
INTRODUCTION
Coronavirus disease 2019 (COVID-19) is a multisystem infection caused by the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) [1]. The COVID-19 pandemic has resulted in an unprecedented response from the scientific community, offering some urgent treatment based on traditional pharmacological chemistry, drug repurposing, and computational screening approaches [2,3]. Drug repurposing of small-molecule antivirals for COVID-19 treatment has been undertaken to identify potential new therapeutics to treat SARS-CoV-2 [4]. Despite the considerable amount of computational and experimental data available for the treatment of COVID-19, only remdesivir, molnupiravir, and nirmatrelvir have been authorized by the U.S. Food and Drug Administration (FDA) as drugs for the treatment of COVID-19 [5–7]. Paxlovid is one of the first approved antiviral drugs for treating COVID-19, developed by Pfizer in 2021 [8–11]. Nirmatrelvir (PF-07321332) is the main ingredient of Paxlovid and inhibits the main protease (Mpro) of virus SARS-CoV-2 (Fig. 1). It is an orally bioavailable drug against SARS-CoV-2 [5,12–15], which also shows antiviral potency against other human coronaviruses [8,16,17].
Nirmatrelvir has a peptide-like structure (Fig. 1), and its co-crystalized complex with the Mpro protease opened up the opportunities for developing a broad family of tri- or tetrapeptide mimetics containing an electrophile capable of interacting with the catalytic cysteine [5,7]. Therefore, the structure of nirmatrelvir is a promising scaffold for the rational design of new, more effective antiviral drugs. Considering its complex multistep synthesis, we utilize an alternative approach based on a computer-aided drug design and prediction of the relationship between the structure of nirmatrelvir derivatives and their binding affinity (BA) for the active site of the Mpro enzyme of SARS-CoV-2.
METHODOLOGY OF RESEARCH
Computer-aided drug discovery has become a powerful tool for streamlining structure-based virtual screening of large chemical databases and offering new opportunities for the cost-effective development of new drug-like ligands [18]. However, this approach is known to be crucially dependent on used chemical spaces. There are several approaches to creating a virtual chemical library of new molecules that are structurally similar to a target structure, such as generating combinatorial libraries from approved drugs and natural products. Another approach is building an evolutionary library starting from a parent scaffold and going through small mutation, fitness scoring, ranking, and selection toward drug-like molecules [19].
Building of virtual evolutionary library
First, we generated a virtual library of nirmatrelvir analogs utilizing an evolutionary algorithm that mimics nature’s evolution of drug-like compounds using DataWarrior software [20]. The procedure was as follows: the algorithm starts with a parent nirmatrelvir (1) (Fig. 1) called the first generation. Next, a series of similar derivatives were built by applying a minor random structural modification of a parent structure. Each new structural modification is then evaluated regarding how much it fits within up to seven customizable fitness criteria, such as molecular weight (Mw), cLogP, polar surface area (PSA) [21], molecular shape and flexibility, and so on (Table 1).
The molecular weight of a drug-like molecule is a crucial parameter that helps to correlate its biological activity with its chemical and physical properties. According to Lipinski’s rule of five [22], for drugs to be orally bioavailable, Mw should be less than 500 g/mol. Nirmatrelvir has Mw = 499 g/mol, so the corresponding fitness criterion was set to be within 490–515 g/mol (see Table 1).
The calculated logarithm of the partition coefficient of a compound between n-octanol and water log(Coctanol/Cwater), referred to as cLogP, is one of the critical fitness criteria. It is a well-established measure of the drug’s hydrophilicity. Low hydrophilicity and, hence, high cLogP leads to poor absorption or permeation. For nirmatrelvir, cLogP equals 0.98, so the corresponding fitness criterion was targeted to be within 0.8–1.2 (Table 1).
The PSA defines the surface sum over all polar atoms, such as oxygen, nitrogen, sulfur, and phosphorus, including attached hydrogens. PSA is a commonly used medicinal chemistry criterion for estimating cell permeability. Drug-like molecules with PSA >140 Å2 are typically poorly permeable into cell membranes. In addition, to penetrate the blood-brain barrier, PSA should be less than 60 Å2. Nirmatrelvir has a PSA of 131 Å2, so this fitness criterion was fixed to be 110–150 Å2.
