
INTRODUCTION
Respiratory influenza viruses continue to pose significant threats to global public health, leading to seasonal outbreaks and occasional pandemics with substantial morbidity and mortality [1]. Nowadays, advanced testing and management of healthcare have helped to maintain the fatality rate low [2]. Despite advancements in vaccination strategies and antiviral treatments, the on-going evolution of influenza viruses and the emergence of drug-resistant strains due to their global prevalence have led to an increase in reported cases by the end of 2023 and continuing into 2024 [3], highlighting the urgent need for novel therapeutic interventions. In recent years, computational approaches have gained prominence in drug discovery efforts, offering efficient and cost-effective means to screen large chemical libraries for potential inhibitors of viral replication. The development of multitarget antiviral agents that can simultaneously inhibit various viral proteins is a promising approach to overcoming resistance and enhancing treatment efficacy[4].
Glycyrrhizic acid (GA), obtained from the roots of the Glycyrrhiza glabra (liquorice) species, is one of the natural substances that has recently gained a lot of attention due to its antiviral potential. GA demonstrates a diverse array of pharmacological properties, encompassing anti-inflammatory, antioxidant, and antiviral effects. Recent research has highlighted its potential against several viruses, such as the human immunodeficiency virus, hepatitis C virus, and severe acute respiratory syndrome coronavirus (SARS-CoV). Furthermore, traditional medical systems assert that GA possesses antiviral capabilities. Previous research has demonstrated its inhibitory properties against a variety of respiratory viruses, including the influenza type A and B viruses. However, the precise molecular mechanisms underlying its antiviral activity, particularly against respiratory influenza viruses, remain incompletely understood.
The objective of this study is to utilize molecular docking to identify the binding affinities and preferred binding sites of GA and key viral protein targets, including the ATP-bound state of BiP (5E84), main protease (Mpro) (6LU7), spike receptor binding domain (6LZG), RNA-dependent RNA polymerase (6M71), spike glycoprotein (6VSB), NSP15 endonuclease (6VWW), Nsp9 RNA-binding protein (6W4B), papain-like protease (6W9C), and neurominadase from H1N1 (5NZ4), followed by MD simulations to examine the stability and dynamic behavior of these interactions. We want to find the best binding conformations and understand how GA’s inhibitory effects on respiratory influenza viruses work at the molecular level by using a wide range of computational methods, such as binding free energy calculations and structural dynamics assessments. We anticipate that the research findings will illuminate GA’s potential as a multitarget antiviral agent against influenza viruses, thereby facilitating future experimental validation and therapeutic development.
The information from this study is very important for the smart design of GA-based medicines and the creation of new multitarget agents to fight respiratory flu viruses by using molecular docking and molecular dynamics simulations (MDSs). Moreover, the computational framework presented herein may serve as a valuable platform for screening other natural compounds and synthetic derivatives with potential antiviral activity, thus contributing to the on-going efforts to mitigate the impact of influenza infections on public health [5–7]. The results may also provide a foundation for the design of novel GA-based therapeutics, offering a promising avenue for the prevention and treatment of influenza infections.
Subsequent MD simulations revealed details on the stability and dynamics of GA-protein complexes throughout time. The simulations demonstrated that GA and the active sites of the spike receptor binding domain (6LZG) exhibited persistent interactions. Hydrophobic and hydrogen bonding interactions primarily contributed to the stability of the binding. Furthermore, estimations of binding free energy supported the favorable binding affinities found in docking studies.
MATERIAL AND METHODS
Molecular docking
Retrieving herbal compounds and preparation of drug library
The 3D structures of GA from liquorice were retrieved from the PubChem database. We used the Structure File Generator (https://cactus.nci.nih.gov/translate/) and Online SMILES Translator to draw and convert compounds with missing.mol/.sdf files [8,9].
The way molecules interact in living things is not fully captured by molecular docking because it is based on the static structures of proteins and ligands. This simplification can affect the accuracy of predicted binding affinities and poses a challenge in interpreting the results as definitive.
Target protein retrieval and reparation
Based on the literature survey, we found that various targets, such as the ATP-bound state of BiP (5E84), main protease (Mpro) (6LU7), spike receptor binding domain (6LZG), RNA-dependent RNA polymerase (6M71), spike glycoprotein (6VSB), NSP15 endonuclease (6VWW), Nsp9 RNA-binding protein (6W4B), papain-like protease (6W9C), and neurominidase from the H1N1 (5NZ4) are major targets to study antiviral activity against respiratory influenza viruses, including SARS-CoV-1 and SARS-CoV-2 [10–14]. Therefore, we retrieved the fasta sequences of the aforementioned targets for antiviral activity against respiratory influenza viruses, such as SARS-CoV-1 and SARS-CoV-2, from the National Centre for Biotechnology information server. We also used the Basic Local Alignment Search tool to search for similar biological sequences on the Protein Data Bank. We then sorted the top 5 to 10 selected sequences based on their improved query coverage, percentage identity, and E-value. We used the PDB databank to obtain the three-dimensional X-ray crystallographic structure of various targets, such as the ATP-bound state of BiP (5E84), main protease (Mpro) (6LU7), spike receptor binding domain (6LZG), RNA-dependent RNA polymerase (6M71), spike glycoprotein (6VSB), NSP15 endonuclease (6VWW), Nsp9 RNA-binding protein (6W4B), papain-like protease (6W9C), and neurominidase from the H1N1 (5NZ4). We validated each target’s accession number using parameters including resolution, mutation, wwPDB validation, co-crystal ligand, and Ramachandran plot [15,16]. Table 1 presents a comparison between the standard values and the recovered protein, which serves as validation for the docking study protein.
