
INTRODUCTION
Indonesia is a rich country in natural resources. Almost all plants that grow on earth can be found in Indonesia [1]. Numerous plants exhibit efficacy as herbal medicines. Herbal medicine also plays an important role in controlling infectious diseases. One of the infectious diseases that has become a global concern in the past 2 years is COVID-19. The handling efforts carried out by both WHO and the Government of Indonesia are to promote vaccination. Massive vaccination efforts have been able to reduce the prevalence of COVID-19 sufferers [2,3]. Even with a noticeable recent decline, COVID-19 continues to exhibit elevated prevalence and mortality rates, further accentuated by the lack of a verified, effective treatment. WHO also reported 1,009,341 COVID-19 cases in the last 28 days until December 24, 2023, an increase of 467,261 cases from the previous period [4]. This emphasizes the crucial need for rigorous and all-encompassing medical strategies to confront the virus.
In vitro, investigations have highlighted the potent inhibition of SARS-CoV-2 infection by both Favipiravir and Remdesivir in standard Vero E6 cells [5]. Multiple studies have assessed various antiviral medications on SARS-CoV-2 patients, such as hydroxychloroquine [6], Lopinavir-Ritonavir, and Ribavirin [7], Remdesivir [8], and Tocilizumab [9]. The FDA in the United States has granted authorization for the use of Nirmatrelvir and Ritonavir (Paxlovid) as a treatment for mild to moderate COVID-19 in both adults and children [10].
Other than the use of synthetic drugs, there is clinical evidence that herbal plants can be used as an alternative therapy for SARS-CoV-2. [11,12]12]. Indonesia also contributes a variety of herbal plants, one of them is the red fruit plant (Pandanus conoideus Lamk) that can inhibit SARS-CoV-2. Red fruit is a typical plant from Papua, Indonesia. Red fruit contains antioxidants, antitumor, immunomodulator, antiparasitic, and anti-HIV [13,14,15]. Previous research reported that flavonoid compounds contained in red fruit have the potential to an anti-SARS-CoV-2 Mpro. The hydrogen bonds formed on the main protease active sites of SARS-CoV-2 Mpro are Asn142 and Cys145 [16].
The potency of antiviral compounds from herbal plants can be predicted through in silico method. This approach enables a quick and precise selection of various compounds. In addition, this approach can also save time and money when compared to the conventional drug discovery stage. Moreover, for the COVID-19 antiviral test, a minimum laboratory standard of Biosafety Level 3 (BSL-3) is required [17]. The molecular docking method approach can be used to design or select compounds that act precisely on target proteins and observe the mechanism of action of these compounds molecularly [18]. This method allows researchers to determine the anti-COVID-19 activity by looking at the binding energy value of the compound’s ability to bind the receptor and the compound engages with the amino acid residues on the target receptor through a specific type of bond [19]. By studying the dynamics of systems in the body, Molecular dynamics (MD) simulations can reveal the ligand-protein’s stability complex [20,21,21,22]21,22].
The stability of ligand-protein complexes in the body is just one aspect of research in the identification of new substances with therapeutic capabilities. In addition, the pharmacokinetic characteristics of these drugs should still be considered. The pharmacokinetic profile of a substance can be seen by estimating its absorption, distribution, metabolism, excretion, and toxicity (ADMET) [23].
Therefore, based on all of the reasons above, this study’s aim is to determine and design flavonoid compounds found in red fruit, and the derivatives that possess the potential to inhibit SARS-CoV-2 Mpro with a stable structure and standard pharmacokinetic profile. The study started with molecular docking followed by MD simulation up to 100 ns and pharmacokinetic prediction.
MATERIAL AND METHODS
Materials
The main instrument employed in this study was a PC with an Intel Core i7-13700KF CPU, and 64 GB of RAM. The OS utilized is Linux, and the primary software installed is Yasara-Structure 23.8.19 [24], Discovery Studio Visualizer (DSV) 21.1.0.20298 [25]. All software settings were left at their default values. Materials utilized include the SARS-CoV-2Mpro crystal structure with PDB ID: 5r7y [26].