In addition to the above criteria, to assess the shape and 3-D conformational similarity of multiple conformers of the candidate molecules, two following algorithms were applied: (i) Pharmacophore Enhanced Shape Alignment “PheSA” and (ii) OrgFunctions Descriptor. These tools allow us to check whether any two molecules may have a compatible protein binding behavior in terms of shape, size, and flexibility (see Table 1).
Structural mutations that best fit these criteria are assigned a higher probability during the generation of new chemical space. The new analogs within the current generation are ranked according to these criteria so that the highest-ranking molecules from this generation are selected to survive and form the parent molecules for the next generation. Following this evolutionary algorithm, up to hundred generations were created with a total number of 1,866 virtual analogs.
Figure 2 shows the 2-D distribution of Mw versus cLogP in the generated library, which reveals a symmetric distribution around a central red point corresponding to a parent nirmatrelvir.
3-D pharmacophore screening
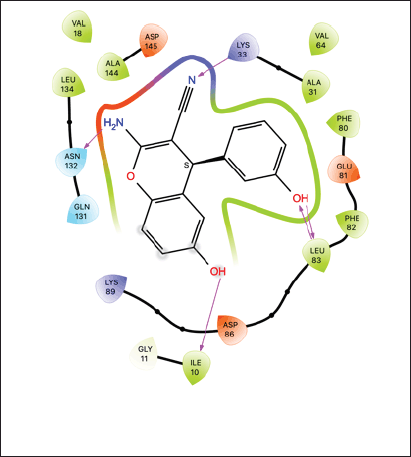
The generated evolutionary library is too large for conventional molecular docking against the Mpro enzyme. Therefore, it was subject to further preprocessing and filtering to match a five-point 3-D pharmacophore model of nirmatrelvir generated using its first available crystal structure in a complex with Mpro (PDB: 7VH8) [23] using LigandScout software developed by Wolber and Langer [24]. A 3-D pharmacophore model is a chemical fingerprint, which takes into account certain chemical features of a ligand, such as hydrogen bond donors, acceptors, lipophilic areas, and positively and negatively ionizable chemical groups, that describe its interaction with the surrounding binding site of a protein. Figure 3 shows the five-point pharmacophore model for nirmatrelvir, which identifies two key pharmacophoric points, such as peripheral trifluoroacetyl group and 6,6-dimethyl-3-azobicyclo[3.1.0]hexane moiety. Such a five-point 3-D pharmacophore has been used to characterize key interactions within the complex nirmatrelvir-Mpro [25]. Finally, applying the pharmacophore-guided screening procedure allowed us to reduce the chemical space of the evolutionary library from 1,866 to 32 active candidates.
Structure-based molecular docking
To evaluate the BA of the 32 selected active hit molecules, we applied structure-based molecular docking calculations using LigandScout software [24]. The LigandScout software uses a built-in version of AutoDock Vina 1.1 [26]. Therefore, to validate the ligand-protease docking algorithm, nirmatrelvir was re-docked, and its geometry was compared with the corresponding X-ray structure of the enzyme-bound ligand with Mpro (PDB: 7VH8), as shown in Figure 4. The molecular docking results for the best-binding conformation of nirmatrelvir and its experimental crystallographic structure are in good agreement, which allows us to use this method for the quantitative analysis of the Mpro affinity of new nirmatrelvir derivatives.
