docked complexes, darker grey regions correlate with stiffer regions. VC: Vaccine construct. [Click here to view] |
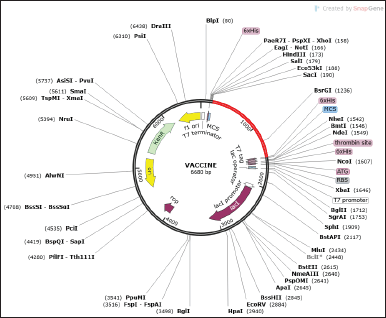 | Figure 8. In silico cloning of the leading VCs. The red fragment designates the inserted vaccine and the pET-28a(+) vector is presented in a black circle. [Click here to view] |
Codon optimization and in silico cloning of VC
Successful cloning and expression of the designed VC are critical for developing a vaccine since a poorly optimized codon could translate to a minimal expression rate in the host. The improved DNAs were 1,311 nucleotides long. A good guanine-cytosine (GC) value ranges between 30 and 70, while the Codon adaptation index (CAI) above 0.5 is good. Our chimeric proteins have good GC and CAI values of 42.94 and 0.99, hinting at a high likelihood of good expression in the prokaryotic host. Finally, the optimized sequence (highlighted in red) was incorporated into the E. coli pET-28a(+) vector for optimal gene expression (Fig. 8).
CONCLUSION
A multi-immune inducer that targets several antigens or various points of the parasite’s entire life cycle may effectively engender robust clinical protection against malaria. To the best of our knowledge, this is the first study to use short regions rather than full-length proteins of experimentally validated vaccine candidates to contrive an antibody-inducing multi-epitope subunit vaccine targeting the blood stage of falciparum malaria. This is important because regions of vaccine candidates that have been experimentally validated to be most immunogenic will be most attractive for mapping immunodominant epitopes capable of being integrated to form a potent subunit vaccine. The VC demonstrated the ability to evoke combined innate and adaptive immune responses via interaction with the TLR-2 and -4, triggering cellular and humoral immunity in addition to forming a stable VC-receptor complex. The limitation of this study is that it is completely performed in silico. Therefore, an experimental study is necessary to determine the full potential of the proposed vaccine as a possible intervention in neutralizing malaria.
ACKNOWLEDGMENT
Covenant Applied Informatics and Communication Africa Centre of Excellence (CApIC-ACE) scholarship to COM is acknowledged. The deference of APC by Covenant University Centre for Research, Innovation and Discovery is appreciated
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took p art in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Arora N, Anbalagan LC, Pannu AK. Towards eradication of malaria: is the WHO’s RTS,S/AS01 vaccination effective enough? Risk Manag Healthc Policy. 2021;14:1033–9. CrossRef
2. Henry NB, Sermé SS, Siciliano G, Sombié S, Diarra A, Sagnon N, et al. Biology of Plasmodium falciparum gametocyte sex ratio and implications in malaria parasite transmission. Malar J. 2019;18:70. CrossRef
3. Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–5. CrossRef
4. Adeyemo AO, Aborode AT, Bello MA, Obianuju AF, Hasan MM, Kehinde DO, et al. Malaria vaccine: the lasting solution to malaria burden in Africa. Ann Med Surg. 2022;79:104031. CrossRef
5. Kanoi BN, Nagaoka H, Morita M, Tsuboi T, Takashima E. Leveraging the wheat germ cell-free protein synthesis system to accelerate malaria vaccine development. Parasitol Int. 2021;80:102224. doi: 10.1016/j.parint.2020.102224
6. Miquel-Clopés A, Bentley EG, Stewart JP, Carding SR. Mucosal vaccines and technology. Clin Exp Immunol. 2019;196:205–14. CrossRef
7. Cantini F, Savino S, Scarselli M, Masignani V, Pizza M, Romagnoli G, et al. Solution structure of the immunodominant domain of protective antigen GNA1870 of Neisseria meningitidis. J Biol Chem. 2006;281:7220–7. CrossRef