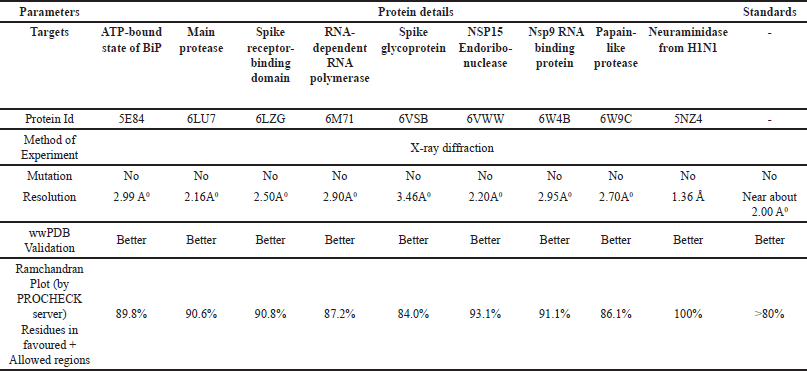 | Table 1. Comparison of standard values with recovered protein for validating docking study protein. [Click here to view] |
The angles from a Ramachandran plot can be used to verify the solution to a crystal structure as well as determine the role of an amino acid in a secondary structure. It also assists with constraining structure prediction simulations and defining energy functions. It displays the protein structure regions that energetically allow backbone dihedral angles against amino acid residues. Table 2 below displays Ramachandran plots for several study targets. The PROCHECK web tool determined the three-dimensional geometry of the protein model, calculated the Ramachandran plot, and generated results for residues in various colored regions, namely red (favorite), yellow (additionally allowed), pale yellow (generously allowed), and whitish yellow (disallowed) [17,18].
 | Table 2. Ramachandran Plots Analysis of Protein Structures Using PROCHECK: comparative assessment for proteins (a) ATP-bound state of BiP (5E84), (b) Main protease (6LU7), (c) Spike receptor-binding domain(6LZG), (d) RNA-dependent RNA polymerase (6M71), (e) Spike glycoprotein (6VSB), (f) NSP15 Endoribo-nuclease (6VWW), (g) Papain-like protease (6W4B), (h) Papain-like protease (6W9C), and (i) Neuraminidase from H1N1 (5NZ4). [Click here to view] |
Every molecule has the potential to be either a macromolecule or a micromolecule; however, it is necessary to optimize and minimize the latter before conducting a docking study. To confirm the binding pocket, we used the protein data bank’s PDB [sum server], a pictorial library of 3D structures for conventional inhibitor interactions with proteins. We completed the proteins with missing residues and synthesized side chains using CHIMAERA v1.16. Subsequently, they underwent optimization and minimization before being included in the docking study. We achieve the optimization of proteins by configuring 1,000 iterations of the Steepest Descent algorithm with a step size of 0.1 A0, followed by 100 iterations of the Conjugate Gradient algorithm with a size of 0.1 A0. All hydrogen atoms, including the ones with slower rates of addition, were included. Protonation statuses were assigned to the histidine residues. Additional fees were applied for both conventional (using the AMBER ff14SB force field) and unconventional (using the AM1-BCC force field) residues. The net charges for all non-standard entities were stabilized to enable the computation of their atomic partial charges using ANTECHAMBER charges. The protein was purified by removing any nonstandard residues, such as water molecules, cocrystal ligands, and superfluous chains, using Biovia Discovery Studio visualizer V21.1.0.20298 after optimization and minimization [19,20].
Grid generation
Auto-Dock Tools, Chimera, and Maestro were used for receptor grid identification. The workspace showed many targets, such as BiP (5E84) bound to ATP, Mpro (6LU7), the spike receptor binding domain (6LZG), RNA-dependent RNA polymerase (6M71), spike glycoprotein (6VSB), NSP15 endonuclease (6VWW), Nsp9 RNA-binding protein (6W4B), papain-like protease (6W9C), and neurominidase from the H1N1 protein (5NZ4). The grid’s volume was determined by utilizing the pocket’s dimensions [21]. The size of the enclosing box was minimized to ensure its compatibility with the protein’s active site and the predicted ligands for docking. Computer-based target identification primarily encompasses the identification of disease-related targets, the identification of binding sites, and the evaluation of drug ability. The ability of the ligand and target protein to interact can be ascertained using the binding site [20]. The grid parameters specified in Table 4 were used to determine the active sites in the docking studies. The problem of low utility or druggability commonly encountered in clinical trials can be effectively mitigated through early druggability assessment of proteins. Moreover, it is necessary to ascertain whether the targeted protein is suitable for therapeutic purposes. Finding binding sites and testing druggability are important for figuring out how proteins work, understanding how cells work, doing molecular docking, and coming up with smart drugs [22–24]. Table 3 below illustrates the active sites for binding and grid generation.
 | Table 3. Analysis of active site binding and grid generation parameters for docking studies. [Click here to view] |
 | Table 4. Grid parameters utilized for determining active sites in docking studies. [Click here to view] |
The size of the enclosing box was minimized to ensure its compatibility with the protein’s active site and the anticipated ligand for docking.
Ligands preparation
The ligand molecules were created using MarvinSketch v21.13 and saved in the 3D MOL2 format. All three compounds underwent processing and optimization using UCSF Chimaera v1.15 with the AM1-BCC semi-empirical force field. The default parameters were used, including 1,000 steps of steepest descent and 100 steps of conjugate gradient [25].