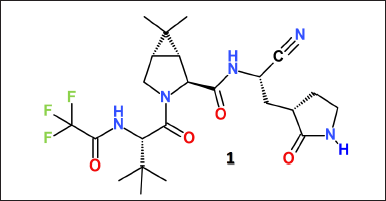 | Figure 1. Molecular structure of nirmatrelvir (PF-07321332). [Click here to view] |
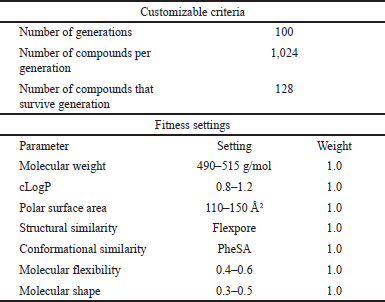 | Table 1. Customizable fitness criteria and corresponding parameters are used to build the virtual evolutionary library of nirmatrelvir`s analogs. [Click here to view] |
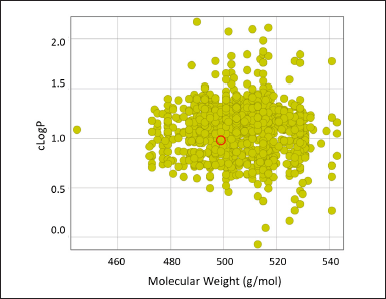 | Figure 2. 2-D plot of distributions of Mw versus cLogP parameters for the generated evolutionary library. [Click here to view] |
Finally, the BA of the 32 selected high-ranked nirmatrelvir analogs was evaluated by conventional molecular docking calculations against the two different X-ray structures of the Mpro enzyme. The first Mpro structure corresponds to the protein taken from its co-crystalized complex with nirmatrelvir (PDB 7VH8). The second Mpro structure corresponds to the ligand-free protein measured at 310 K (PDB 7MHK) [27]. Finally, our in silico screening procedure allowed us to identify up to eight hits with a BA higher than that of a parent nirmatrelvir.
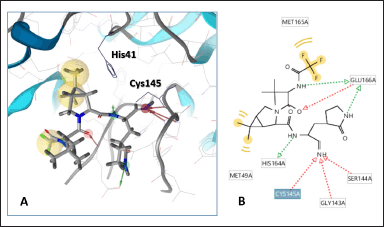 | Figure 3. (A) 3-D five-point pharmacophore model of nirmatrelvir derived from its crystal structure in a complex with Mpro (PDB: 7VH8). Highlighted residues His41 and Cys145 belong to the catalytic dyad of the active site of Mpro. (B) Scheme of intermolecular interactions of nirmatrelvir with key surrounding residues of Mpro. [Click here to view] |
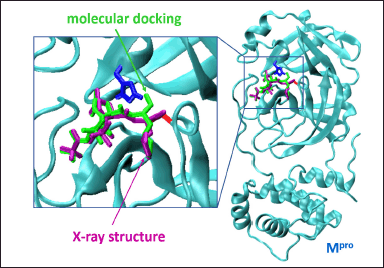 | Figure 4. Comparison of the results of molecular docking of nirmatrelvir with its X-ray structure in the complex with Mpro. The geometry of nirmatrelvir incorporation into the Mpro main protease molecule of the SARS-CoV-2 virus (PDB: 7VH8). The catalytic amino acid residues His41 and Cys145 of the active center of the protease are shown in blue or red. The binding mode of the ligand estimated by the molecular docking is shown in green, whereas the X-ray structure is given in purple. [Click here to view] |
COMPUTATIONAL METHODS
OSIRIS DataWarrior software, version 5.5.0., was used to generate an evolutionary library and to calculate the physicochemical properties of ligands [20].
MarvinSketch was used for drawing, displaying, and characterizing chemical structures (MarvinSketch 23.1., 2023, ChemAxon, https://chemaxon.com/, accessed on 1 February 2023).