8. Chaudhuri R, Ahmed S, Ansari FA, Singh HV, Ramachandran S. MalVac: database of malarial vaccine candidates. Malar J. 2008;7:184. CrossRef
9. Talukdar S, Zutshi S, Prashanth KS, Saikia KK, Kumar P. Identification of potential vaccine candidates against Streptococcus pneumoniae by reverse vaccinology approach. Appl Biochem Biotechnol. 2014;172:3026–41. CrossRef
10. Monterrubio-López GP, González-Y-Merchand JA, Ribas-Aparicio RM. Identification of novel potential vaccine candidates against tuberculosis based on reverse vaccinology. BioMed Res Int. 2015;2015:1–16. CrossRef
11. Burns AL, Dans MG, Balbin JM, de Koning-Ward TF, Gilson PR, Beeson JG, et al. Targeting malaria parasite invasion of red blood cells as an antimalarial strategy. FEMS Microbiol Rev. 2019;43:223–38. CrossRef
12. Draper SJ, Angov E, Horii T, Miller LH, Srinivasan P, Theisen M, et al. Recent advances in recombinant protein-based malaria vaccines. Vaccine. 2015;33:7433–43. CrossRef
13. Ogutu BR, Apollo OJ, McKinney D, Okoth W, Siangla J, Dubovsky F, et al. Blood stage malaria vaccine eliciting high antigen-specific antibody concentrations confers no protection to young children in Western Kenya. PLoS One. 2009;4:e4708. CrossRef
14. Ferreira MU, da Silva Nunes M, Wunderlich G. Antigenic diversity and immune evasion by malaria parasites. Clin Vaccine Immunol. 2004;11:987–95. CrossRef
15. Mahajan RC, Farooq U, Dubey ML, Malla N. Genetic polymorphism in Plasmodium falciparum vaccine candidate antigens. Indian J Pathol Microbiol. 2005;48:429–38.
16. Matuschewski K. Vaccines against malaria-still a long way to go. FEBS J. 2017;284:2560–8. CrossRef
17. Stiepel RT, Batty CJ, MacRaild CA, Norton RS, Bachelder E, Ainslie KM. Merozoite surface protein 2 adsorbed onto acetalated dextran microparticles for malaria vaccination. Int J Pharm. 2021;593:120168. CrossRef
18. Takala SL, Coulibaly D, Thera MA, Batchelor AH, Cummings MP, Escalante AA, et al. Extreme polymorphism in a vaccine antigen and risk of clinical malaria: implications for vaccine development. Sci Transl Med [Internet]. 2009 [cited 2022 Sep 30];1:2ra5. CrossRef
19. Frimpong A, Kusi KA, Ofori MF, Ndifon W. Novel strategies for malaria vaccine design. Front Immunol. 2018;9:2769. CrossRef
20. Adedeji EO, Ogunlana OO, Fatumo S, Beder T, Ajamma Y, Koenig R, et al. Anopheles metabolic proteins in malaria transmission, prevention and control: a review. Parasit Vectors. 2020;13:465. CrossRef
21. Espinoza J. Malaria resurgence in the Americas: an underestimated threat. Pathogens. 2019;8:11. CrossRef
22. Chandramohan D, Zongo I, Sagara I, Cairns M, Yerbanga RS, Diarra M, et al. Seasonal malaria vaccination with or without seasonal malaria chemoprevention. N Engl J Med. 2021;385:1005–17. CrossRef
23. Penny MA, Verity R, Bever CA, Sauboin C, Galactionova K, Flasche S, et al. Public health impact and cost-effectiveness of the RTS,S/AS01 malaria vaccine: a systematic comparison of predictions from four mathematical models. Lancet. 2016;387:367–75. CrossRef
24. World Health Organization. World malaria report 2019 [Internet]. Geneva, Switzerland: World Health Organization; 2019 [cited 2022 Sep 23]. Available from: https://apps.who.int/iris/handle/10665/330011
25. Coulibaly D, Kone AK, Traore K, Niangaly A, Kouriba B, Arama C, et al. PfSPZ-CVac malaria vaccine demonstrates safety among malaria-experienced adults: a randomized, controlled phase 1 trial. EClinicalMedicine. 2022;52:101579. CrossRef