Methods
The structural data for the SARS-CoV-2 main protease (PDB ID: 5r7y) was sourced from the protein data bank [26]. This structure underwent a preparation phase involving the removal of water molecules and ions using Yasara-structure tools. While the native ligand, JFM (N-(2-phenylethyl) methanesulfonamide), remained for benchmarking the potential of herbal bioactive compounds as competitors, water molecules, and ions were eliminated. To validate the docking methodology, 100 redocking iterations of the native ligand were conducted utilizing supplementary plug-ins within the Yasara-structure.
There have been 225 compound modifications performed. Each compound design of quercetin 3’-glucoside derivatives can be seen in Supplementary Table 1. Figure 1 depicts the lead compound of quercetin 3’-glucoside. Preparation begins with the separation of the protein and its native ligand. The next stage of preparation produces 5r7y_receptor.sce and 5r7y_ligand.yob files. All compound modification changes were energy-reduced, before moving on to the molecular docking stage.
Molecular docking
Yasara-structure, employing a force field scoring function approach, serves as the basis for the docking method utilized to calculate the binding energy value. The docking process is performed using macro command (dock_run.mcr) which is available in Yasara-structure. The grid box was created with a radius of 5.0 Å around the native ligand. The Vina docking method within the Yasara-structure was utilized to compute binding energy and identify receptor residues involved in the interaction. Upon finishing the docking process, the outcomes were stored in the PDB (.pdb) file format. Subsequently, the post-docking data underwent analysis and visualization using DSV [25].
MD simulation
Yasara-structure was employed for conducting molecular dynamics simulations on a Linux system, leveraging the md_runmembrane.mcr macro command for preparation, minimization, equilibration, and production phases. During the preparation phase, crucial parameters encompassed a cubic simulation cell containing water solvent, cell configuration with periodic boundaries, a membrane box size 30 Å larger than the protein, and a water box size 20 Å larger than the protein. Additionally, 0.9% NaCl ions were introduced, employing the AMBER14 forcefield. The simulation was set at pH 7.4, a temperature of 300 K, and a pressure of 1 bar [27,28]. Executed at a speed of 2.5 fs, the simulation data were automatically saved in .sim file format, lasting for 100 ns. Post-simulation, the md_analyze.mcr macro command in Yasara-structure was utilized for determining RMSD and RMSF values, while the BEcalculation.mcr macro command evaluated the binding free energy from the MD simulation results.
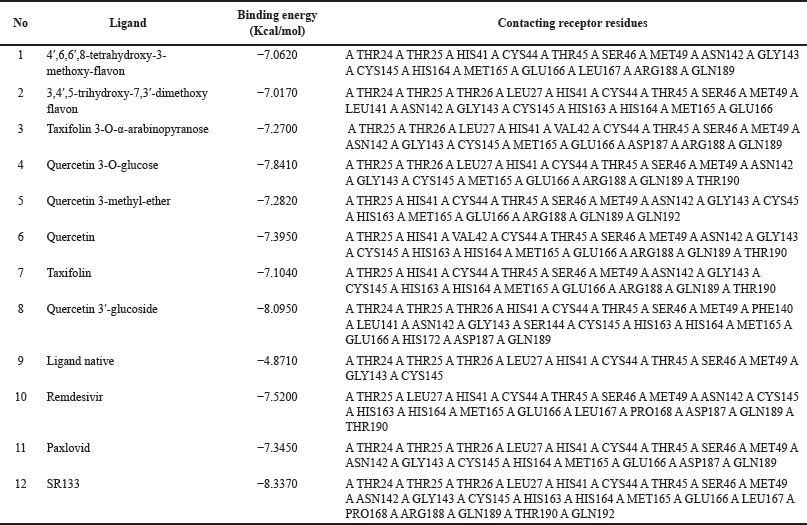 | Table 1. Binding energy value and contacting receptor residue from docking result. [Click here to view] |
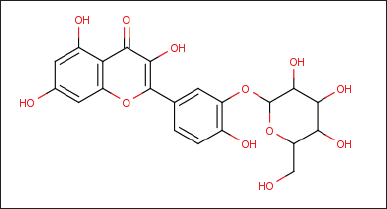 | Figure 1. Lead compound of quercetin 3’-glucoside. [Click here to view] |
The molecular dynamics analysis revealed the binding free energy to be exceptionally stable, subsequently undergoing rigorous internal validation. This validation process encompassed an extensive assessment conducted across 1,000 redocking iterations, enabling a comparison of docked postures for the calculation of RMSD values concerning the docked ligand poses [27,28]28]. The first step in this redocking process involves using the target_prep.mcr macro command to dissociate the protein from the ligand. Subsequently, the redocking procedure can be iteratively repeated 1,000 times using the dock_run_1,000.mcr macro command. The RMSD value obtained can be used to determine the outcome of this validation.