Molecular docking of target protein with ligands
Once the ligands and proteins were obtained, their structures were converted to the pdbqt format using an internal bash script created with AutoDock Tools 1.5.6 for ligands and ADFRsuit for proteins. This script allowed all the rotatable bonds of the ligands to rotate freely while considering the receptor as stiff [26]. For docking studies, we utilized the AutoDock Vina 1.2.3, with 0.375 A spacing between grid points. The grid box was precisely positioned at the active site of the enzyme with great accuracy, enabling the program to explore potential interaction sites between the ligands and the receptor. Alternative arrangements were deemed the standard [24,27]. The XYZ center has coordinates (X*Y*Z), and the grid box has dimensions of 20 * 20 * 20 A0. The CPU parameter was set to 23, the exhaustiveness parameter was set to 32, the number of modes parameter was set to 9, and the energy range parameter was set to 3. The redockings were executed using identical settings as the previous dockings.
Visualization
The results received from Autodock Vina processing were used to create a complex utilizing the Biovia Discovery Studio visualizer. Maestro 12.3 (academic version) and LigPlus 1.2 were utilized to generate 2D and 3D pictures of complexes [27]. The interactions and binding energies of the test substances were evaluated and compared to those of conventional inhibitors.
The pharmacodynamics of the chemical were investigated using adsorption, distribution, metabolism, and excretion. SWISS-ADME (http://www.swissadme.ch/) is a website that allows users to sketch their potential ligand or drug molecule and provides parameters like lipophilicity, water solubility, and drug-likeness rules [28]. Predicting chemical toxicity is a crucial aspect of medication discovery. The toxicity analysis was conducted using the pkCSM (http://biosig.unimelb.edu.au/pkcsm/prediction) and PROTOX (http://tox.charite.de/protox_II/) web servers [25]. It predicts the level of tolerance of the small molecule when injected into human and animal models. Several toxicological consequences, such as AMES toxicity, LD50, maximal resistance dose for humans, and hepatotoxicity, are being considered [28].
MDSs
Simulation set-up
We examined 6LZG’s dock complexes with standard Ribavirin and test drug GA using MD simulations on a Desmond 2020.3 computer [25]. We implemented this system in a 10 × 10 × 10-period boundary salvation box using the OPLS-2005 force field [29–32] and an explicit solvent model with SPC water molecules [33]. Neutralizing the charge with 0.15 M NaCl solutions created an environment akin to living organisms. The protein-ligand complexes underwent retraining after equilibration with an NVT ensemble for 10 ns. After the previous phase, an NPT ensemble performed a brief (12 ns) equilibration and minimization. The NPT ensemble was established using the Nose-Hoover chain coupling approach [34] with the same 27°C temperature, 1.0 ps relaxation period, and 1 bar of pressure employed throughout all of the simulations. We set the timestep to 2 fs. To regulate pressure, we used a barostat approach based on the Martyna–Tuckerman–Klein chain coupling system with a relaxation duration of 2 ps [35]. The particle mesh Ewald technique [36] was used to calculate long-range electrostatic interactions with a 9-coulomb interaction radius. The RESPA integrator was used to compute the bonded forces of each trajectory at a time step of 2 fs. The final manufacturing run time for 6LZG_Apo, 6LZG_Ribavirin, and 6LZG_GA was 100 ns. The final production run was finished in 100 ns per unit. To check how consistent the MD was, we found the root mean square deviation (RMSD), the radius of gyration (Rg), the root mean square fluctuation (RMSF), and the hydrogen amount (H-bonds). The stability was monitored using simulations.
Binding free energy analysis
Molecular mechanics and generalized The Born surface area technique (MM-GBSA) was used to calculate the binding free energies of 6LZG_Apo, 6LZG_Ribavirin, and 6LZG_GA complexes. In the last fifty frames of the simulation trajectory, the MM-GBSA binding free energy was estimated using the Python script thermal mmgsba.py and the VSGB solvation model, as well as the OPLS_2005 force field and a one-step sample size. To calculate the binding free energy of MM-GBSA (kcal/mol), the distinct energy modules of columbic, covalent, hydrogen bond, van der Waals, self-contact, lipophilic, and ligand and protein solvation were aggregated using the additivity concept. You may figure out how much Gbind costs by inserting the following numbers into the equation [34].
? Gbind = Δ GMM + Δ GSolv – Δ GSA
where
ΔGbind designates the binding free energy,
ΔGMM designates the difference between the free energies of ligand-protein complexes and the total energies of protein and ligand in isolated form,
ΔGSolv designates the difference in the GSA solvation energies of the ligand-receptor complex and the sum of the solvation energies of the receptor and the ligand in the unbound state,
ΔGSA designates the difference in the surface area energies for the protein and the ligand.
The accuracy of MD simulations is dependent on the force field parameters used to model the molecular interactions. Although widely validated force fields are employed, they still represent approximations and may not capture all the nuances of real molecular behavior.
RESULT AND DISCUSSION
Molecular docking result
We used a method known as structure-based molecular docking to find a substance that can potentially inhibit respiratory influenza viruses, including SARS-CoV-1 and SARS-CoV-2. Using an in silico method, we screened GA obtained from liquorice and compared it to the commercially available antiviral drug Ribavirin. Some of the proteins we chose for virtual screening were BiP in its ATP-bound state (5E84), Mpro, a main protease (6LU7), a spike receptor binding domain (6LZG), RNA-dependent RNA polymerase (6M71), spike glycoprotein (6VSB), NSP15 endonuclease (6VWW), papain-like protease (6W9C), and neurominidase from the H1N1 virus (5NZ4). In silico toxicology studies proved that GA was safe, and bioactivity predictions suggest that it might work by stopping proteases or enzymes from doing their job. This method involved testing the effectiveness of GA, as well as a commercially available antiviral drug called ribavirin. These compounds were chosen because they have shown therapeutic potential against various infectious diseases, including their ability to fight against malaria and other viral diseases.
