
INTRODUCTION
Recently, the novel coronavirus (COVID-19) has led to millions of deaths throughout the world (WHO, 2019b). Multiple preventive approaches have been suggested to avoid the infection of COVID-19, like social distancing, regular cleaning the hands with sanitizer, using masks to cover mouth and nose in a crowd, and regular health check-ups (WHO, 2019a). Furthermore, investigations are ongoing in order to identify new drug molecules in the management of COVID-19, which may still take time as the drug development process is quite complicated and is time-consuming. Hence, it is necessary to identify alternative sources against COVID-19 that can be processed via traditional data mining.
Plectranthus amboinicus is a Mexican mint that belongs to the family Lamiaceae and has been reported for its multiple pharmacological activities, including anti-viral activities (Arumugam et al., 2016). Further, multiple extracts of P. amboinicus have been investigated against herpes and HIV (Arumugam et al., 2016). Likewise, P. amboinicus is also used to manage respiratory problems and is also a potent anti-inflammatory agent (Arumugam et al., 2016; Gurgel et al., 2009) that possesses anti-inflammatory activity in virus-infected tissues. The literature studies suggest the necessity of investigating P. amboinicus against COVID-19 as this infection is associated with respiratory problems and inflammation.
Hence, the present study utilized in silico tools to investigate the reported bioactives from P. amboinicus against three reported targets of COVID-19, i.e., PLpro, 3clpro, and spike protein, followed by the effect of bioactives on probably regulated pathways using the Gene ontology (GO) analysis of possibly modulated protein-based targets.
MATERIALS AND METHODS
Compounds and their targets
Phytoconstituents present in P. amboinicus were retrieved from the ChEBI database (https://www.ebi.ac.uk/chebi/) along with their simplified molecular-input line-entry system (SMILES). The 2D structure of retrieved compounds is shown in Figure 1. The 3D structures of each compound were obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) in a .sdf format and converted into a .pdb format using Discovery Studio 2019 (Dassault Systèmes BIOVIA, 2019). The protein-based target of each compound was retrieved from DIGEP-Pred (Lagunin et al., 2013) at the cutoff value of 0.7 probable activity.
Druglikeness score of compounds
The druglikeness score of each bioactives was predicted using MolSoft (https://molsoft.com/mprop/) which was based on molecular weight, the number of hydrogen bond donors/acceptors, and MolLogP as explained by Lipinksi’s Rule of Five (Lipinski, 2004).
In silico molecular docking
Ligand preparation
The .sdf 3D files of the ligand were converted into .pdb files using Discovery Studio 2019 (Dassault Systèmes BIOVIA, 2019). The energy of individual molecules was minimized using Merck Molecular Force Field 94 (mmff94) (Halgren, 1996) and then converted into a .pdbqt format.
Macromolecule preparation
The 3D structures of 3CLpro (PDB: 6LU7), PLpro (PDB: 4M0W), and spike proteins [homology modeled target, accession number: AVP78042.1 as query sequence, and PDB: 6VSB as a template using the Swiss Model (Schwede et al., 2003)] were obtained from the RCSB database which was in a complex with other heteromolecules. These heteromolecules were removed and saved in .pdb files using Discovery Studio 2019 and then converted into a .pdbqt format as a macromolecule for docking.
 | Figure 1. 2D structure of (a) 11,20-dihydroxysugiol, (b) 11-hydroxysugiol, (c) plectranthol A, (d) plectranthol B, and (e) abietatriene. [Click here to view] |
Molecular docking
Molecular docking was carried out using AutoDock 4 (Morris et al., 2009). After docking, 10 different ligand poses were obtained. The ligand pose scoring the lowest binding energy (kcal/mol) was chosen to visualize the ligand–protein interaction using Discovery Studio 2019.
Enrichment analysis
The combined list of modulated genes was queried in STRING (Szklarczyk et al., 2019) and the modulated pathways were identified by checking the Reactome database (www.reactome.org). The interaction of bioactives, their targets, and respective pathways were constructed using Cytoscape (Shannon et al., 2003). The node size and the map were used to analyze the network interaction using a network analyzer.