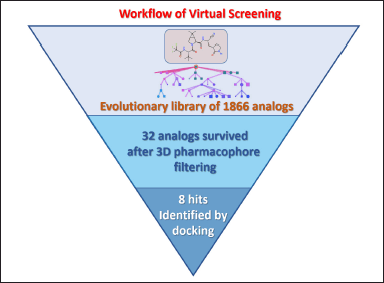 | Figure 5. A workflow of virtual screening of nirmatrelvir analogs. [Click here to view] |
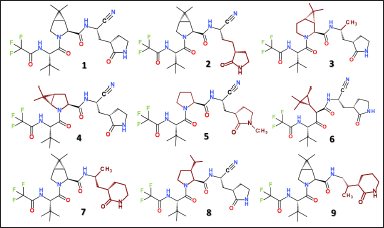 | Figure 6. Molecular structure of nirmatrelvir (1) and its designed analogs, revealing the higher BA. Mutated fragments are colored in wine. [Click here to view] |
Molecular docking was performed for selected inhibitors against the Mpro protease of SARS-CoV-2. The spatial structure of the Mpro protease was taken from the Protein Data Bank (PDB ID: 7VH8) [23]. The ligand and water molecules were removed. Semi-flexible molecular docking calculations were performed using the AutoDock Vina 1.1 [26] built into the LigandScout software (version 4.4.9) [24], in which the receptor was kept rigid, and the ligand molecules were conformationally flexible. For each ligand, three independent runs were performed. The best docking mode corresponds to the highest ligand BA. Molecular graphics and visualization were performed using LigandScout 4.4.9 and VMD 1.9.3 [28].
RESULT AND DISCUSSION
Identifying novel (i.e., never-before-seen) drug-like compounds, relying upon in silico screening of “in stock” and “on demand” virtual libraries of compounds, is a critical and rate-limiting step in drug discovery [29–32]. Therefore, alternative strategies have also been suggested based on generating new chemical spaces, originating from already approved drugs and following its consequent hit-to-lead optimization [33,30,34,19,35–37]. Herein, we report structure-based optimization of new noncovalent inhibitors against the coronavirus SARS-CoV-2 Mpro, starting from the FDA-approved drug nirmatrelvir. The X-ray structure of Mpro-bound nirmatrelvir was used as a targeting scaffold to guide the optimization procedure. A series of structural mutations of nirmatrelvir were suggested, accompanied by adjustments of corresponding physicochemical properties, such as Mw, cLogP, and PSA, respectively. Our virtual screening workflow and hit-selecting protocols of the best-binding analogs of nirmatrelvir are summarized in Figure 5.
The generated evolutionary library composed of 1,866 nirmatrelvir-like compounds was optimized by seven physicochemical fitness criteria (Table 1). However, filtering this library through the five-point 3-D pharmacophore model of nirmatrelvir (Fig. 3) discarded the vast majority of generated structures, while only an exceedingly small subset of 32 top-scoring compounds survived for further analysis.
To evaluate the BA of these 32 active hit candidates against Mpro, we applied conventional molecular docking calculations using LigandScout software. Our docking results demonstrate that up to eight designed analogs revealed a BA higher than a parent nirmatrelvir did, as summarized in Figure 6 and Table 2.
Energetic characteristics of the protease-ligand interaction and some physicochemical properties of hit-analogs are summarized in Table 2. The following parameters were selected as BA criteria: (1) BA estimated by the AutoDock Vina docking algorithm. This parameter is very common and allows a direct comparison with the previously published BAs of a large number of ligands revealing the inhibitory activity against SARS-CoV-2 coronavirus proteases [38–42]. (2) Binding enthalpy (BE) for the protein–ligand complex calculated using the MMF94 empirical force field. This parameter is calculated as the difference between the potential energy of the complex and the energy of the starting substances. (3) Binding affinity score (BAS) calculated by the LigandScout software package. BAS takes into account both the protein-ligand BE and the environmental effect. This parameter is considered the most representative and was therefore chosen for the selection of hit ligands [43].
The calculated value of the binding enthalpy for nirmatrelvir equals −10.4 kcal/mol, which agrees well with the experimentally determined value of isothermal titration calorimetry −10.75 kcal/mol [44]. The experimental free energy of nirmatrelvir binding to Mpro is −11.2 kcal/mol, which is somewhat underestimated to be −7.2 kcal/mol by AutoDock Vina.