Primary screening showed that GA, when docked with different 9 targets, had a docking score ranging from −9.6 to −7 kcal/mol (Table 5). When docked against the receptor Spike receptor-binding domain (6LZG), GA has the highest docking score of −10.307, which is higher than the standard ribavirin’s docking score of −6.18 with the same target (Table 5).
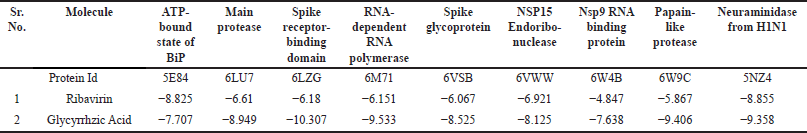 | Table 5. Intermolecular ligand interactions and docking score with the ATP-bound state of BiP, Main protease, Spike receptor-binding domain, RNA-dependent RNA polymerase, Spike glycoprotein, NSP15 endoribonuclease, Nsp9 RNA binding protein, Papain-like protease, and Neuraminidase from the H1N1 using LigPlot v1.4.5, PLIP server, Maestro V12.8, and Biovia Discovery studio visualizer. [Click here to view] |
The 2D interaction diagram (Fig. 1) describes the interaction between the bioactive phytoconstituent (GA) docked against the ATP-bound state of BiP (5E84), main protease (Mpro) (6LU7), spike receptor binding domain (6LZG), RNA-dependent RNA polymerase (6M71), spike glycoprotein (6VSB), NSP15 endonuclease (6VWW), Nsp9 RNA-binding protein (6W4B), papain-like protease (6W9C), and neurominidase from the H1N1 (5NZ4), respectively.
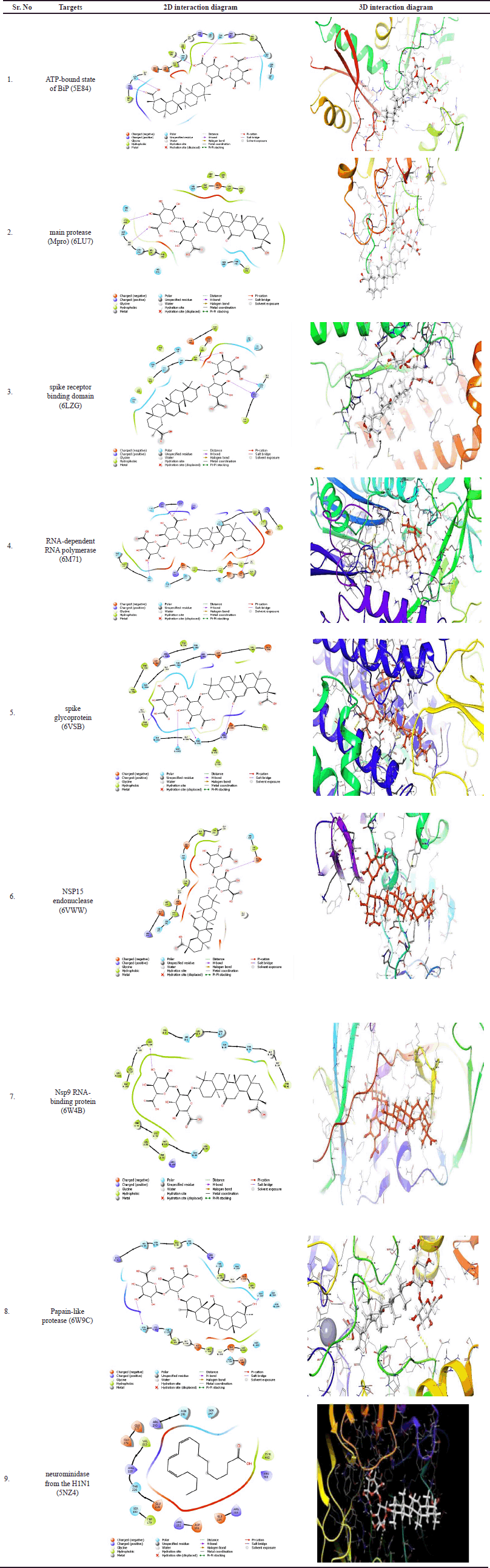 | Figure 1. The 2D and 3D interactions of GA phyto-ligands with the protein targets (a) ATP-bound state of BiP (5E84), (b) main protease (6LU7), (c) spike receptor-binding domain (6LZG), (d) RNA-dependent RNA polymerase (6M71), (e) spike glycoprotein (6VSB), (f) NSP15 Endoribo-nuclease (6VWW), (g) papain-like protease (6W4B), (h) papain-like protease (6W9C), and (i) neuraminidase from H1N1 (5NZ4). [Click here to view] |
The diagram described different proteins that react with GA in docked poses. SARS-CoV-2 MTase complexes share SAM binding residues with red circles in the diagrams.
Figure 1 depicts the 2D and 3D interactions of GA phytoligands with the different 9 protein targets. Molecular docking analysis found that the phytochemical GA had the highest binding energy of −10.307 Kcal/Mol against the spike receptor-binding domain with solitary hydrogen bond with LYS positioned. The binding energy of standard Ribavarine was reported to be −8.855 Kcal/Mol against Neuraminidase from the H1N1 target. The hydrophobic interaction is a crucial factor in the binding of ligands and targets. The docking results revealed that GA had the highest binding energy against various targets, including several targets, such as the ATP-bound state of BiP (5E84), main protease (Mpro) (6LU7), the spike receptor binding domain (6LZG), RNA-dependent RNA polymerase (6M71), spike glycoprotein (6VSB), NSP15 endonuclease (6VWW), Nsp9 RNA-binding protein (6W4B), papain-like protease (6W9C), and neurominidase from the H1N1 (5NZ4).