Pharmakokinetic prediction
pkCSM ADMET was utilized to forecast the pharmacokinetic profiles of selected ligands, evaluating both the quality and safety of these compounds. The generation of SMILES strings for each ligand involved the conversion of PDB ligand files into SMI format using DSV, enabling comprehensive analysis.
RESULTS AND DISCUSSION
Molecular docking
Molecular docking can predict the structure of ligand-protein complexes. Docking simulations can illustrate how a drug candidate attaches to its target protein to determine its potential affinity and activity [29]. As an outcome of this docking simulation, the docking score was utilized to gauge the binding energy value, reflecting the strength of interaction between the ligand and the receptor [30].
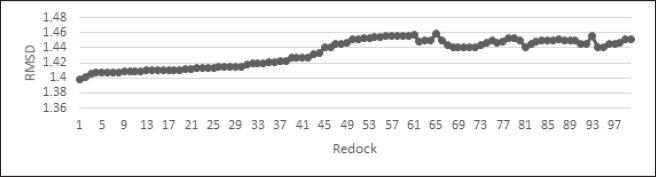 | Figure 2. RMSD value for redocking 100 times. [Click here to view] |
The crystal structure with PDB ID 5r7y is employed as the target for the SARS-CoV-2 Mpro due to scientific evidence indicating that compounds interacting with this protein hold potential as antiviral agents. As a crucial component of SARS-CoV-2 Mpro, the 5r7y protein serves as a significant focus in the quest for antiviral drug development against the virus. Through computational analyses and laboratory investigations, compounds exhibiting strong binding affinity to the 5r7y protein have demonstrated inhibitory effects against SARS-CoV-2 Mpro, presenting themselves as promising candidates for combating SARS-CoV-2 infection. Scientific research underscores the pivotal role of the 5r7y protein in the replication cycle of the SARS-CoV-2 virus, thus inhibiting its activity holds the potential to impede viral development. Consequently, leveraging the 5r7y protein as a target for inhibiting SARS-CoV-2 Mpro represents a promising strategy in the endeavor to develop antiviral medications to combat COVID-19 [26,31].
Based on Supplementary Table 1, design of new compound is intended to increase the value of the docking score, binding energy, and contact with receptor residue. The substituting groups are electron-donating and electron-withdrawing groups. Furthermore, the reason for using the groups is also based on the substitution of isosteres groups. This is done because the substitution of isosteres groups can increase the resulting docking score of the ligand-protein complex [32].
The validation of the docking methodology involved 100 times redocking against the native ligand. This validation method produced a delta RMSD value of less than 1.5 (Fig. 2). Therefore, the docking method to be used is proven to be valid.
After the docking approach was determined to be valid, the next step was to dock all flavonoid compounds discovered in red fruit. In addition, we employed remdesivir and paxlovid as positive controls. The binding energy value of flavonoid compounds from the best docking results is then used as a lead compound to design new compounds. Table 1 displays the binding energy and contacting receptor residue values for red fruit flavonoids, remdesivir, paxlovid, and the design of a new compound derivative of quercetin 3′-glucoside as the best docking results.
Based on the docking results of flavonoid compounds found in red fruit, the quercetin 3′-glucoside compound had the highest binding energy value. This follows the previous research which declares that flavonoid (quercetin 3′-glucoside) has the potential to inhibit SARS-CoV-2 Mpro [16]. Therefore, we further used this compound as a lead compound in designing new compounds. A total of 225 proposed compounds were docked showing that the design compound with code number 133 (SR133) had a better binding energy value than the native ligand, lead compound, or reference ligands (remdesivir and paxlovid) (Fig. 3). This docking stage uses 999 iterations of each ligand (Table 1). Based on the table, it can be seen that SR133 has a better binding energy value than other ligands. The free binding energy of the complex is −8.3370 Kcal/mol. This table also provides a visual representation of the contacting receptor residues between ligands and proteins.