Figure 7 shows the typical binding modes of nirmatrelvir and its hit-analogs at the active site of the Mpro enzyme. In conventional docking, all the inhibitors utilized a noncovalent mode of action. Notably, analogs 2-6 bind to the Mpro pocket by its N-terminal trifluoroacetamide moiety. The results of Table 2 and Figure 7 indicate that minor structural modifications of the parent nirmatrelvir 1, such as the replacement of 3-ethyl-2-oxopyrrolidine by 3-propyl-2-oxopyrrolidine in 2, lead to an increase of BE and BAS from −13.6 to −22.3, respectively. It suggests the possibility of further modification on these sites to increase the binding strength with Mpro.
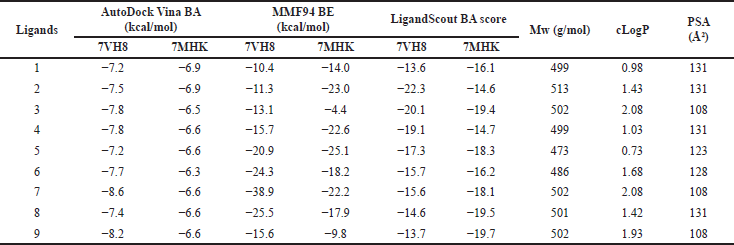 | Table 2. Molecular docking results of nirmatrelvir and its analogs against two different X-ray structures (PDB ID: 7VH8 and 7MHK) and their physicochemical parameters. [Click here to view] |
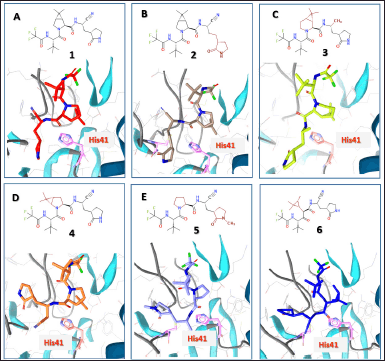 | Figure 7. Best-binding modes of nirmatrelvir (1) and its analogs at the active site of the Mpro enzyme estimated by molecular docking calculations. [Click here to view] |
A characteristic structural feature of the inhibitor binding with the active center of Mpro is the similar location of the trifluoroacetylamino fragment, which is observed in derivatives 1-6 presented in Figure 7. Thus, the overall BA of these inhibitors is increased due to structural modifications of the opposite part of the molecule. The main structural modifications that occurred in inhibitors 2-9 are located in two fragments of the nirmatrelvir molecule, namely, in the azabicyclo[3.1.0]hexane and oxopyrrolidine fragments (Fig. 7).
Figure 8A and C show that derivatives 7 and 9 increase in BAS from −13.6 to −15.6 and −13.7 (Table 2), respectively. A characteristic structural feature of both derivatives is the replacement of the 2-oxopyrrolidine ring with a 2-oxopiperidine ring. However, such a modification obviously does not contribute to increasing the antiviral effect of nirmatrelvir derivatives.
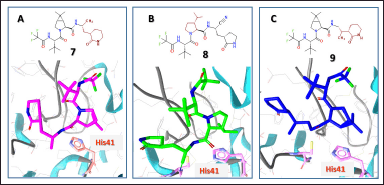 | Figure 8. Binding modes of some nirmatrelvir’s analogs at the active site of the Mpro enzyme estimated by molecular docking calculations. [Click here to view] |
Finally, it has been reported that a conventional docking procedure utilizing an enzyme structure from a corresponding co-crystalized protein-ligand complex is somewhat affected by the structural plasticity of an enzyme and the presence of a bulky ligand bound at the active center [45–47]. Therefore, the selected 32 hit analogs were also re-docked against the ligand-free room-temperature X-ray structure of Mpro (PDB 7MHK), as summarized in Figure S1 and Table 2. These results suggest that the ligand binding to the Mpro structure, measured at 310 K, is characterized by some decrease in the absolute BA. However, our molecular docking demonstrates that, in terms of BAS, the most identified hit analogs, except 2 and 4, revealed promising binding parameters.