RESULTS
Compounds and their targets
Five different bioactives, i.e., 11, 20-dihydroxysugiol, 11-hydroxysugiol, plectranthol A, plectranthol B, and abietatriene, were recorded as major bioactives in the ChEBI database for the keyword “P. amboinicus”. These molecules were identified to downregulate multiple proteins, i.e., vitamin D3 receptor (VDR) was downregulated by all compounds, retinoic acid receptor alpha (RARα) by plectranthol B and CD83 by abietatriene.
Druglikeness score of compounds
Plectranthol B was predicted to possess the highest druglikeness score, i.e., 1.47, with a molecular weight of 536.28, the number of hydrogen bond acceptors (7) and donors (3), and MolLogP of 7.18. The details of each compound for their druglikeness score are summarized in Table 1.
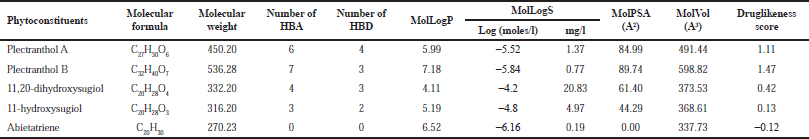 | Table 1. Druglikeness score of bioactives from P. amboinicus. [Click here to view] |
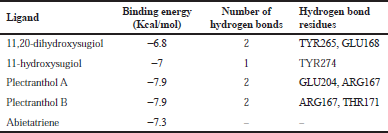 | Table 2. Binding energy with PLpro (PDB: 4M0W). [Click here to view] |
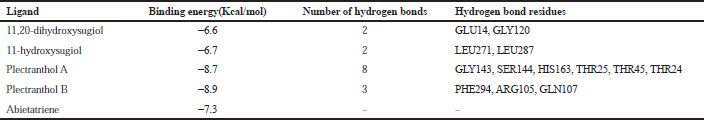 | Table 3. Binding energy with 3CLpro (PDB: 6LU7). [Click here to view] |
 | Table 4. Binding energy with spike protein. [Click here to view] |
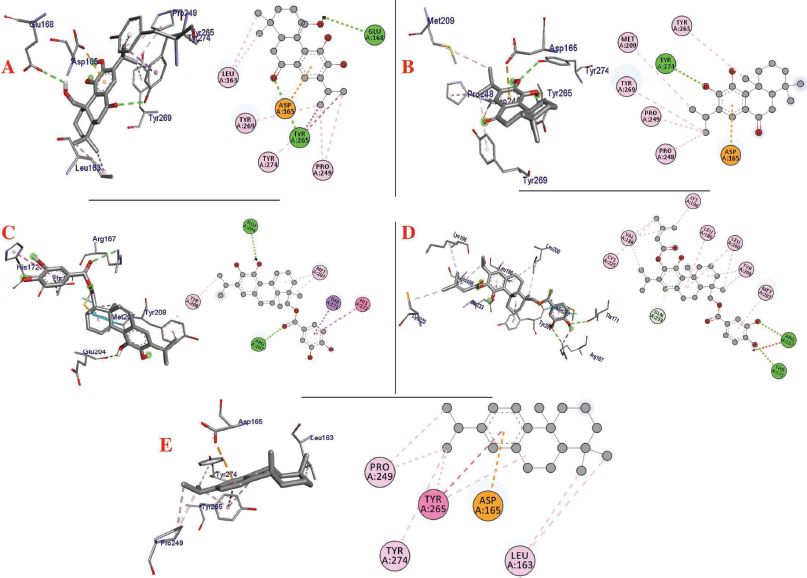 | Figure 2. Interaction of (a) 11,20-dihydroxysugiol, (b) 11-hydroxysugiol, (c) plectranthol A, (d) plectranthol B, and (e) abietatriene with PLpro (PDB: 4M0W). [Click here to view] |
