
INTRODUCTION
The coronavirus disease 2019 (COVID-19) outbreak was first reported at Wuhan, China, in December 2019 (Du Toit, 2020), is caused by the severe acute respiratory coronavirus 2 (SARS-CoV-2) infections. It has resulted in over millions of infected cases and hundreds of thousands of deaths worldwide in just a matter of few months. This pandemic has severely paralyzed the global economy for several months and has almost exhausted many healthcare service facilities around the world. The high transmission rate of SARS-CoV-2 makes it further difficult to contain. Although the fatality rates of the Middle East respiratory syndrome coronavirus (MERS-CoV) and severe acute respiratory syndrome coronavirus (SARS) are higher than SARS-CoV-2, the transmission of the SARS-CoV-2 virus is much faster than that of the previous human coronaviruses, resulting to even higher death rates (Cascella et al., 2020). SARS-CoV-2 belongs to the subfamily Coronavirinae, family Coronaviridae, order Nidovirales, and genus Betacoronavirus. Coronaviruses are enveloped viruses containing positive sense single-stranded RNA as genetic material. It has approximately 26–32 kb, containing six open reading frames (ORFs): ORF1ab, ORF3a, ORF6, ORF7a, ORF8, and ORF10, wherein ORF1 encodes for 16 nonstructural proteins (NSP1–16) and four structural proteins which include spike (S), envelope (E), membrane (M), and nucleocapsid (N) (Ahmed et al., 2020; Lu et al., 2020). A study showed that T-cell responses elicited by the structural proteins of SARS are more immunogenic than its NSP (Li et al., 2008).
Phylogenetic studies showed that SARS-CoV-2 shared 80.0% sequence identity with SARS (Gralinski and Menachery, 2020; Zhou et al., 2020). A proteomic analysis conducted to compare the sequences of SARS-CoV-2 with SARS showed that proteins in SARS-CoV-2 share 95%–100% identity with SARS (Xu et al., 2020), except for ORF8 and ORF10 which have no homologous proteins in SARS (Chan et al., 2020). Among these proteins is the nucleocapsid (N). Being the most abundant and highly conserved protein in coronaviruses, it shares 90.52% identity with SARS (Xu et al., 2020). Similarities between these two coronaviruses can be used as an advantage in designing drugs and vaccines against SARS-CoV-2. In this regard, the therapeutic agents used to treat SARS can be potentially used as COVID-19 treatment (Chen et al., 2020).
Recently, several COVID-19 vaccines have finished phase III clinical trials; however, these cannot guarantee an outright success upon administration to larger and more diverse populations around the world. Studies conducted to develop drugs and vaccines are far from over until SARS-CoV-2 is finally tamed in most populations worldwide. Witnessing the devastating effects of COVID-19, the rapid development of antiviral drugs and vaccines is imperative. Data mining and immunoinformatics have efficiently hastened and reduced the cost required in experimental immunology to be able to identify vaccine candidates and biomarkers (Oyarzún et al., 2016; Vaishnav et al., 2015). Herein, this work aims to develop a candidate SARS-CoV-2 vaccine using immunoinformatics tools for the efficient identification of potential vaccines from experimentally validated T-cell (CD4+ and CD8+) epitopes of SARS which were retrieved from the viral sequence database.
METHODOLOGY
Identification of experimentally validated SARS T-Cell epitopes
Experimentally validated SARS CD4+ and CD8+ epitopes were retrieved from the Immune Epitope Database and Analysis Resource (IEDB). Epitopes were restricted to “severe acute respiratory syndrome-related coronavirus [human coronavirus (strain SARS)]” with “humans” as the host, specifically binding to “MHC class I” for CD8+ epitopes or “MHC class II” for CD4+ epitopes and “T-cell assays” with only positive results included.