The mean docking score values, ΔG (Kcal/mol), and the likelihood of binding to the active site are displayed in Table 5.
Target prediction
The GA was investigated for molecular interactions with antiviral targets (Table 6). It has the highest binding affinity shows hydrogen bonds with ten residues and shows Hydrophobic Interactions with one and two residues, respectively.
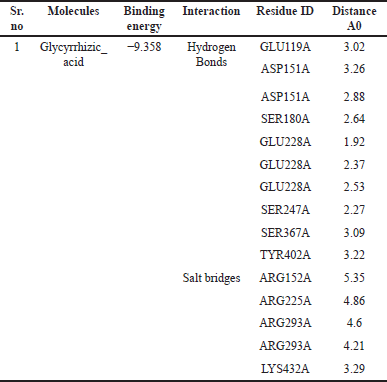 | Table 6. The interaction and distance between inhibitors and critical amino acids of nine different targets in docking complexes were assessed using the LIGPLOT software. [Click here to view] |
MDSs result
Stability of GA-protein complexes
Simulations based on MDs and experiments were employed to investigate the stability and convergence of the Cα-backbone of 6LZG_Apo, 6LZG_Ribavirin, and 6LZG_GA protein-ligand complexes. Figure 2A represents the RMSD of three different proteins: 6LZG_Apo, 6LZG_Ribavirin, and 6LZG_GA, over a time span of 100 nanoseconds (ns). RMSD is a common metric in structural biology and MDs, used to measure the average distance between the atoms of superimposed molecules. High RMSD values typically indicate significant conformational changes from a reference structure, while lower RMSD values suggest that the structure is relatively similar to the reference. 6LZG_GA exhibits an increase in RMSD till 75 ns, which then stabilizes till the end of the simulation with an average RMSD of 2.51 Å. This could indicate a significant structural change or rearrangement that then stabilizes. The RMSD values for the Apo state were found stable throughout the simulation with an average RMSD of 2.62 Å. While 6LZG_Ribavirin shows a gradual increase over time of 52 ns and then stabilizes till the end of 100 ns with an average RMSD of 2.47 Å, suggesting continuous conformational changes or deviations from the reference structure and then get stabilizes. All three Protein states show RMSD below 3 Å indicating stable states while comparing, the Apo state displays slightly higher fluctuations in RMSD values than standard Ribavirin and GA throughout the simulation. The stability and dynamics of each combination can be inferred from their RMSD trajectories. The 6LZG_Ribavirin, and 6LZG_GA, after an initial drift from the reference structure, seem to maintain a consistent structure. The Apo state might be the less stable among the three due to its dynamic nature, with significant conformational changes. The complexes 6LZG_Ribavirin, and 6LZG_GA demonstrate similar behaviors in terms of their structural deviations from a reference. 6LZG_Apo state undergoes consistent changes, while 6LZG_Ribavirin and 6LZG_GA shows late stabilization. Figure 2B presents the RMSF values of residues for three protein-ligand complexes: 6LZG_Apo, 6LZG_Ribavirin, and 6LZG_GA. The RMSF is a measure of the average deviation of a set of atoms (in this case, protein residues) over time from a reference position, typically used in MDs to determine the flexibility or variability of specific residues in a protein structure. 6LZG_Apo displays variable fluctuations throughout the residue indices. Notably, there are sharp peaks around residue 116–120, indicating specific regions of high flexibility. Overall, 6LZG_Ribavirin has moderately high fluctuations with pronounced peaks at residues 116–121, 137–138, 272, and 408–410, suggesting these regions might be particularly dynamic or flexible when interacting with Ribavirin. The complex 6LZG_GA shows significant fluctuations, especially around residues 83 and 117–120. The higher the RMSF value for a specific residue, the greater its deviation from the reference, indicating increased flexibility or movement. From graph 1B, all three complexes show increased flexibility at 116–120, which is the region of the protein that is naturally more mobile because of the presence of loop regions. After critical observation, it has been found that fluctuations happen at residues in the loop regions which are not found in the binding site. Figure 2C illustrates the Rg for three complexes: 6LZG_Apo, 6LZG_Ribavirin, and 6LZG_GA, plotted against simulation time in nanoseconds (ns). The Rg measures the distribution of a set of points in space, and in the context of MDs, it offers insights into the compactness or spatial distribution of a protein molecule over time. Range of RG values 6LZG_Apo, 6LZG_Ribavirin, and 6LZG_GA are 24.33–28.79, 24.41–33.34, and 24.46–28.32, respectively, which indicates protein with GA has a more compact structure than the other two states while protein with Ribavirin has less compact structure. A larger Rg generally implies a more expanded or less compact protein conformation, whereas a smaller Rg suggests a more compact or folded state. All three complexes demonstrate dynamic behavior throughout the simulation, as evidenced by changes in their Rg values. These fluctuations provide insights into the protein’s conformational changes, potentially influenced by the compounds’ binding. The results can be a foundation for understanding the structural effects of these compounds on 6LZG. Figure 2D represents the number of hydrogen bonds formed between the protein 6LZG and two different ligands (Ribavirin, and GA) over a time course of 100 ns. Hydrogen bonds play a crucial role in molecular recognition, protein-ligand interactions, and stabilizing the molecular complex. The strength and number of hydrogen bonds can be indicative of the ligand’s binding affinity to the protein. The number of hydrogen bonds in 6LZG_Ribavirin shows the range of 0–6 throughout the 100 ns simulation time. The relatively low count suggests either transient or weak bonding or perhaps the molecule interacts through other non-hydrogen bond mechanisms. 6LZG_GA complex reveals a higher and more consistent hydrogen bond count than the 6LZG_Ribavirin interaction. The bond count fluctuates mainly between 0 and 8, indicating a stronger and more stable interaction between 6LZG and Glycyrrhizic acid. The stability and duration of a protein-ligand complex can be gauged by the consistency and number of hydrogen bonds. A higher count suggests a potentially stronger interaction, which might result in higher binding affinity. The data provides valuable insights into the ligand binding behavior, which could be instrumental in understanding their potential therapeutic effects or drug design strategies.