26. Datoo MS, Natama HM, Somé A, Bellamy D, Traoré O, Rouamba T, et al. Efficacy and immunogenicity of R21/Matrix-M vaccine against clinical malaria after 2 years’ follow-up in children in Burkina Faso: a phase 1/2b randomised controlled trial. Lancet Infect Dis. 2022;22(12):1728–36. CrossRef
27. Hussain A, Yang H, Zhang M, Liu Q, Alotaibi G, Irfan M, et al. mRNA vaccines for COVID-19 and diverse diseases. J Controlled Release. 2022;345:314–33. CrossRef
28. Martinelli DD. In silico vaccine design: a tutorial in immunoinformatics. Healthc Anal. 2022;2:100044. CrossRef
29. Rappuoli R. Reverse vaccinology. Curr Opin Microbiol. 2000;3:445–50. CrossRef
30. Vakili B, Eslami M, Hatam GR, Zare B, Erfani N, Nezafat N, et al. Immunoinformatics-aided design of a potential multi-epitope peptide vaccine against Leishmania infantum. Int J Biol Macromol. 2018;120:1127–39. CrossRef
31. Sayed SB, Nain Z, Khan MSA, Abdulla F, Tasmin R, Adhikari UK. Exploring Lassa virus proteome to design a multi-epitope vaccine through immunoinformatics and immune simulation analyses. Int J Pept Res Ther. 2020;26:2089−107. doi: CrossRef
32. Ali M, Pandey RK, Khatoon N, Narula A, Mishra A, Prajapati VK. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci Rep. 2017;7:9232. CrossRef
33. Rahmani A, Baee M, Rostamtabar M, Karkhah A, Alizadeh S, Tourani M, et al. Development of a conserved chimeric vaccine based on helper T-cell and CTL epitopes for induction of strong immune response against Schistosoma mansoni using immunoinformatics approaches. Int J Biol Macromol. 2019;141:125–36. CrossRef
34. Pritam M, Singh G, Swaroop S, Singh AK, Singh SP. Exploitation of reverse vaccinology and immunoinformatics as promising platform for genome-wide screening of new effective vaccine candidates against Plasmodium falciparum. BMC Bioinform. 2019;19:468. CrossRef
35. Pritam M, Singh G, Swaroop S, Singh AK, Pandey B, Singh SP. A cutting-edge immunoinformatics approach for design of multi-epitope oral vaccine against dreadful human malaria. Int J Biol Macromol. 2020;158:159–79. CrossRef
36. Albrecht-Schgoer K, Lackner P, Schmutzhard E, Baier G. Cerebral malaria: current clinical and immunological aspects. Front Immunol. 2022;13:863568. CrossRef
37. World Health Organization. World malaria report 2021 [Internet]. Geneva, Switzerland: World Health Organization; 2021 [cited 2022 Sep 23].
38. Bahl A. PlasmoDB: the Plasmodium genome resource. A database integrating experimental and computational data. Nucleic Acids Res. 2003;31:212–5. CrossRef
39. Morita M, Takashima E, Ito D, Miura K, Thongkukiatkul A, Diouf A, et al. Immunoscreening of Plasmodium falciparum proteins expressed in a wheat germ cell-free system reveals a novel malaria vaccine candidate. Sci Rep. 2017;7:46086. CrossRef
40. Nagaoka H, Kanoi BN, Jinoka K, Morita M, Arumugam TU, Palacpac NMQ, et al. The N-terminal region of Plasmodium falciparum MSP10 is a target of protective antibodies in malaria and is important for PfGAMA/PfMSP10 interaction. Front Immunol. 2019;10:2669. CrossRef
41. Nagaoka H, Sasaoka C, Yuguchi T, Kanoi BN, Ito D, Morita M, et al. PfMSA180 is a novel Plasmodium falciparum vaccine antigen that interacts with human erythrocyte integrin associated protein (CD47). Sci Rep. 2019;9:5923. CrossRef
42. Saha S, Raghava GPS. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct Funct Bioinform. 2006;65:40–8. CrossRef
43. Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017;45:W24–9. CrossRef
44. Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007;8:4. CrossRef
45. Dimitrov I, Naneva L, Doytchinova I, Bangov I. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics. 2014;30:846–51. CrossRef