Retrieval of SARS-CoV-2 protein sequences, processing, and alignment
All SARS-CoV-2 amino acid sequences with corresponding proteins in the list of retrieved SARS epitopes were acquired from the Virus Pathogen Database and Analysis Resource (ViPR). Sequences collected from humans, starting from December 2019 to August 2020, were retrieved. Only those with complete genomes were included, and the minimum coding sequence (CDS) limit was employed per protein. Identical sequences were also removed. The list of retrieved sequences were clustered using a 0.99 cut-off in CD-HIT suite (http://weizhongli-lab.org/cdhit_suite/cgi-bin/index.cgi?cmd=cd-hit) to obtain a set of representative sequences per protein. The representative sequences were then aligned using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) to obtain multiple sequence alignments. Protein Variability Server was used to identify highly conserved sequences in SARS-CoV-2 proteins. Shannon variability threshold H ≤ 0.5 was used. Sequence variability was masked using the first sequence in the alignment as a reference. Fragments with ≥9 residues were selected. All acquired SARS epitopes having 100% overlap with the retrieved SARS-CoV-2 reference protein sequences were retained for further evaluation.
Identification of MHC binding, cross-reactivity, and population coverage (PC)
Restricted MHC I alleles binding to CD8+ epitopes were identified in silico using MHC I Binding Tool NetMHCcons in IEDB which predicts binding to any known MHC I molecule integrating three well-known methods: PickPocket, NetMHC, and NetMHCpan, for more accurate results (Karosiene et al., 2012). In screening for binding alleles, the most frequent MHC I alleles (Weiskopf et al., 2013) were used with IC50 < 500 nmol/dm3, which classifies epitopes as good binders (Jensen et al., 2018). To identify MHC II alleles binding to CD4+ epitopes, SMM-align (NetMHCII) method in IEDB MHC II Binding Tool was used. This was reported to outperform other state-of-the-art MHC II prediction algorithms (Nielsen et al., 2007). The most frequent MHC II alleles (Greenbaum et al., 2011) with IC50 < 500 nmol/dm3 were used. To avoid potential cross-reactivity in humans, protein–protein BLAST (BLASTp) was utilized to identify epitopes with a significant match to known human protein sequences. The PC for each set of CD4+ and CD8+ epitopes was estimated using the PC tool in IEDB. In addition, PC in areas where infection rates of SARS-CoV-2 are high, including the USA, Brazil, Mexico, France, India, Iran, Spain, and Russia, was also calculated. This tool helps to efficiently maximize the PC of epitopes while minimizing the number of epitopes that must be included in a vaccine (Bui et al., 2006). Candidate CD4+ and CD8+ vaccine epitopes were selected to give optimal worldwide PC.
Docking and molecular dynamics simulation of MHC-epitope complexes
Selected epitopes were docked with the most common MHC alleles in the list. The Protein Data Bank (PDB) crystal structures of HLA-DRB1*01:01 (PDB-ID: 3PDO), HLA-A*02:03 (PDB-ID: 3OX8), HLA-B*44:02 (PDB-ID: 3L3D), HLA-B*53:01 (1A1O), and HLA-A*02:06 (PDB-ID: 3OXR) were retrieved from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB). GalaxyPepDock server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=PEPDOCK) was utilized to dock epitopes with cleaned MHC crystal structures: GAALQIPFAMQMAYRF (S), MAYRFNGIGVTQNVLY (S), and QALNTLVKQLSSNFGAI (S) with 3PDO; RLNEVAKNL (S), VLNDILSRL (S), GMSRIGMEV (N), LLLDRLNQL (N) with 3OX8; MEVTPSGTWL (N) with 3L3D; RRPQGLPNNTASWFT (N) with 1A1O; and LQLPQGTTL (N) with 3OXR. Docked structures were further refined in GalaxyRefineComplex server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=COMPLEX). Binding energy and dissociation constant were calculated in the PRODIGY webserver at 37°C. This tool uses intermolecular contacts and noninterface surface properties for predictive models (Xue et al., 2016). The stability of the interaction of an epitope with its MHC allele was further evaluated using molecular dynamics simulation, wherein the root mean square deviation (RMSD) plot per residue was obtained. This process was carried out in the MDWeb server (http://mmb.irbbarcelona.org/MDWeb/) using C-alpha Brownian dynamics in 100 ps time, with 0.01 ps time change, 3.8 Ǻ distance between alpha carbon atoms, 10 steps output frequency, and 40 kcal/mol Ǻ2 force constant. This method used GROMACS MD setup with solvation, employing the force-field Amber-99sb (Hospital et al., 2012).