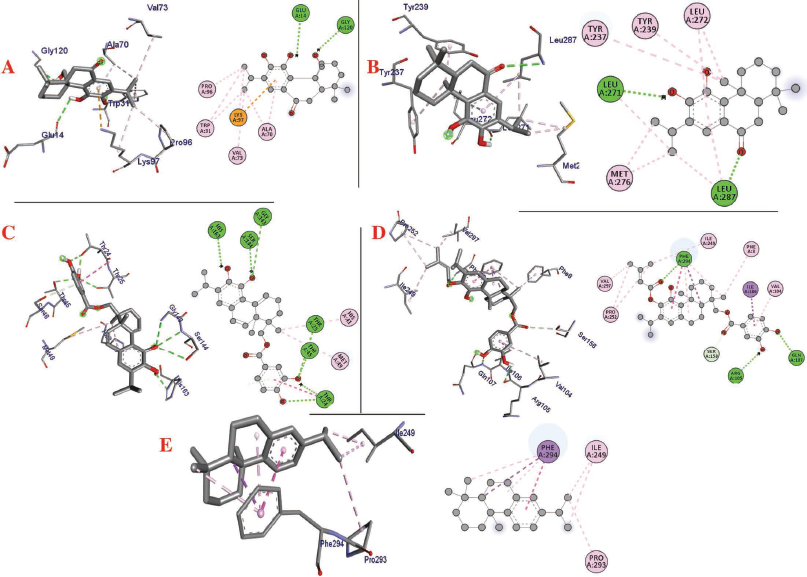 | Figure 3. Interaction of (a) 11,20-dihydroxysugiol, (b) 11-hydroxysugiol, (c) plectranthol A, (d) plectranthol B, and (e) abietatriene with 3CLpro (PDB: 6LU7). [Click here to view] |
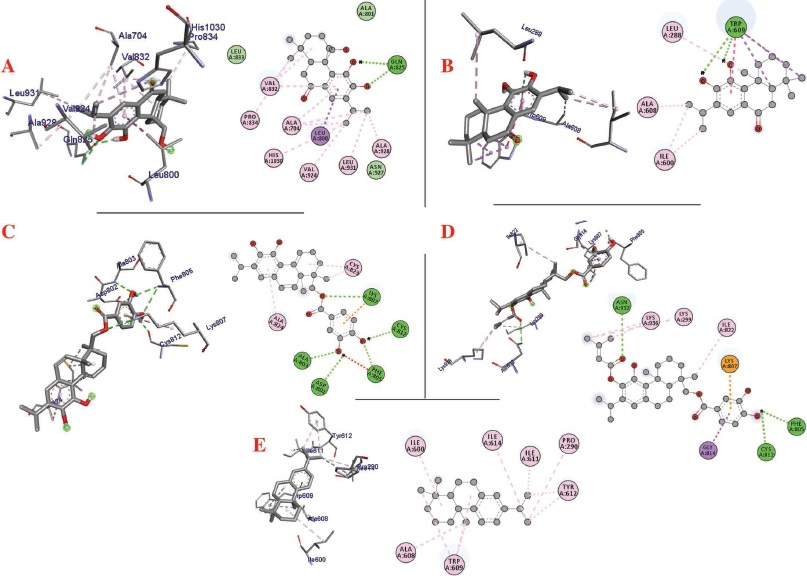 | Figure 4. Interaction of (a) 11,20-dihydroxysugiol, (b) 11-hydroxysugiol, (c) plectranthol A, (d) plectranthol B, and (e) abietatriene with Spike protein. [Click here to view] |
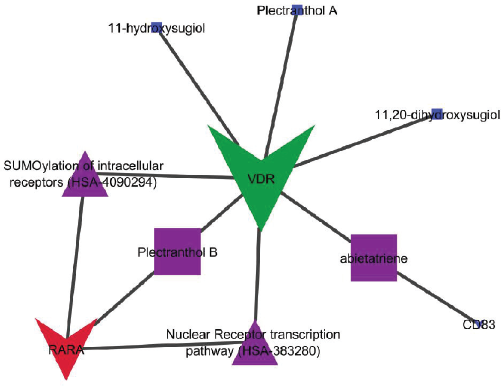 | Figure 5. Network interactions of bioactives with respected proteins and modulated pathways. [Click here to view] |
In silico molecular docking
The molecular docking process identified plectranthol A and plectranthol B to possess the highest binding affinities, i.e., −7.9 kcal/mol with PLpro, with an equal number of hydrogen bond interactions. Although plectranthol B showed the highest binding affinity (−8.9 kcal/mol) with 3CLpro, plectranthol A shared the highest number of hydrogen bond interactions, i.e., eight with 3CLpro. Similarly, plectranthol A was predicted to possess the highest binding affinity (−8.4 kcal/mol) and hydrogen bond interactions with spike protein. The binding affinity of an individual ligand with each compound is summarized in Table 2–4. The 3D interactions of each ligand molecule with each PLpro, 3CLpro, and spike protein are shown in Figures 2–4 respectively.