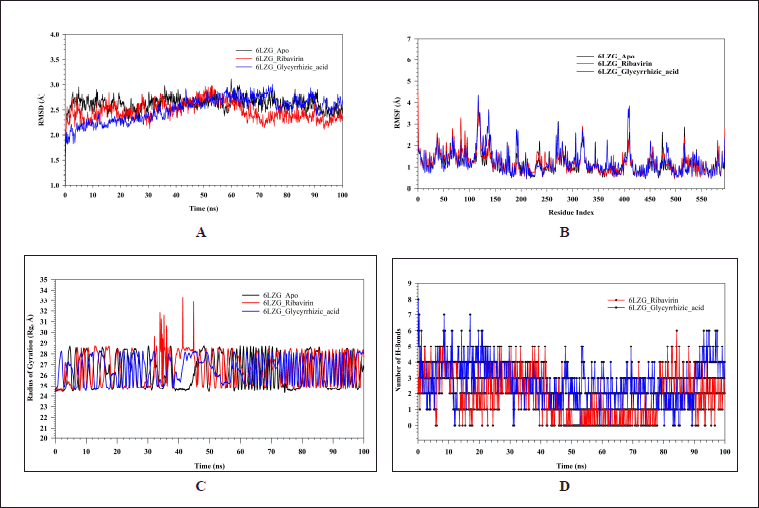 | Figure 2. MD simulation analysis of 100 ns trajectories of (A) Cα backbone of 6LZG_Apo (black), 6LZG_Ribavirin (red), and 6LZG_GA (blue), (B) RMSF of Cα backbone of 6LZG_Apo (black), 6LZG_Ribavirin (red), and 6LZG_GA (blue). (C) The radius of Rg of Cα backbone of 6LZG_Apo (black), 6LZG_Ribavirin (red), and 6LZG_GA (blue). (D) Formation of hydrogen bonds in 6LZG_Ribavirin (red), and 6LZG_GA (blue). [Click here to view] |
Molecular mechanics generalized born surface area (MM-GBSA) calculations
The Table 7 showcases the binding free energy in the form of MM-GBSA components for protein-ligand complexes 6LZG_Ribavirin, and 6LZG_GA, as calculated by MD-simulation. The table represents the binding energies (in kcal/mol) of two ligands (Ribavirin and GA) to a protein (6LZG). The energy values are categorized based on various contributions such as lipid, van der Waals (vdW), Coulombic, hydrogen bond, solvent effect (SolvGB), and covalent energies. Overall binding energies (ΔG_bind) of 6LZG_GA have the most negative binding energy, suggesting the strongest binding affinity among the two ligands. Ribavirin has somewhat comparable energy, but weaker binder than GA. Lipophilic binding energies (ΔG_bindLipo) for all ligands show negative values, indicating favorable lipophilic interactions. Ribavirin exhibits the most significant lipophilic interaction. van der Waals binding energies (ΔG_bindvdW) shows positive values for both the ligands suggesting weaker or repulsive vdW interactions. Coulombic Binding Energies (ΔG_bindCoulomb) for all ligands demonstrate negative values, indicating favorable electrostatic interactions. GA shows the strongest Coulombic interaction. Hydrogen bonding energies (ΔG_bindHbond) for all ligands exhibit strong hydrogen bond contributions. GA shows the most significant hydrogen bonding energy. The positive values of the solvent effect (ΔG_bindSolvGB) indicate an unfavorable contribution from the solvent effect upon binding. Both ligands have the same unfavorable solvent contribution. Covalent binding energies (ΔG_bindCovalent) were found to be negative for all ligands indicating favorable covalent interactions However, GA has stronger covalent interactions than Ribavirin. GA seems to have the strongest overall binding affinity for 6LZG, based on its most negative overall binding energy. This strong binding can be attributed to its significant lipophilic, Coulombic, hydrogen bonding, and covalent interactions
 | Table 7. Binding free energy components for the 6LZG_Ribavirin, and 6LZG_GA, calculated by MM-GBSA. [Click here to view] |
CONCLUSION
Summary of findings
In this study, we explored the potential of GA as a multitarget antiviral agent against respiratory influenza viruses using molecular docking and MDs simulations. Our findings indicate that GA exhibits strong binding affinities to key influenza viral proteins, including spike receptor-binding domain (6LZG), papain-like protease (6W9C), neurominadase from H1N1 (5NZ4), and the RNA-dependent RNA polymerase (RdRp) complex. The docking results suggest that GA can effectively interact with the active sites of these proteins, potentially disrupting crucial stages of the viral life cycle such as viral entry, replication, and release.
MD simulations further confirmed the stability of the GA-protein complexes, demonstrating robust interactions characterized by significant hydrogen bonding and hydrophobic contacts. Binding free energy calculations supported the favorable binding affinities observed in the docking studies, highlighting the potential of GA to serve as an effective inhibitor of multiple influenza viral targets.