CONCLUSION
Herein, we carried out a computer-aided drug design intending to identify new analogs of nirmatrelvir capable of a higher antiviral potency against the coronavirus SARS-CoV-2. First, we generated a new virtual library composed of 1,866 nirmatrelvir analogs, which was further preprocessed and filtered against a 3-D pharmacophore model of a parent nirmatrelvir, reducing the chemical space to 32 active analogs. Second, conventional structure-based molecular docking against the two different enzyme structures allowed us to identify up to eight nirmatrelvir analogs with high-ranked binding characteristics. The selected hits target the Mpro enzymes of SARS-CoV-2 with a higher BA than a parent nirmatrelvir. We found that the main structural modifications that increase the overall inhibitory effect of novel analogs are localized in the azabicyclo[3.1.0]hexane and 2-oxopyrrolidine fragments. Finally, the suggested workflow of the rational design of new antiviral noncovalent inhibitors based on the scaffold of approved drugs is a promising, extremely low-cost, and time-efficient approach for developing new potential pharmaceutical ingredients for the treatment of COVID-19.
AUTHOR CONTRIBUTIONS
K.O.L was responsible for the conceptualization, formal analysis, investigation, methodology, and writing of the original draft. A.S.S contributed to the investigation and methodology. A.K, V.V.I, T.L, S.M.K, and O.N.K were responsible for methodology, writing of the original draft, and the review and editing of the article.
FINANCIAL SUPPORT
K.O.L., A.K., V.V.I., S.M.K., and O.N.K. acknowledge Grant ? 42/0062 (2021.01/0062) “Molecular design, synthesis and screening of new potential antiviral pharmaceutical ingredients for the treatment of infectious diseases COVID-19” from the National Research Foundation of Ukraine.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest in relation to this research, whether financial, personal, authorship, or otherwise, that could affect the research and its results presented in this paper.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. La Monica G, Bono A, Lauria A, Martorana A. Targeting SARS-CoV-2 main protease for treatment of COVID-19: covalent inhibitors structure–activity relationship insights and evolution perspectives. J Med Chem. 2022;65(19):12500–34. CrossRef
2. da Silva SJR, do Nascimento JCF, Germano Mendes RP, Guarines KM, Targino Alves da Silva C, da Silva PG, et al. Two years into the COVID-19 pandemic: lessons learned. ACS Infect Dis. 2022;8(9):1758–814. CrossRef
3. Mousavi H, Zeynizadeh B, Rimaz M. Green and efficient one-pot three-component synthesis of novel drug-like furo[2,3-d]pyrimidines as potential active site inhibitors and putative allosteric hotspots modulators of both SARS-CoV-2 MPro and PLPro. Bioorg Chem. 2023;135:106390. CrossRef
4. Galindez G, Matschinske J, Rose TD, Sadegh S, Salgado-Albarrán M, Späth J, et al. Lessons from the COVID-19 pandemic for advancing computational drug repurposing strategies. Nat Comput Sci. 2021;1(1):33–41. CrossRef
5. Allais C, Connor CG, Do NM, Kulkarni S, Lee JW, Lee T, et al. Development of the commercial manufacturing process for Nirmatrelvir in 17 months. ACS Centr Sci. 2023;9(5):849–57. doi: https://doi.org/10.1021/acscentsci.3c00145 CrossRef
6. Fischer C, Feys JR. SARS-CoV-2 Mpro inhibitors: achieved diversity, developing resistance and future strategies. Future pharmacol. 2023;3(1):80–107. CrossRef
7. Lamb YN. Nirmatrelvir plus ritonavir: first approval. Drugs. 2022;82(5):585–91. CrossRef
8. Blair HA. Nirmatrelvir plus ritonavir in COVID-19: a profile of its use. Drug Therap Perspect. 2023;39(2):41–7. CrossRef
9. Halford B. The path to Paxlovid. ACS Cent Sci. 2022;8(4):405–7. CrossRef