46. Gupta A, Mir SS, Saqib U, Biswas S, Vaishya S, Srivastava K, et al. The effect of fusidic acid on Plasmodium falciparum elongation factor G (EF-G). Mol Biochem Parasitol. 2013;192:39–48. CrossRef
47. Mehla K, Ramana J. Identification of epitope-based peptide vaccine candidates against enterotoxigenic Escherichia coli: a comparative genomics and immunoinformatics approach. Mol Biosyst. 2016;12:890–901. CrossRef
48. Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48:W449–54. CrossRef
49. Alam A, Khan A, Imam N, Siddiqui MF, Waseem M, Malik MZ, et al. Design of an epitope-based peptide vaccine against the SARS-CoV-2: a vaccine-informatics approach. Brief Bioinform. 2021;22:1309–23. CrossRef
50. Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007;8:424. CrossRef
51. McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–5. CrossRef
52. Källberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, et al. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7:1511–22. CrossRef
53. Raman S, Vernon R, Thompson J, Tyka M, Sadreyev R, Pei J, et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins Struct Funct Bioinform. 2009;77:89–99. CrossRef
54. Rodrigues-da-Silva RN, Martins da Silva JH, Singh B, Jiang J, Meyer EVS, Santos F, et al. In silico identification and validation of a linear and naturally immunogenic B-cell epitope of the Plasmodium vivax malaria vaccine candidate merozoite surface protein-9. PLoS One. 2016;11:e0146951. CrossRef
55. Ko J, Park H, Heo L, Seok C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012;40:W294–7. CrossRef
56. Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–10. CrossRef
57. Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–9. CrossRef
58. Iheagwam FN, Ogunlana OO, Chinedu SN. Model optimization and in silico analysis of potential dipeptidyl peptidase IV antagonists from GC-MS identified compounds in Nauclea latifolia leaf extracts. Int J Mol Sci. 2019;20:5913. CrossRef
59. Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, Sette A, et al. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008;9:514. CrossRef
60. Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The proteomics protocols handbook [Internet]. Totowa, NJ: Humana Press; 2005 [cited 2022 Oct 1]. pp. 571–607.
61. Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, et al. The ClusPro web server for protein–protein docking. Nat Protoc. 2017;12:255–78. CrossRef
62. Kozakov D, Beglov D, Bohnuud T, Mottarella SE, Xia B, Hall DR, et al. How good is automated protein docking?: automated protein docking. Proteins Struct Funct Bioinform. 2013;81:2159–66. CrossRef
63. Rapin N, Lund O, Castiglione F. Immune system simulation online. Bioinformatics. 2011;27:2013–4. CrossRef
64. Rapin N, Lund O, Bernaschi M, Castiglione F. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS One. 2010;5:e9862. CrossRef
65. Sarkar B, Ullah MA, Araf Y, Rahman MS. Engineering a novel subunit vaccine against SARS-CoV-2 by exploring immunoinformatics approach. Inform Med Unlocked. 2020;21:100478. CrossRef
66. López-Blanco JR, Aliaga JI, Quintana-Ortí ES, Chacón P. iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res. 2014;42:W271–6. CrossRef
67. Grote A, Hiller K, Scheer M, Munch R, Nortemann B, Hempel DC, et al. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005;33:W526–31. CrossRef
68. Ito D, Takashima E, Yamasaki T, Hatano S, Hasegawa T, Miura K, et al. Antibodies against a Plasmodium falciparum RON12 inhibit merozoite invasion into erythrocytes. Parasitol Int. 2019;68:87–91. CrossRef
69. Amlabu E, Mensah-Brown H, Nyarko PB, Akuh O, Opoku G, Ilani P, et al. Functional characterization of Plasmodium falciparum surface-related antigen as a potential blood-stage vaccine target. J Infect Dis. 2018;218:778–90. CrossRef