RESULTS AND DISCUSSION
Identification of SARS-CoV-2 epitopes
A total of 70 SARS T-cell epitopes (43 CD4+ and 27 CD8+) were retrieved from IEDB. The set of 43 CD4+ consists of five ORF3a, four N, and 34 S epitopes, while the set of 27 CD8+ consists of two ORF3a, 15 N, and 10 S epitopes. Reported close homology of protein sequences between SARS and SARS-CoV-2 has been the basis of this work in seeking putative T-cell epitopes which can be potentially used as an ensemble of the vaccine against SARS-CoV-2. To identify which among these retrieved SARS epitopes overlap with SARS-CoV-2, unique protein sequences of ORF3a, N, and S in SARS-CoV-2 were retrieved from ViPR. Overall, 438 ORF3a, 529 N, and 899 S sequences were isolated from different countries worldwide, starting from December 2019 to August 2020. These sequences were clustered, aligned, and highly conserved reference sequence for each protein was obtained. One important factor to consider in designing a vaccine to avoid epitope immune escape is to focus on highly conserved sequences so that more effective immunity can be developed against a broader range of SARS-CoV-2 isolates around the world. To determine the highly conserved reference sequence for each protein in this study, Shannon’s variability threshold ≤ 0.5 was used (Molero-Abraham et al., 2013; Shannon, 1997). From 70 retrieved SARS epitopes, only 17 epitopes (4 CD4+ and 13 CD8+) have 100% overlap with the highly conserved reference SARS-CoV-2 sequences. These were further evaluated (Tables 1 and 2) for cross-reactivity with the human proteome. Among all blasted CD4+ epitopes, GAALQIPFAMQMAYRF has the highest E-value = 1.5e−1 with percentage identity (66.67%), while 3 CD8+ epitopes have 100% identity: LALLLLDRL (E-value = 3.3e−1), ILLNKHIDA (E-value = 1.9e−1), and ALNTPKDHI (E-value = 5.4e−1). Matches with E-values < 1.0e−30 can be cross-reactive in some allergic individuals (Hileman et al., 2002). In this work, E-values of epitopes blasted against the human protein in databases are > 1; thus, at best, these epitopes have poor matches and are less likely to cause autoimmune reactions in humans.
Identification of MHC binding, cross-reactivity, and PC
The MHC alleles binding to the 17 identified SARS-CoV-2 epitopes were obtained from IEDB. Table 3 shows epitopes with their corresponding MHC allele binders at IC50 ≤ 500 nM. CD8+ and CD4+ epitopes with the highest global PC are VLNDILSRL (67.81%) and GAALQIPFAMQMAYRF (75.40%), respectively. Candidate SARS-CoV-2 vaccines were finally selected to include the minimum number of epitopes that can give the optimal worldwide % PC for each set of CD4+ and CD8+ epitopes. Results turned to include 10 epitopes (7 CD8+ and 3 CD4+): RLNEVAKNL (S), VLNDILSRL (S), GMSRIGMEV (N), LLLDRLNQL (N), MEVTPSGTWL (N), RRPQGLPNNTASWFT (N), LQLPQGTTL (N), GAALQIPFAMQMAYRF (S), MAYRFNGIGVTQNVLY (S), and QALNTLVKQLSSNFGAI (S). The estimated worldwide % PC for the set of CD4+ and CD8+ epitopes are 81.81% and 89.49%, respectively. The PC for the set of CD8+ candidate epitopes in known areas with high SARS-CoV-2 mortality rates include 91.45% in the USA, 80.59% in Brazil, 71.59 % in India, 86.61% in Mexico, 93.97% in France, 90.85% in Iran, 80.2% in Spain, and 85.94% in Russia. For the set of CD4+ epitopes, PC are 88.1% in USA, 63.8% in Brazil, 74.99 % in India, 55.54% in Mexico, 88.54% in France, 64.22% in Iran, 80.51% in Spain, and 77.62% in Russia. Since not all the identified MHC II alleles in this work were available and included in the calculation of the IEDB tool, the estimated % PC for the set of CD4+ epitopes could be larger in reality. These results show that the 10 epitopes identified in this work are potential SARS-CoV-2 vaccines which can be utilized as an immunotherapeutic agent in many areas severely affected by COVID-19.