Hence, GA, which adheres to Lipinski’s rule of five, could be developed as a potent inhibitor of the ATP-bound state of BiP, main protease, spike receptor-binding domain, RNA-dependent RNA polymerase, spike glycoprotein, NSP15 Endoribonuclease, Nsp9 RNA binding protein, papain-like protease, and neuraminidase from the H1N1 receptor. All of these are significant targets for respiratory influenza viruses such as SARS-CoV-1 and SARS-CoV-2. Consequently, the multitarget approach of GA could offer a strategic advantage in overcoming the limitations posed by the high mutation rates and drug resistance commonly associated with influenza viruses. These computational insights lay a solid foundation for future experimental validation and development of GA-based therapeutic strategies. By targeting multiple viral proteins simultaneously, GA holds promise as a novel antiviral agent for the treatment and prevention of influenza infections, potentially contributing to more effective and durable antiviral therapies.
Future directions
In conclusion, our study underscores the potential of GA as a multitarget inhibitor of respiratory influenza viruses, warranting further investigation and development in the field of antiviral drug research. The study relies on computational methods such as molecular docking and MDSs, which, while powerful, cannot fully replicate the complexity of biological systems. The results provide predictions that need experimental validation to confirm the actual biological activity of GA against influenza viruses.
CONTRIBUTION STATEMENT OF AUTHOR
All listed authors contributed significantly to the article’s conception and composition.
AUTHOR CONTRIBUTIONS
All authors made significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data. Additionally, Aarati R. Supekar, Dr. Santosh S. Bhujbal participated in the crafting of the article and contributed to its critical revision for significant intellectual content. All authors consented to submit their work to the current journal, provided final approval of the published version, and agreed to be responsible for all aspects of the work. All authors are authorized to be authors in accordance with the International Committee of Medical Journal Editors (ICMJE) requirements and guidelines.
FINANCIAL SUPPORT
This research did not received particular grants from public, commercial, or non-profit funding sources or entities.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Ul Islam A, Serseg T, Benarous K, Ahmmed F, Kawsar SMA. Synthesis, antimicrobial activity, molecular docking and pharmacophore analysis of new propionyl mannopyranosides. J Mol Struct [Internet]. 2023;1292(135999):135999. doi: CrossRef
2. Edouard Mathieu, Hannah Ritchie, Lucas Rodés-Guirao, Cameron Appel, Charlie Giattino, Joe Hasell, et al. “Coronavirus Pandemic (COVID-19)”. 2020. Published online at OurWorldInData.org. Available from: ‘https://ourworldindata.org/coronavirus’ [Online Resource]
3. World Health organisation; [cited 2024 May 25]. Available from https://www.who.int/publications/m/item/covid-19-epidemiological-update-15-march-2024 [Online Resource]
4. Abu R, Hasan MM, Aeyas A, Chowdhury FI, Khandaker MU. In-silico approach to designing effective antiviral drugs against SARS-CoV-2 and SARS-CoV-1 from reported phytochemicals: a quality improvement study. Ann Med Surg. 2023;85(7):3446–60. doi: CrossRef
5. Serseg T, Benarous K, Serseg M, Rehman HM, El Bakri Y, Goumri-Said S. Discovery of inhibitors against SARS-CoV-2 associated fungal coinfections via virtual screening, ADMET evaluation, PASS, molecular docking, dynamics and pharmacophore studies. Arab J Basic Appl Sci [Internet]. 2022;29(1):337–50. doi: CrossRef
6. Mamedov NA, Egamberdieva D. Phytochemical constituents and pharmacological effects of licorice: a review. In: Ozturk M, Hakeem K, editors. Plant and Human Health. Cham, Switzerland: Springer; 2019. vol. 3, 1–21. doi: CrossRef
7. Wahab S, Annadurai S, Abullais SS, Das G, Ahmad W, Ahmad MF, et al. Glycyrrhiza glabra (licorice): a comprehensive review on its phytochemistry, biological activities, clinical evidence and toxicology. Plants [Internet]. 2021;10(12):2751. doi: CrossRef
8. Chandra A, Chaudhary M, Qamar I, Singh N, Nain V. In silico identification and validation of natural antiviral compounds as potential inhibitors of SARS-CoV-2 methyltransferase. J Biomol Struct Dyn [Internet]. 2022;40(14):6534–44. doi: CrossRef
9. Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des [Internet]. 2013;27(3):221–34. doi: CrossRef
10. Padmika W, Uthpali M, Nimanthi J. Molecular docking and ADMET based study to identify potential phytochemical inhibitors for papain like protease of SARS-CoV-2 (preprint). doi: CrossRef
11. Olawale F, Iwaloye O, Elekofehinti OO. Virtual screening of natural compounds as selective inhibitors of polo-like kinase-1 at C-terminal polo box and N-terminal catalytic domain. J Biomol Struct Dyn. 2022 Dec 26;40(24):13606–24. doi: CrossRef
12. Pitsillou E, Liang J, Ververis K, Lim KW, Hung A, Karagiannis TC. Identification of small molecule inhibitors of the deubiquitinating activity of the SARS-CoV-2 papain-like protease: In silico molecular docking studies and in vitro enzymatic activity assay. Front Chem [Internet]. 2020;8:623971. doi: CrossRef
13. Anggiresti K, El H, Puspita A, Noor R. Silico study of secondary metabolites in dendrobium spp. as SARS-CoV-2 antivirus on main protease (Mpro). J Riset Biol dan Aplikasinya. 2022;4(1):19–25. doi:10.26740/jrba.v4n1. doi: CrossRef
14. Pandit M, Latha N. In silico studies reveal potential antiviral activity of phytochemicals from medicinal plants for the treatment of COVID-19 infection. Res Square. 2020. doi: CrossRef
15. Mishra A, Pathak Y, Choudhir G, Kumar A, Mishra SK, Tripathi V. Abstract 712: anticancer natural compounds as potential inhibitors of novel coronavirus (COVID19) main protease: an in-silico study. Cancer Res [Internet]. 2021;81(13_Supplement):712. doi: CrossRef
16. Hisham Shady N, Youssif KA, Sayed AM, Belbahri L, Oszako T, Hassan HM, et al. Sterols and triterpenes: antiviral potential supported by in-silico analysis. Plants [Internet]. 2020;10(1):41. doi: CrossRef
17. Shah A, Patel V, Parmar B. Discovery of some antiviral natural products to fight against novel Coronavirus (SARS-CoV-2) using an in silico approach. Comb Chem High Throughput Screen [Internet]. 2021;24(8):1271–80.