70. Arumugam TU, Takeo S, Yamasaki T, Thonkukiatkul A, Miura K, Otsuki H, et al. Discovery of GAMA, a Plasmodium falciparum merozoite micronemal protein, as a novel blood-stage vaccine candidate antigen. Infect Immun. 2011;79:4523–32. CrossRef
71. Garcia-Senosiain A, Kana IH, Singh SK, Chourasia BK, Das MK, Dodoo D, et al. Peripheral merozoite surface proteins are targets of naturally acquired immunity against malaria in both India and Ghana. Infect Immun. 2020;88:e00778–19. CrossRef
72. Nagaoka H, Kanoi BN, Ntege EH, Aoki M, Fukushima A, Tsuboi T, et al. Antibodies against a short region of PfRipr inhibit Plasmodium falciparum merozoite invasion and PfRipr interaction with Rh5 and SEMA7A. Sci Rep. 2020;10:6573. CrossRef
73. Patel SD, Ahouidi AD, Bei AK, Dieye TN, Mboup S, Harrison SC, et al. Plasmodium falciparum merozoite surface antigen, PfRH5, elicits detectable levels of invasion-inhibiting antibodies in humans. J Infect Dis. 2013;208:1679–87. CrossRef
74. Richards JS, Arumugam TU, Reiling L, Healer J, Hodder AN, Fowkes FJI, et al. Identification and prioritization of merozoite antigens as targets of protective human immunity to Plasmodium falciparum malaria for vaccine and biomarker development. J Immunol. 2013;191:795–809. CrossRef
75. Roussilhon C, Oeuvray C, Müller-Graf C, Tall A, Rogier C, Trape JF, et al. Long-term clinical protection from falciparum malaria is strongly associated with IgG3 antibodies to merozoite surface protein 3. PLoS Med. 2007;4:e320. CrossRef
76. Daubersies P, Thomas AW, Millet P, Brahimi K, Langermans JAM, Ollomo B, et al. Protection against Plasmodium falciparum malaria in chimpanzees by immunization with the conserved pre-erythrocytic liver-stage antigen 3. Nat Med. 2000;6:1258–63. CrossRef
77. Ghosn S, Chamat S, Prieur E, Stephan A, Druilhe P, Bouharoun-Tayoun H. Evaluating human immune responses for vaccine development in a novel human spleen cell-engrafted NOD-SCID-IL2rγNull mouse model. Front Immunol. 2018;9:601. CrossRef
78. Toure-Balde A, Perlaza BL, Sauzet JP, Ndiaye M, Aribot G, Tall A, et al. Evidence for multiple B- and T-cell epitopes in Plasmodium falciparum liver-stage antigen 3. Infect Immun. 2009;77:1189–96. CrossRef
79. Prieur E, Druilhe P. The malaria candidate vaccine liver stage antigen-3 is highly conserved in Plasmodium falciparum isolates from diverse geographical areas. Malar J. 2009;8:247. CrossRef
80. Bahl V, Chaddha K, Mian SY, Holder AA, Knuepfer E, Gaur D. Genetic disruption of Plasmodium falciparum merozoite surface antigen 180 (PfMSA180) suggests an essential role during parasite egress from erythrocytes. Sci Rep. 2021;11:19183. CrossRef
81. Singh SP, Srivastava D, Mishra BN. Genome-wide identification of novel vaccine candidates for Plasmodium falciparum malaria using integrative bioinformatics approaches. 3 Biotech. 2017;7:318. CrossRef
82. Silveira ELV, Dominguez MR, Soares IS. To B or not to B: understanding B cell responses in the development of malaria infection. Front Immunol. 2018;9:2961. CrossRef
83. Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology. 2018;154:394–406. CrossRef
84. Lata KS, Kumar S, Vaghasia V, Sharma P, Bhairappanvar SB, Soni S, et al. Exploring Leptospiral proteomes to identify potential candidates for vaccine design against leptospirosis using an immunoinformatics approach. Sci Rep. 2018;8:6935. CrossRef
85. Weber CA, Mehta PJ, Ardito M, Moise L, Martin B, De Groot AS. T cell epitope: friend or foe? Immunogenicity of biologics in context?. Adv Drug Deliv Rev. 2009;61:965–76. CrossRef