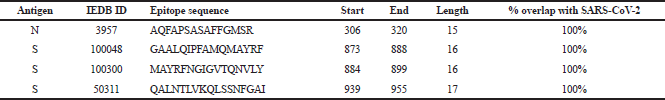 | Table 1. SARS CD4+ epitopes retrieved from IEDB. [Click here to view] |
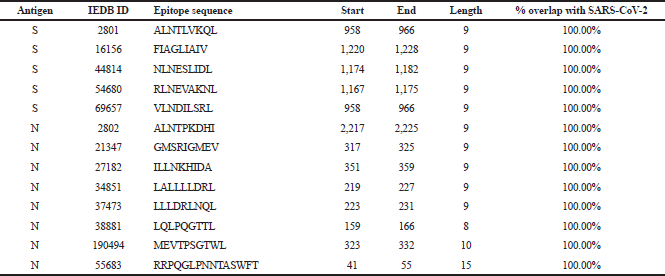 | Table 2. SARS CD4+ epitopes retrieved from IEDB. [Click here to view] |
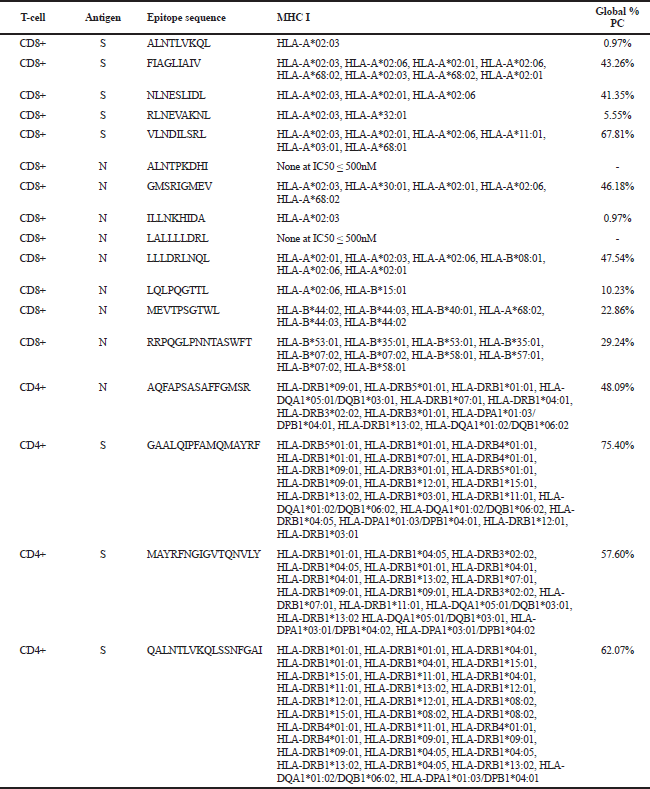 | Table 3. SARS-CoV-2 epitopes. [Click here to view] |
Docking and molecular dynamics simulation of MHC-epitope complexes
Retrieved MHC structures were docked and refined with their corresponding epitopes. Epitope-MHC docked models with the lowest energy were chosen as representative docked structures. Figure 1 shows the structures of GAALQIPFAMQMAYRF (A), MAYRFNGIGVTQNVLY (B), and QALNTLVKQLSSNFGAI (C) docked with HLA-DRB1*01:01; RLNEVAKNL (D), VLNDILSRL (E), GMSRIGMEV (F), and LLLDRLNQL (G) docked with HLA-A*02:03; MEVTPSGTWL docked with HLA-B*44:02 (H); RRPQGLPNNTASWFT docked with HLA-B*53:01 (I); and LQLPQGTTL docked with HLA-A*02:06 (J). The figures show that all docked epitopes are bound within the epitope-binding groove of each MHC molecule. To support data on efficient binding of epitopes with their predicted MHC alleles, binding energy (ΔG) and dissociation constant (Kd) were calculated for each epitope-MHC docked complex (Table 4). The lower the value of binding energy and dissociation constant, the more stable and stronger the binding of the epitope with the MHC structure. All epitope-MHC docked structures have negative ΔG values for binding energy, which indicate that the complex formation is favorable and stable (Paul et al., 2014; Sebastián et al., 2017). Dissociation constants calculated for the docked structures in this study fall within the known Kd range of most peptide-MHC binding structures (Chang et al., 2006) which further provide evidence on the stability of identified candidate vaccine epitopes binding to their MHC alleles. Among