10. Marzi M, Vakil MK, Bahmanyar M, Zarenezhad E. Paxlovid: mechanism of action, synthesis, and in silico study. BioMed Res Intern. 2022;2022:7341493. CrossRef
11. Owen DR, Allerton CMN, Anderson AS, Aschenbrenner L, Avery M, Berritt S, et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374(6575):1586–93. CrossRef
12. Dawood AA. The efficacy of Paxlovid against COVID-19 is the result of the tight molecular docking between Mpro and antiviral drugs (nirmatrelvir and ritonavir). Adv Med Sci. 2023;68(1):1–9. CrossRef
13. Duveau DY, Thomas CJ. The remarkable selectivity of nirmatrelvir. ACS Pharm Transl Sci. 2022;5(6):445–7. CrossRef
14. Mahase E. Covid-19: Pfizer’s paxlovid is 89% effective in patients at risk of serious illness, company reports. BMJ. 2021;375:n2713. CrossRef
15. Yang KS, Leeuwon SZ, Xu S, Liu WR. Evolutionary and structural insights about potential SARS-CoV-2 evasion of nirmatrelvir. J Med Chem. 2022;65(13):8686–98. CrossRef
16. Li J, Wang Y, Solanki K, Atre R, Lavrijsen M, Pan Q, et al. Nirmatrelvir exerts distinct antiviral potency against different human coronaviruses. Antiviral Res. 2023;211:105555. CrossRef
17. Petrakis V, Rafailidis P, Trypsianis G, Papazoglou D, Panagopoulos P. The antiviral effect of nirmatrelvir/ritonavir during COVID-19 pandemic real-world data. Viruses. 2023;15(4):976. CrossRef
18. Sadybekov AV, Katritch V. Computational approaches streamlining drug discovery. Nature. 2023;616(7958):673–85. CrossRef
19. Lyu J, Irwin JJ, Shoichet BK. Modeling the expansion of virtual screening libraries. Nat Chem Biol. 2023;19:712–18. doi: https://doi.org/10.1038/s41589-022-01234-w CrossRef
20. Sander T, Freyss J, von Korff M, Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model. 2015;55(2):460–73. CrossRef
21. Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem. 2000;43(20):3714–7. CrossRef
22. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev. 2001;46(1):3–26. CrossRef
23. Zhao Y, Fang C, Zhang Q, Zhang R, Zhao X, Duan Y, et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Prot Cell. 2021;13(9): 689–93. CrossRef
24. Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model. 2005;45(1):160–9. CrossRef
25. Hayek-Orduz Y, Vásquez AF, Villegas-Torres MF, Caicedo PA, Achenie LEK, González Barrios AF. Novel covalent and non-covalent complex-based pharmacophore models of SARS-CoV-2 main protease (Mpro) elucidated by microsecond MD simulations. Sci Rep. 2022;12(1):14030. CrossRef
26. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–61. CrossRef
27. Ebrahim A, Riley BT, Kumaran D, Andi B, Fuchs MR, McSweeney S, et al. The temperature-dependent conformational ensemble of SARS-CoV-2 main protease (Mpro). IUCrJ. 2022;9(5):682–94. CrossRef
28. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–8. CrossRef
29. Chatterjee A, Walters R, Shafi Z, Ahmed OS, Sebek M, Gysi D, et al. Improving the generalizability of protein-ligand binding predictions with AI-Bind. Nat Commun. 2023;14(1):1989. CrossRef
30. Gentile F, Fernandez M, Ban F, Ton AT, Mslati H, Perez CF, et al. Automated discovery of noncovalent inhibitors of SARS-CoV-2 main protease by consensus Deep Docking of 40 billion small molecules. Chem Sci. 2021;12(48):15960–74. CrossRef
31. Joshi RP, Schultz KJ, Wilson JW, Kruel A, Varikoti RA, Kombala CJ, et al. AI-Accelerated design of targeted covalent inhibitors for SARS-CoV-2. J Chem Inf Model. 2023;63(5):1438–53. CrossRef
32. Zahrychuk HY, Gladkov ES, Kyrychenko AV, Poliovyi DO, Zahrychuk OM, Kucher TV, et al. Structure-based rational design and virtual screening of valsartan drug analogs towards developing novel inhibitors of Angiotensin II type 1 receptor. Biointerface Res Appl Chem. 2023;13(5):440.