86. Bemani P, Amirghofran Z, Mohammadi M. Designing a multi-epitope vaccine against blood-stage of Plasmodium falciparum by in silico approaches. J Mol Graph Model. 2020;99:107645. CrossRef
87. Kurup SP, Butler NS, Harty JT. T cell-mediated immunity to malaria. Nat Rev Immunol. 2019;19:457–71. CrossRef
88. Bonam SR, Rénia L, Tadepalli G, Bayry J, Kumar HMS. Plasmodium falciparum malaria vaccines and vaccine adjuvants. Vaccines. 2021;9:1072. CrossRef
89. Shanmugam A, Rajoria S, George AL, Mittelman A, Suriano R, Tiwari RK. Synthetic toll like receptor-4 (TLR-4) agonist peptides as a novel class of adjuvants. PLoS One. 2012;7:e30839. CrossRef
90. Pandey RK, Bhatt TK, Prajapati VK. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci Rep. 2018;8:1125. CrossRef
91. Maharaj L, Adeleke VT, Fatoba AJ, Adeniyi AA, Tshilwane SI, Adeleke MA, et al. Immunoinformatics approach for multi-epitope vaccine design against P. falciparum malaria. Infect Genet Evol. 2021;92:104875. CrossRef
92. Aldakheel FM, Abrar A, Munir S, Aslam S, Allemailem KS, Khurshid M, et al. Proteome-wide mapping and reverse vaccinology approaches to design a multi-epitope vaccine against Clostridium perfringens. Vaccines. 2021;9:1079. CrossRef
93. Singh H, Jakhar R, Sehrawat N. Designing spike protein (S-Protein) based multi-epitope peptide vaccine against SARS COVID-19 by immunoinformatics. Heliyon. 2020;6:e05528. CrossRef
94. Khan SN, Ali R, Khan S, Rooman M, Norin S, Zareen S, et al. Genetic diversity of polymorphic marker merozoite surface protein 1 (Msp-1) and 2 (Msp-2) genes of Plasmodium falciparum isolates from malaria endemic region of Pakistan. Front Genet. 2021;12:751552. CrossRef
95. Ashgar SS, Faidah H, Bantun F, Jalal NA, Qusty NF, Darwish A, et al. Integrated immunoinformatics and subtractive proteomics approach for multi-epitope vaccine designing to combat S. pneumoniae TIGR4. Front Mol Biosci. 2023;10:1212119. CrossRef
96. Zhang NZ, Huang SY, Zhou DH, Xu Y, He JJ, Zhu XQ. Identification and bioinformatic analysis of a putative calcium-dependent protein kinase (CDPK6) from Toxoplasma gondii. Genet Mol Res GMR. 2014;13:10669–77. CrossRef
97. Khatoon N, Pandey RK, Prajapati VK. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci Rep. 2017;7:8285. doi: 10.1038/s41598-017-08842-w.
98. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–91. CrossRef
99. Ferdous S, Kelm S, Baker TS, Shi J, Martin ACR. B-cell epitopes: discontinuity and conformational analysis. Mol Immunol. 2019;114:643–50. CrossRef
100. Solanki V, Tiwari M, Tiwari V. Prioritization of potential vaccine targets using comparative proteomics and designing of the chimeric multi-epitope vaccine against Pseudomonas aeruginosa. Sci Rep. 2019;9:5240. CrossRef
101. Iheagwam FN, Israel EN, Kayode KO, De Campos OC, Ogunlana OO, Chinedu SN. GC-MS analysis and inhibitory evaluation of Terminalia catappa leaf extracts on major enzymes linked to diabetes. Evid Based Complement Alternat Med. 2019;2019:6316231. CrossRef
102. Onile OS, Musaigwa F, Ayawei N, Omoboyede V, Onile TA, Oghenevovwero E, et al. Immunoinformatics studies and design of a potential multi-epitope peptide vaccine to combat the fatal visceral leishmaniasis. Vaccines. 2022;10:1598. CrossRef
103. Van Aalten DMF, De Groot BL, Findlay JBC, Berendsen HJC, Amadei A. A comparison of techniques for calculating protein essential dynamics. J Comput Chem. 1997;18:169–81. CrossRef
104. Kovacs JA, Chacón P, Abagyan R. Predictions of protein flexibility: first-order measures. Proteins Struct Funct Bioinform. 2004;56:661–8. CrossRef
SUPPLEMENTARY MATERIAL
The supplementary material can be accessed at the journal's website: Link Here [https://japsonline.com/admin/php/uploadss/4324_pdf.pdf].