the three CD4+ vaccine epitopes, QALNTLVKQLSSNFGAI has the greatest binding affinity and forms the most stable complex with HLA-DRB1*01:01 as indicated by the lowest Kd and binding energy. For CD8+ candidate vaccines, RRPQGLPNNTASWFT has the greatest binding affinity and the most stable complex formed with HLA-B*53:01. To further investigate the stability of epitope-MHC docked structures, molecular dynamics simulation was conducted to obtain the RMSD plot of each complex. Figure 2 shows the RMSD plots for GAALQIPFAMQMAYRF (A), MAYRFNGIGVTQNVLY (B), and QALNTLVKQLSSNFGAI (C) docked with HLA-DRB1*01:01; RLNEVAKNL (D), VLNDILSRL (E), GMSRIGMEV (F), and LLLDRLNQL (G) docked with HLA-A*02:03; MEVTPSGTWL docked with HLA-B*44:02 (H); RRPQGLPNNTASWFT docked with HLA-B*53:01 (I); and LQLPQGTTL docked with HLA-A*02:06 (J). All residues from each complex have RMSD ≤ 1.0 Å, which indicates the best interaction between the docked ligand and protein (Fu et al., 2018).
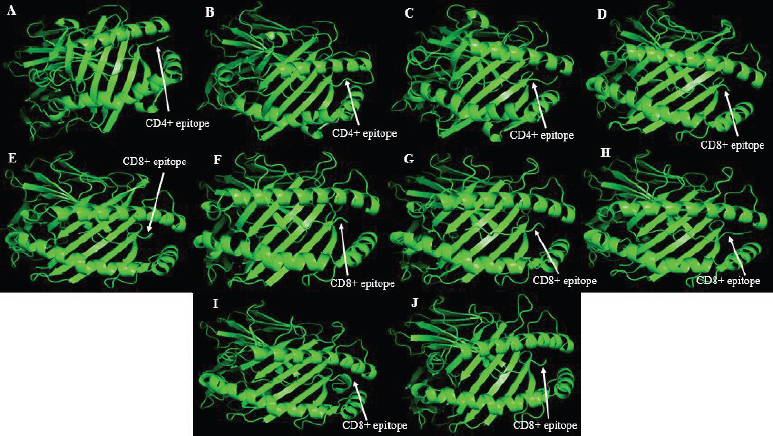 | Figure 1. Structures of 10 candidate vaccine epitopes docked with their respective MHC alleles as viewed in PyMOL. [Click here to view] |
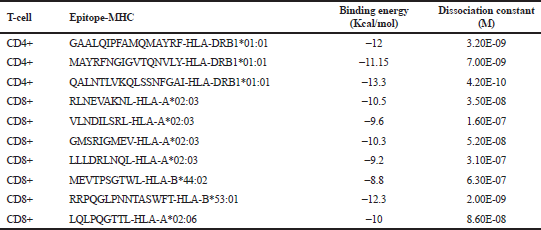 | Table 4. Ten candidate vaccine epitopes against SARS-CoV-2. [Click here to view] |
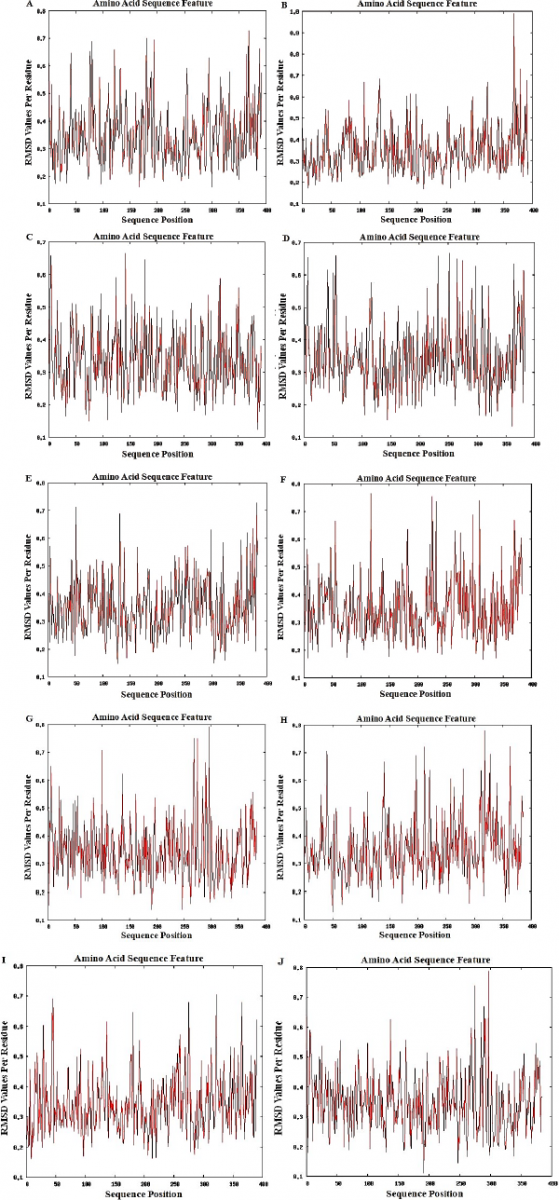 | Figure 2. The RMSD plots for the ten epitope-MHC docked structures (A to J) show that all values per residue are less than one which indicate the stability of all the complexes. [Click here to view] |
CONCLUSION
Reports on closely similar protein sequences of SARS with SARS-CoV-2 have been the basis of this work in seeking putative T-cell epitopes which can be potentially used as an ensemble of vaccine against SARS-CoV-2. This work identified 10 candidate vaccine epitopes having 100% overlap with the highly conserved sequences