33. Brewitz L, Dumjahn L, Zhao Y, Owen CD, Laidlaw SM, Malla TR, et al. Alkyne derivatives of SARS-CoV-2 main protease inhibitors including nirmatrelvir inhibit by reacting covalently with the nucleophilic cysteine. J Med Chem. 2023;66(4):2663–80. CrossRef
34. Hou N, Shuai L, Zhang L, Xie X, Tang K, Zhu Y, et al. Development of highly potent noncovalent inhibitors of SARS-CoV-2 3CLpro. ACS Cent Sci. 2023;9(2):217–27. CrossRef
35. Pozzi C, Vanet A, Francesconi V, Tagliazucchi L, Tassone G, Venturelli A, et al. Antitarget, anti-SARS-CoV-2 leads, drugs, and the drug discovery–genetics alliance perspective. J Med Chem. 2023;66(6):3664–702. CrossRef
36. Saramago LC, Santana MV, Gomes BF, Dantas RF, Senger MR, Oliveira Borges PH, et al. AI-Driven discovery of SARS-CoV-2 main protease fragment-like inhibitors with antiviral activity in vitro. J Chem Inf Model. 2023;63(9):2866–80. CrossRef
37. Snizhko AD, Kyrychenko AV, Gladkov ES. Synthesis of novel derivatives of 5,6,7,8-tetrahydro-quinazolines using of α-aminoamidines and in silico screening of their biological activity. Int J Mol Sci. 2022;23(7):3781. CrossRef
38. Al-Khafaji K, Al-Duhaidahawi D, Taskin Tok T. Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. J Biomol Struct Dyn. 2020;39(9):3387–95. CrossRef
39. Ghahremanpour MM, Tirado-Rives J, Deshmukh M, Ippolito JA, Zhang CH, Cabeza de Vaca I, et al. Identification of 14 known drugs as inhibitors of the main protease of SARS-CoV-2. ACS Med Chem Lett. 2020;11(12):2526–33. CrossRef
40. Llanos MA, Gantner ME, Rodriguez S, Alberca LN, Bellera CL, Talevi A, et al. Strengths and weaknesses of docking simulations in the SARS-CoV-2 era: the main protease (Mpro) case study. J Chem Inf Model. 2021;61(8):3758–70. CrossRef
41. Mengist HM, Dilnessa T, Jin T. Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Front Chem. 2021;9:622898. CrossRef
42. Thangavel N, Albratty M. Pharmacophore model-aided virtual screening combined with comparative molecular docking and molecular dynamics for identification of marine natural products as SARS-CoV-2 papain-like protease inhibitors. Arab J Chem. 2022;15(12):104334. CrossRef
43. Evseeva LV, Ivanov VV, Karpina VR, Kovalenko SS, Kuznetsov IE, Langer T, et al. The virtual screening application for searching potential antiviral agents to treat COVID-19 disease. J Org Pharm Chem. 2020;18(2):3–15. CrossRef
44. Kneller DW, Li H, Phillips G, Weiss KL, Zhang Q, Arnould MA, et al. Covalent narlaprevir- and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat Commun. 2022;13(1):2268. CrossRef
45. Kneller DW, Phillips G, O’Neill HM, Jedrzejczak R, Stols L, Langan P, et al. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat Commun. 2020;11(1):3202. CrossRef
46. Pathak N, Chen YT, Hsu YC, Hsu NY, Kuo CJ, Tsai HP, et al. Uncovering flexible active site conformations of SARS-CoV-2 3CL proteases through protease pharmacophore clusters and COVID-19 drug repurposing. ACS Nano. 2021;15(1):857–72. CrossRef
47. Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368(6489):409. CrossRef
SUPPLEMENTARY MATERIAL
 | Figure S1. Best-binding modes of nirmatrelvir (1) and its analogs at the active site of the root-temperature Mpro enzyme structure estimated by molecular docking calculations [Click here to view] |