 | Figure 3. Docking for SR133 results. [Click here to view] |
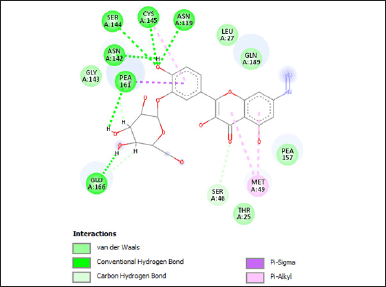 | Figure 4. The visualization of the ligan-protein complex after MD simulation. [Click here to view] |
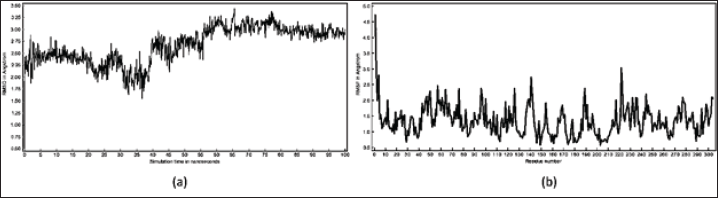 | Figure 5. (a) RMSD (Å) vs time (ns) relationship graph; (b) RMSF vs residue number relationship graph. [Click here to view] |
MD simulation
The ligand-protein complex’s stability can be determined via MD simulation. The MD simulation data was subsequently employed to calculate both the RMSD and free-binding energy values. Figure 4 illustrates the visualization of the ligand-protein complex. The authors chose to conduct molecular dynamic simulations at 300 K because this temperature is commonly used in MD simulations to represent room temperature, and it allows for comparison with a wide range of other studies. The impact of this temperature on their results is important to consider, as it may affect the conformational space sampled by the protein and the stability of the system. While enzymes are bioactive at body temperature (~310 K), the use of 300 K is often justified by the fact that the conformational space sampled by the protein is not significantly different from that at 310 K. The thermal equilibrium reached at 300 K ensures a consistent kinetic energy distribution, crucial for accurately representing system behavior. Moreover, simulating at this temperature facilitates direct comparison with experimental data, enhancing validation and interpretation of simulation outcomes, especially in fields where experiments are commonly conducted at or around room temperature [33,34]34]. Furthermore, simulations conducted at elevated temperatures were prevalent in the early stages of MD development, yet they resulted in unrealistic trajectories, highlighting the need for their integration with runs performed at room temperature [35].
The visual representation highlights the presence of crucial hydrogen bonds with key amino acid residues (Asn142 and Cys145), essential for inhibiting SARS-CoV-2 Mpro. Prior research has emphasized that the active site of SARS-CoV-2 Mpro encompasses hydrogen bonds specifically with the primary amino acid residues Asn142 and Cys145 [36]. Furthermore, there exist supplementary hydrogen bonds involving Asn119, Ser144, Pea161, and Glu166 collectively contributing to an augmented potential for inhibiting this virus.
The MD simulation findings indicated stabilization of the RMSD value after 2 ns. Calculations of the delta RMSD value were conducted every 5 ns, consistently yielding values less than 1 Å (Fig. 5a). Moreover, the system exhibited a free binding energy value of −5.7040 Kcal/mol, signifying the most stable during the computation of binding free energy (Fig. 6). According to thermodynamics, if a ligand-receptor complex was formed that has a lower potential energy, then interconnection between the ligand and the receptor can occur [37].
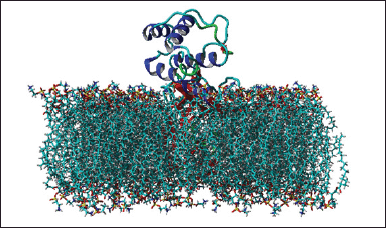 | Figure 6. The system with the highest level of stability. [Click here to view] |
 | Figure 7. (a) Interaction between SR133 and Asn142. (b) Interaction between SR133 and Cys145. [Click here to view] |
The experiment was subsequently expanded to include measuring the RMSF values of amino acid residues. This described the interaction’s stability throughout the simulation, as measured at each residue position based on the fluctuations that occur. Figure 5b illustrates a graphical depiction of the correlation between the RMSF value and the corresponding residue number. Amino acid residue fluctuations occur as a result of ligand-induced binding, where increased fluctuation values indicate increased residue flexibility and thus indicate reduced stability in the amino acid residue interactions formed [38]. The suggested ligand demonstrated high interaction stability within the main catalytic site of SARS-CoV-2Mpro, evident from its low RMSF values.