Enrichment analysis
The combined GO analysis of modulated targets identified two pathways to be majorly regulated with special reference to the Reactome pathway database, i.e., SUMOylation of intracellular receptors (HSA-4090294) and nuclear receptor transcription pathway (HSA-383280): modulated RARα and VDR. The interaction of each bioactive with its targets and modulated protein is shown in Figure 5.
DISCUSSION
This study aimed to investigate the bioactives from P. amboinicus against the three targets of COVID-19, i.e., PLpro, 3CLpro, and spike protein, and their possible role in modulating multiple pathways. Furthermore, this study also calculated the probable druggability characteristics based on the Rule of five. Traditional medicines possess an important role in the management of infectious (Pinn, 2001) and non-infectious diseases (Duyu et al., 2020; Hughes et al., 2015; Khanal and Patil, 2019; Khanal and Patil, 2020a, 2020b; Patil et al., 2019; Rodrigues et al., 2020a, 2020b). Multiple approaches have been made to investigate the bioactives from traditional medicines using in silico approaches which often help to identify the probable lead hit molecules (Duyu et al., 2020; Khanal et al., 2019a, 2019b, 2019c; Patil et al., 2019; Rodrigues et al., 2020a, 2020b; Ternikar et al., 2020). Nonetheless, approaches have been made to investigate the probable anti-viral agents against COVID-19 using molecular docking, network pharmacology, and enrichment analysis (Balkrishna et al., 2020; Khanal et al., 2020a, 2020b, 2020c). Hence, in the present study, we aimed to investigate the bioactives of P. amboinicus against COVID-19.
Initially, the bioactives of P. amboinicus were retrieved from the ChEBI database and their proteins and targets were predicted, which were further queried in STRING; they also identified the SUMOylation of intracellular receptors (HSA-4090294) and nuclear receptor transcription pathway (HSA-383280) as modulated pathways. The previous report identified the involvement of viral infections with the SUMOylation of IRF3 and IRF7, leading to the downregulation of type I Interferon gene expression (Kubota et al., 2008). In the present study, the combined action of 11,20-dihydroxysugiol, 11-hydroxysugiol, plectranthol A, plectranthol B, and abietatriene was identified to regulate the SUMOylation of intracellular receptors by modulating RARα and VDR. Furthermore, the controversial reports with vitamin D and associated genes in viral infection (Lee, 2020) need to be investigated.
In the present study, all the bioactives were identified to downregulate RARα, except abietatriene. However, previous reports reflect the inhibition of retinoid-associated receptors that may lead to prone viral infections (Song et al., 2018). Furthermore, a previous study reflects the anti-viral potency of P. amboinicus (Arumugam et al., 2016); however, anti-viral efficacy of individual bioactives needs to be still investigated. This result reflects the controversial investigation of lead bioactives against viral infection treatment from P. amboinicus. The outcome of this controversial result could be due to two main reasons. Firstly, the secondary metabolites from P. amboinicus are not sufficient enough to regulate the multiple genes/targets. Secondly, the cutoff point of target prediction could be high against the viral infection as the study is set at a minimum 70% similarity index.
Additionally, 3CLpro, PLpro, and spike protein are involved in the manipulation of the ubiquitin system, processing of pp1a and pp1ab into the replicase proteins, and utilization angiotensin-converting enzyme 2 as a receptor to enter inside the cell (Bhoj and Chen, 2009; Li et al., 2003; Lindner et al., 2005). Based on the binding affinity of bioactives from P. amboinicus against these three bioactives, we identified all the phytoconstituents to act over those proteins, except abietatriene as it did not possess any hydrogen bond interactions compared to the rest of the molecules. This reflects the abietatriene-3CLpro/PLpro/spike protein complex as unstable compared to other bioactives in the biological system. In the docking studies, the highest binding affinity (−7.9 kcal/mol) of plectranthol A and B was predicted with PLpro via the two hydrogen bond interactions, i.e., -OH...GLU204, =O...ARG167 for plectranthol A and -OH...ARG167, -O...THR171 for plectranthol B. Likewise, plectranthol B (−8.9 kcal/mol) was predicted to have the interaction with 3CLpro via =O...PHE294, -OH...ARG105, and -O...GLN107. Furthermore, plectranthol A (−8.4 kcal/mol) was predicted to have the highest binding affinity with spike protein by interacting with -O...LYS807, -OH...CYS812, -O... PHE805, -OH...ASP802, and -O...ALA803. The compound abietatriene had no any hydrogen bond interactions with PLpro, 3CLpro, and spike protein which could be due to the absence of a functional group in the molecules.