of S and N in SARS-CoV-2 and possessing large PC worldwide and for areas with known high SARS-CoV-2 mortality rates. The proposed set of epitopes include RLNEVAKNL, VLNDILSRL, GMSRIGMEV, LLLDRLNQL, MEVTPSGTWL, RRPQGLPNNTASWFT, LQLPQGTTL, GAALQIPFAMQMAYRF, MAYRFNGIGVTQNVLY, and QALNTLVKQLSSNFGAI. The evaluations of binding energy, binding affinity, and molecular dynamics simulations showed stability of docked epitopes, with their respective MHC alleles, which were identified in this study. Overall, results from this work suggest that the identified epitopes can be used to form a vaccine against SARS-CoV-2, which are nonetheless anticipated to be further examined in vivo.
ACKNOWLEDGEMENTS
The author would like to thank the referees who shared their time, and expertise in reviewing this manuscript; and the Polytechnic University of the Philippines for its support.
CONFLICT OF INTEREST
The author has no conflict of interest.
FUNDING
None.
REFERENCES
Ahmed SF, Quadeer AA, McKay MR. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses, 2020; 12(3):254. CrossRef
Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics, 2006; 7: 153. CrossRef
Cascella M, Rajnik M, Cuomo A, Dulebohn SC, Di Napoli R. Features, evaluation, and treatment of coronavirus (COVID-19). In: StatPearls. StatPearls Publishing, Treasure Island, FL, 2020.
Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S, Yuen KY. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect, 2020; 9(1):221–36. CrossRef
Chang ST, Ghosh D, Kirschner DE, Linderman JJ, Peptide length-based prediction of peptide–MHC class II binding. Bioinformatics, 2006; 22(22)15:2761–7. CrossRef
Chen Y, Liu Q, Guo D. Review Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol, 2020; 92(4):418–23. CrossRef
Du Toit A. Outbreak of a novel coronavirus. Nat Rev Microbiol, 2020; 18(3):123.