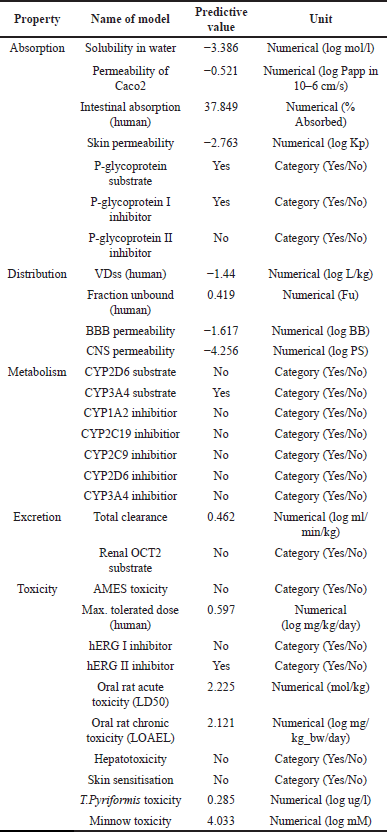 | Table 2. ADMET prediction of SR133. [Click here to view] |
During the visual analysis of the MD simulation report, hydrogen bonds were observed forming between the ligand’s H atom and Asn142 residue, as well as between the ligand’s O atom and Cys145. The calculated bond distances were 2.184 Å and 2.936 Å, respectively (Fig. 7). According to the criteria where a donor and/or acceptor within ≤3.5 Å indicate potential hydrogen bond formation [39], these interactions were noted. Consequently, this system was purposefully chosen and meticulously examined for validation. By conducting internal validation through 1,000 ligand-receptor redocking iterations, each with 25 iterations, a comprehensive set of 1,000 datasets featuring RMSD values under 2 Å was generated. The detailed RMSD values for these datasets are available in Supplementary Table 2.
Pharmacokinetics prediction
The best-suggested chemical has undergone pharmacokinetic prediction, and the outcome was ADMET prediction for the SR133 ligand (Table 2). Based on their solubility properties, drugs can be categorized as follows. A drug has high solubility if its LogS value is greater than −2, slightly soluble between −2 and −4, and insoluble below −4. SR133 has good solubility, as seen from the findings of the ADMET prediction study. The potent ligand exhibited strong oral absorption and skin penetration, as identified by HIA values >30% and Log Kp for skin permeability < −2.5. In addition, the ligand was not registered as a P-glycoprotein II inhibitor [40].
Ligand distribution was excellent when the log Vdss value was −1.44 log l/kg and the plasma’s free fraction exceeded 20%. The higher the free fraction, the higher the medicine’s effectiveness, requiring a reduced amount of drug per dosage. Additionally, the ligand only barely penetrated the blood barrier (logBB < −1 and logPS < −3), suggesting that it will not have direct impacts on the central nervous system. It may be claimed that the ligand does not interact with the metabolism of other medications because it neither acts as a substrate nor inhibits cytochrome P450 [27]. SR133 has a total clearance of 0.462, and the ligand is not an OCT2 substrate. The maximum SR133 dosage that is tolerated for humans is 0.597 log mg/kg/day. Table 2 also displays the ligand’s acute and long-term toxicity to rats when administered orally. The ligand was non-irritating and non-hepatotoxic.
CONCLUSION
The molecular docking results of flavonoid compounds from red fruit reveal that quercetin 3’-glucoside exhibits the highest binding energy value of −8.0950 Kcal/mol. Additionally, this compound interacts with critical receptor residues, such as Asn142 and Cys145 located in the active site of the SARS-CoV-2 Mpro, further justifying its selection as a lead compound for designing novel derivatives. Moreover, a successful design process has yielded a total of 225 new compounds. Based on molecular docking results, molecular dynamics simulation, and pharmacokinetic prediction, it is evident that quercetin 3′-glucoside derivatives [7-diazenyl-3,5-dihydroxy-2-(4-hydroxy-3-{(3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl)oxy}phenyl)-4H-chromen-4-one], show considerable promise as SARS-CoV-2Mpro inhibitors. The docking result reveals that this novel compound exhibits the highest binding energy value of −8.3370 kcal/mol. Additionally, it forms hydrogen bonds with key amino acid residues, namely Asn142 and Cys145. Molecular dynamic simulations further confirm the stability of its structure. Moreover, the pharmacokinetic evaluation indicates that this compound possesses a favorable adsorption, distribution, metabolism, excretion, and toxicity profile, positioning it as a promising candidate for drug development.