The calculation of the druglikeness score helps to understand the oral bioavailability of drug-like compounds if administered orally. This theory was proposed by Lipinksi, which is based on molecular weight, number of hydrogen bond donors, number of hydrogen bond acceptors, and LogP value of the compounds (Lipinski, 2004). Furthermore, P. amboinicus belongs to the traditional medicine system and is consumed orally. Hence, we attempted to investigate the ChEBI-recorded bioactives for druglikeness score in which 80% of bioactives secured positive druglikeness scores, reflecting their probability to get absorbed in the gastrointestinal tract.
CONCLUSION
The present study investigated the reported bioactives in the ChEBI database against three targets of COVID-19 using in silico molecular docking and system biology approach which identified plectranthol A and B as lead hits. However, the findings of the presents are based on computer simulations which need to be further investigated via experimental studies.
CONFLICT OF INTEREST
All the authors declare that they have no conflicts of interest for this work.
FUNDING
None.
REFERENCES
Arumugam G, Swamy MK, Sinniah UR. Plectranthus amboinicus (Lour.) spreng: botanical, phytochemical, pharmacological and nutritional significance. Molecules, 2016; 21(4):369. CrossRef
Balkrishna A, Subarna P, Singh J, Varshney A. Withanone from withania somnifera may inhibit novel coronavirus (COVID-19) entry by disrupting interactions between viral S-protein receptor binding domain and host ACE2 receptor. Res Sq. 2020; CrossRef
Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature, 2009; 458(7237):430–7. CrossRef
Dassault Systèmes BIOVIA. Discovery studio. Dassault Systèmes, San Diego, CA, 2019.
Duyu T, Khatib NA, Khanal P, Patil BM, Hullatti KK. Network pharmacology-based prediction and experimental validation of Mimosa pudica for Alzheimer's disease. J Phytopharmacol, 2020; 9(1):46–53. CrossRef
Gurgel AP, da Silva JG, Grangeiro ARS, Oliveira DC, Lima CMP, da Silva ACP, Oliveira RAG, Souza IA. In vivo study of the anti-inflammatory and antitumor activities of leaves from Plectranthus amboinicus (Lour.) Spreng (Lamiaceae). J Ethnopharmacol, 2009; 125(2):361–3. CrossRef
Halgren TA. Merck molecular force field. I. basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem, 1996; 17:490–519. CrossRef
Hughes GD, Aboyade OM, Beauclair R, Mbamalu ON, Puoane TR. Characterizing herbal medicine Use for noncommunicable diseases in Urban South Africa. Evid Based Complement Alternat Med, 2015; 2015:736074. CrossRef
Khanal P, Duyu T, Patil BM, Dey Y, Pasha I, Kavalapure RS. In silico screening of JAK-STAT modulators from the antiviral plants of Indian traditional system of medicine with the potential to inhibit 2019 novel coronavirus. Res Sq, 2020a. CrossRef
Khanal P, Mandar BK, Magadum P, Patil BM, Hullatti KK. Insilico docking study of Limonoids from Azadirachta indica with pfpk5: a novel anti-malarial target for Plasmodium falciparum. Indian J Pharm Sci, 2019a; 81(2):326–32. CrossRef
Khanal P, Mandar BK, Patil BM, Hullatti KK. In silico antidiabetic screening of borapetoside C, cordifolioside A and magnoflorine. Indian J Pharm Sci, 2019c; 81(3):550–55. CrossRef
Khanal P, Patil BM. Gene set enrichment analysis of alpha-glucosidase inhibitors from ficus benghalensis. Asian Pac J Trop Biomed, 2019; 9(6):263–70. CrossRef
Khanal P, Patil BM, Mandar BK, Dey YN, Duyu T. Network pharmacology-based assessment to elucidate the molecular mechanism of anti-diabetic action of Tinospora cordifolia. Clin Phytosci, 2019b; 5(1):35. CrossRef