Fu Y, Zhao J, Chen Z. Insights into the molecular mechanisms of protein-ligand interactions by molecular docking and molecular dynamics simulation: a case of oligopeptide binding protein. Comput Math Methods Med, 2018; 2018:1–12. CrossRef
Gralinski LE, Menachery VD. Return of the coronavirus: 2019-nCoV. Viruses, 2020; 12(2):135. CrossRef
Greenbaum J, Sidney J, Chung J, Brander C, Peters B, Sette A. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics, 2011; 63:325–35. CrossRef
Hileman RE, Silvanovich A, Goodman RE, Rice EA, Holleschak G, Astwood JD, Hefle SL. Bioinformatic methods for allergenicity assessment using a comprehensive allergen database. Int Arch Allergy Immunol, 2002; 128:280–91. CrossRef
Hospital A, Andrio P, Fenollosa C, Cicin-Sain D, Orozco M, Gelpí JL. MDWeb and MDMoby: an integrated web-based platform for molecular dynamics simulations. Bioinformatics, 2012; 28(9):1278–9. CrossRef
Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, Sette A, Peters B, Nielsen M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology, 2018; 154(3):394–406. CrossRef
Karosiene E, Lundegaard C, Lund O, Nielsen M. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics, 2012; 64:177–86. CrossRef
Li CK, Wu H, Yan H, Ma S, Wang L, Zhang M, Tang X, Temperton NJ, Weiss RA, Brenchley JM, Douek DC, Mongkolsapaya J, Tran BH, Lin CL, Screaton GR, Hou JL, McMichael AJ, Xu XN. T cell responses to whole SARS coronavirus in humans. J Immunol, 2008; 181(8):5490–500. CrossRef
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y, Ma X, Zhan F, Wang L, Hu T, Zhou H, Hu Z, Zhou W, Zhao L, Chen J, Meng Y, Wang J, Lin Y, Yuan J, Xie Z, Ma J, Liu WJ, Wang D, Xu W, Holmes EC, Gao GF, Wu G, Chen W, Shi W, Tan W. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet (London, England), 2020; 395:565–74. CrossRef
Molero-Abraham M, Lafuente EM, Flower DR, Reche PA. Selection of conserved epitopes from hepatitis C virus for pan-populational stimulation of T-cell responses. Clin Dev Immunol, 2013; 2013:601943. CrossRef
Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics, 2007; 8:238. CrossRef
Oyarzún P, Kobe B. Recombinant and epitope-based vaccines on the road to the market and implications for vaccine design and production. Hum Vaccin Immunother, 2016; 12(3):763–7. CrossRef
Paul BK, Ghosh N, Mukherjee S. Binding interaction of a prospective chemotherapeutic antibacterial drug with β-lactoglobulin: results and challenges. Langmuir, 2014; 30(20):5921–9. CrossRef
Sebastián M, Serrano A, Velázquez-Campoy A, Medina M. Kinetics and thermodynamics of the protein-ligand interactions in the riboflavin kinase activity of the FAD synthetase from Corynebacterium ammoniagenes. Sci Rep, 2017; 7:7281. CrossRef
Shannon, CE. The mathematical theory of communication. 1963. MD Comput. 1997; 14(4):306–17. CrossRef
Vaishnav N, Gupta A, Paul S, John GJ. Overview of computational vaccinology: vaccine development through information technology. J Appl Genet, 2015; 56(3):381–91. CrossRef
Weiskopf D, Angelo MA, de Azeredo EL, Sidney J, Greenbaum JA, Fernando AN, Broadwater A, Kolla RV, De Silva AD, de Silva AM, Mattia KA, Doranz BJ, Grey HM, Shresta S, Peters B, Sette A. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8+ T cells. Proc Natl Acad Sci U S A, 2013; 110(22):E2046–53. CrossRef
Xu J, Zhao S, Teng T, Abdalla AE, Zhu W, Xie L, Wang Y, Guo X. Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses, 2020; 12(2):244. CrossRef
Xue LC, Rodrigues JP, Kastritis PL, Bonvin AM, Vangone A. PRODIGY: a web server for predicting the binding affinity of protein-protein complexes. Bioinformatics, 2016; 32(23):3676–8. CrossRef
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 2020; 579(7798): 270–3. CrossRef