ACKNOWLEDGEMENTS
The Authors acknowledge the scholarship provided by “Indonesia Endowment Funds for Education (LPDP) and Center for Higher Education Funding (BPPT)”, and gratefully acknowledge all research facilities provided by the Austrian-Indonesian Center for Computational Chemistry, Department of Chemistry, Faculty of Mathematics and Natural Sciences Universitas Gadjah Mada. This study was supported by Final Project Recognition Grant Universitas Gadjah Mada Number 5075/UN1.P.II/Dit-Lit/PT.01.01/2023.
AUTHOR CONTRIBUTIONS
The study protocol was approved by the Ethics Committee of the Research Organization for Agriculture and Food, National Research and Innovation Agency, Indonesia with [Approval No. 66/Kpts/OT.050/H.4.3/XI/2020]. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Minister of Health of the Republic of Indonesia Number: 381 concerning the 2007 National Traditional Medicine Policy, Jakarta, Indonesia: Minister of Health.
2. Covid19.go.id [Internet]. Indonesia: Covid-19 response task force, Situasi COVID-19 di Indonesia. [updated 2023 Jun 27; cited 2023 Jul 15]. Available from https://covid19.go.id/id/
3. Covid19.who.int [Internet]. World Health Organization: WHO Coronavirus (Covid-19) Dashboard, [cited 2023 August 4]. Available from https://covid19.who.int/
4. Data.who.int [Internet]. WHO Coronavirus (COVID-19) dashboard > Cases [Dashboard], [cited 2024 Jan 2]. Available from https://data.who.int/dashboards/covid19/cases
5. Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–71. CrossRef
6. Mahévas M, Tran VT, Roumier M, Chabrol A, Paule R, Guillaud C, et al. Clinical efficacy of hydroxychloroquine in patients with covid-19 pneumonia who require oxygen: observational comparative study using routine care data. BMJ. 2020;369:m1844. CrossRef
7. Hung IFN, Lung KC, Tso EYK, Liu R, Chung TWH, Chu MY, et al. Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet. 2020;395:1695–704. CrossRef
8. Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395:1569–578. CrossRef
9. Xu X, Han M, Li T, Sun W, Wang D, Fu B, et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci. 2020;117:10970–975. CrossRef
10. FDA.gov [Internet] U.S. Food & Drugs Administration: Fact sheet for healthcare providers: emergency authorization for Paxlovid, [cited 2022 Feb 23]. Available from https://www.fda.gov/
11. Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci. 2003;100:13190–195. CrossRef
12. Ren J, Zhang AH, and Wang XJ. Traditional Chinese medicine for COVID-19 treatment. Pharmacol Res. 2020;155:104743. CrossRef
13. Felle ZR, Wijayanti MA, and Supargiyono. The effect of pandanus conoideus lamk extract to the serum level of TNF-α, IL-10 and parasitemia of plasmodium berghei infected in Mice. Trop Med J. 2013;3:39–47.
14. Tafor D, Djunaidi A, Wasityastuti W, and Solikhah EN. Tumor necrosis factor-alpha (TNF-alpha) and intercellular adhesion molecule-1 (ICAM-1) expression of plasmodium berghei infected swiss mice treated with red fruit (Pandanus Conoideus Lam) ethanol extract. Trop Med J. 2013;3:155–65.