Khanal P, Patil BM, Chand J, Naaz Y. Anthraquinone Derivatives as an Immune Booster and their Therapeutic Option Against COVID-19. Nat Prod Bioprospect. 2020;10(5):325–335. CrossRef
Khanal P, Patil BM, Pasha I, Dey YN, Chand S. Withanolides from Withania somnifera as an immune booster and their therapeutic option against COVID-19. Res Sq, 2020b. CrossRef
Khanal P, Patil BM. Gene ontology enrichment analysis of α-amylase inhibitors from duranta repens in diabetes mellitus. J Diabetes Metab Disord, 2020b. CrossRef
Khanal P, Patil BM. α-Glucosidase inhibitors from Duranta repens modulate p53 signaling pathway in diabetes mellitus. Adv Tradit Med, 2020a; 20:427–38. CrossRef
Kubota T, Matsuoka M, Chang TH, Tailor P, Sasaki T, Tashiro M, Kato A, Ozato K. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J Biol Chem, 2008; 283(37):25660–70. CrossRef
Lagunin A, Ivanov S, Rudik A, Filimonov D, Poroikov V. DIGEP-pred: web service for in silico prediction of drug-induced gene expression profiles based on structural formula. Bioinformatics, 2013; 29:2062–3. CrossRef
Lee C. Controversial effects of vitamin D and related genes on viral infections, pathogenesis, and treatment outcomes. Nutrients, 2020; 12(4):962. CrossRef
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. Angiotensinconverting enzyme 2 is a functional receptor for the SARS coronavirus. Nature, 2003; 426(6965):450–4. CrossRef
Lindner HA, Fotouhi-Ardakani N, Lytvyn, Lachance P, Sulea T, Ménard R. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J Virol, 2005; 79(24):15199–208. CrossRef
Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol, 2004;1(4):337–41. CrossRef
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ AutoDock4 and autoDockTools4: automated docking with selective receptor flexibility. J Comput Chem, 2009; 30:2785–91. CrossRef
Patil VS, Biradar PR, Attar V, Khanal P. In silico docking analysis of active biomolecules from cissus quadrangularis L. against PPAR-γ. Indian J Pharm Educ. 2019; 53(3S):S332–7. CrossRef
Pinn G. Herbal medicine in infectious disease. Aust Fam Physician, 2001; 30(7):681–4.
Rodrigues J, Hullatti KK, Jalalpure S, Khanal P. In-vitro Cytotoxicity and in silico molecular docking of alkaloids from Tiliacora acuminata. Indian J Pharm Educ, 2020a; 54(2S):S295–300. CrossRef
Rodrigues J, Hullatti KK, Khanal P. In silico and in vitro cytotoxicity profile of hydroalcoholic extract/fraction(s) of Pachygone ovata. J Appl Pharm Sci, 2020b; 10(05):135–41. CrossRef
Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003; 31(13):3381–5. CrossRef
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res, 2003; 13:2498–504. CrossRef
Song M, Sun Y, Tian J, He W, Xu G, Jing Z, Li W. Silencing retinoid X receptor alpha expression enhances early-stage hepatitis B virus infection in cell cultures. J Virol, 2018; 92(8):e01771–17. CrossRef
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering CV. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res, 2019; 47:D607–13. CrossRef
Ternikar SG, Patil MB, Pasha I, Khanal P. Gene set enrichment analysis of α-amylase and α-glucosidase inhibitors of Cassia glauca. J Diabetes Metab Disord, 2020. CrossRef
WHO. Coronavirus disease (COVID-19) advice for the public. 2019a. Available via: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/advice-for-public. (Accessed 03 June 2020).
WHO. Rolling updates on coronavirus disease (COVID-19). 2019b Available via: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/events-as-they-happen. (Accessed 03 June 2020).