18. Jain J, Kumari A, Somvanshi P, Grover A, Pai S, Sunil S. In silico analysis of natural compounds targeting structural and nonstructural proteins of chikungunya virus. F1000Res [Internet]. 2017;6:1601. doi: CrossRef
19. Singh S, Sk MF, Sonawane A, Kar P, Sadhukhan S. Plant-derived natural polyphenols as potential antiviral drugs against SARS-CoV-2 via RNA-dependent RNA polymerase (RdRp) inhibition: an in-silico analysis. J Biomol Struct Dyn [Internet]. 2021;39(16):6249–64. doi: CrossRef
20. Gurung AB, Ali MA, Lee J, Farah MA, Al-Anazi KM. Unravelling lead antiviral phytochemicals for the inhibition of SARS-CoV-2 Mpro enzyme through in silico approach. Life Sci [Internet]. 2020;255(117831):117831. doi: CrossRef
21. Tripathi V, Mishra A, Pathak Y, Kumar A, Mishra S. Natural compounds as potential inhibitors of SARS-CoV-2 main protease: an in-silico study. Asian Pac J Trop Biomed [Internet]. 2021;11(4):155. doi: CrossRef
22. Liao J, Wang Q, Wu F, Huang Z. In silico methods for identification of potential active sites of therapeutic targets. Molecules [Internet]. 2022;27(20):7103. doi: CrossRef
23. Gupta SS, Kumar A, Shankar R, Sharma U. In silico approach for identifying natural lead molecules against SARS-COV-2. J Mol Graph Model [Internet]. 2021;106(107916):107916. doi: CrossRef
24. Rathinavel T, Thangaswamy S, Ammashi S, Kumarasamy S. Virtual screening of COVID-19 drug from three Indian traditional medicinal plants through in silico approach. Res J Biotechnol. 2020;15(10):124–40.
25. Abdolmaleki S, Ganjalikhany MG. Binding affinity optimization and structural evaluation of designed antibodies against PD-1 and PD-L1 as critical immune checkpoints involved in cancer treatment. Res Sq [Preprint]. 2024doi: CrossRef
26. Kazmi I, Al-Abbasi FA, AlGhamdi SA, Alghamdi AM, Zeyadi M, Sheikh RA, et al. Influence of rosiridin on streptozotocin-induced diabetes in rodents through endogenous antioxidants-inflammatory cytokines pathway and molecular docking study. J Biomol Struct Dyn [Internet]. 2023;8(12)1–16. doi: CrossRef
27. Shaw DE, Maragakis P, Lindorff-Larsen K, Piana S, Dror RO, Eastwood MP, et al. Atomic-level characterization of the structural dynamics of proteins. Science [Internet]. 2010;330(6002):341–6. doi: CrossRef
28. Chaturvedi M, Nagre K, Yadav JP. In silico approach for identification of natural compounds as potential COVID 19 main protease (Mpro) inhibitors. Virusdisease [Internet]. 2021;32(2):325–9. doi: CrossRef
29. Bowers KJ, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE, Chow E, et al. Molecular dynamics---Scalable algorithms for molecular dynamics simulations on commodity clusters. In: Proceedings of the 2006 ACM/IEEE conference on Supercomputing—SC ’06. New York, NY: ACM Press; 2006. doi: CrossRef
30. Chow E, Rendleman CA, Dror RO, Hughes DH, Gullingsrud J, Sacerdoti FD, et al. Desmond performance on a cluster of multicore processors. DE Shaw Res Tech Rep. 2008;28(7).
31. Shivakumar D, Williams J, Wu Y, Damm W, Shelley J, Sherman W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J Chem Theory Comput [Internet]. 2010;6(5):1509–19. doi: CrossRef
32. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys [Internet]. 1983;79(2):926–35. doi: CrossRef
33. Martyna GJ, Tobias DJ Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys 1994;101:4177–89. doi: CrossRef
34. Martyna GJ, Klein ML, Tuckerman M. Nose-hoover chains-the canonical ensemble via continuous dynamics. J Chem Phys. 1992;97:2635–43. doi: CrossRef
35. Toukmaji AY, John A. Board. Ewald summation techniques in perspective: a survey. Comput Phys Commun [Internet]. 1996;95(2–3):73–92. doi: CrossRef
36. Joshi T, Joshi T, Sharma P, Mathpal S, Pundir H, Bhatt V, et al. In silico screening of natural compounds against COVID-19 by targeting Mpro and ACE2 using molecular docking. Eur Rev Med Pharmacol Sci [Internet]. 2020;24(8):4529–36. doi: CrossRef