15. Tambaip T, Br Karo M, Hatta M, Dwiyanti R, Natzir R, Nasrum Mas M, et al. Immunomodulatory effect of orally red fruit (Pandanus conoideus) extract on the expression of CC chemokine receptor 5 mRNA in HIV patients with antiretroviral therapy. Res J Immunol. 2018;11:15–21. CrossRef
16. Umar, AbdK. Flavonoid compounds of buah merah (Pandanus conoideus Lamk) as a potent SARS-CoV-2 main protease inhibitor: in silico approach, Futur. J Pharm Sci. 2021;7:158. CrossRef
17. Prieto-Martínez FD, Arciniega M, and Medina-Franco JL. Acoplamiento molecular: avances recientes y retos, TIP Rev. Espec. en Ciencias Químico-Biológicas. 2018;21:65–87. CrossRef
18. Lin X, Li X, and Lin X. A review on applications of computational methods in drug screening and design. Molecules. 2020;25:1375. CrossRef
19. Rachmania RA, Hariyanti H, Zikriah R, and Sultan A. Studi In Silico senyawa alkaloid herba bakung putih (Crinum Asiaticum L.) pada penghambatan enzim siklooksigenase (COX). Jurnal Kimia VALENSI. 2018;4:124–36. CrossRef
20. Masone D, and Grosdidier S. Collective variable driven molecular dynamics to improve protein–protein docking scoring. Comput Biol Chem. 2014;49:1–6. CrossRef
21. Childers MC, and Daggett V. Insights from molecular dynamics simulations for computational protein design. Mol Syst Des Eng. 2017;2:9–33. CrossRef
22. Ahmed M, Sadek MM, Abouzid KA, and Wang F, In silico design: extended molecular dynamic simulations of a new series of dually acting inhibitors against EGFR and HER2, J Mol Graph Model. 2013;44:220–31. CrossRef
23. Shivanika C, Kumar D, Ragunathan V, Tiwari P, Sumitha A. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J Biomol Struct Dyn. 2022;40:585–611. CrossRef
24. Krieger E, Koraimann G, Vriend G. Increasing the precision of comparative models with YASARA NOVA-a self-parameterizing force field. Proteins Struct Funct Genet. 2002;47(3):1–10. CrossRef
25. Biovia. Dassault Systèmes, Discovery studio visualizer, V21.1.0.20298. San Diego, CA:Dassault Systèmes; 2020.
26. Douangamath A, Fearon D, Gehrtz P, Krojer T, Lukacik P, Owen CD. et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat Commun. 2020;11:5047. CrossRef
27. Nugraha G, Istyastono EP. Virtual target construction for structure-based screening in the discovery of histamine H2 receptor ligands. Int J Appl Pharm. 2021;13:239–41. CrossRef
28. Nugraha G, Pranowo HD, Mudasir M, & Istyastono EP. Virtual target construction for discovery of human histamine H4 receptor ligands employing a structure-based virtual screening approach. Int J Appl Pharm. 2022;14(4):213–18. CrossRef
29. Lengauer T, and Rarey M. Computational methods for biomolecular docking. Curr Opin Struct Biol. 1996;6(3):402–6. CrossRef
30. Nurhidayah M, Fadilah F, Arsianti A, Bahtiar A. Identification of Fgfr inhibitor as St2 receptor/interleukin-1 receptor-like 1 inhibitor in chronic obstructive pulmonary disease due to exposure to E-cigarettes by network pharmacology and a molecular docking prediction. Int J App Pharm. 2022;14:256–66. CrossRef
31. Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–93. CrossRef
32. Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem. 2011;54(8):2529–91. CrossRef
33. Kubitzki MB, and de Groot BL. Molecular dynamics simulations using temperature-enhanced essential dynamics replica exchange. Biophys J. 2007;92(12):4262–70. CrossRef
34. Jung J, Kobayashi C, and Sugita Y. Optimal temperature evaluation in molecular dynamics simulations with a large time step. J Chem Theory Comput. 2018;15(1):84–94. CrossRef
35. Hospital A, Goni JR, Orozco M, and Gelpi JL. Molecular dynamics simulations: advances and applications. Adv Appl Bioinform Chem. 2015;8:37–47. CrossRef
36. Arora S, Lohiya G, Moharir K, Shah S, and Yende S. Identification of potential flavonoid inhibitors of the SARS-CoV-2 main protease 6YNQ: a molecular docking study. Digital Chinese Med. 2020;3:239–48. CrossRef
37. Schneider G. “De novo molecular design”. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co; 2014.
38. Chaudhary N and Aparoy P. Deciphering the mechanism behind the varied binding activities of COXIBs through molecular dynamic simulations, MM-PBSA binding energy calculations and per-residue energy decomposition studies, J Biomol Struct Dyn. 2017;35(4):868–82. CrossRef
39. Zhang QY, and Aires-de-Sousa J. Random forest prediction of mutagenicity from empirical physicochemical descriptors. J Chem Inf Model. 2007;47(1):1–8. CrossRef
40. Pires DE, Blundell TL, and Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58(9):4066–72. CrossRef