
INTRODUCTION
Influenza, caused by influenza viruses, is an acute respiratory infection of a potentially deadly disease that infects hundreds of millions of people each year around the globe [1]. The burden of influenza exists despite available vaccines and with the current coronavirus disease 2019 infections, which complicates health care systems and indicates the urgent need for effective anti-influenza drugs.
Influenza virus consists of four genera, i.e., types A, B, C, and D [2], in which the Influenza A virus (IAV) is the most pathogenic to humans among the four genera. Oseltamivir (OTV), also known as Tamiflu, is an antiviral medication that is commonly used to treat and prevent influenza A and B infections. It blocks the activity of NA, which allows the virus to spread from infected to healthy cells. It can help reduce the severity and duration of the illness, as well as the risk of complications such as pneumonia.
However, recent findings have indicated that drug resistance to OTV emerges owing to mutations in NA. In vitro tests of OTV against IAV revealed the existence of mutations including E119A, E119D, E119G, H274Y, I117T, I117V, I117V-E119A, K150N, N294S, R292K, V116A, and Y252H, which resulted in decreased efficacy of OTV [3–9]. The present study attempted to understand, at the molecular level, how these mutations affect OTV binding affinity by employing computational methods such as homology modeling, molecular docking, MDS, and energy decomposition analysis (EDA) [10].
Mihajlovic and Mitrasinovic [11] studied mutations at His274 which slightly enhanced or reduced the susceptibility of OTV to NA. The effect of H274Y mutation in drug–target interactions was also studied by Karthick and Ramanathan [12], which implied the stable binding of the OTV with wild-type (WT) NA as compared to mutant-type NA. In both studies, molecular docking and short MDS were used. Nguyen et al. [13] used the smooth reaction path generation method to calculate binding free energies and found that the binding free energies of NA A/H5N1 in WT and H274Y and N294S mutants with OTV show good agreement with experimental results. To the best of our knowledge, there is no previous study on OTV resistance that covers more comprehensive residue mutations and longer MDSs than our present work. The present study would reveal the atomistic details about designing OTV analogs having better activity toward the NA of IAV.
COMPUTATIONAL METHOD
Homology modeling, molecular docking (Glide XP), MDS, EDA, and molecular mechanics with generalized born and surface area solvation (MM-GBSA) calculations were used to study the binding of OTV to WT and mutant proteins as described in other papers [14,15].
Protein structure preparation
The NA structure in complex with OTV was obtained from PDB ID 2HU4, resolution of 2.50 Å [16]. The structure preparation followed the previous computational protocol [17]. Point mutations E119A, E119D, E119G, H274Y, I117T, I117V, I117V-E119A, K150N, N294S, R292K, V116A, and Y252H were used in the WT system.
OTV was prepared using Maestro and then docked with extra precision into WT, E119A, E119D, E119G, H274Y, I117T, I117V, I117V-E119A, K150N, N294S, R292K, V116A, and Y252H protein structures [18].
MDS, analysis, MM-GBSA binding energy calculations
Each protein–ligand system was used to construct MDS systems using the OPLS_2005 force field [19]. Na+ ions were added to a NaCl concentration of 0.15 M NaCl to neutralize the systems and were used to model the protein–ligand systems. All MDSs were performed for 200 ns using the Desmond simulation package [20].
The system relaxation was carried out based on previous work [17,21–23]. The analysis of simulation results was computed over the entire trajectory using the Desmond trajectory clustering tool [17,24].
MM-GBSA binding energies were predicted for each protein–ligand system as described in the previous works [10,17,25].
Energy decomposition analyses
The EDA calculations were conducted using the absolutely localized molecular orbitals (ALMO)-EDA scheme with the ωB97M-V functional and the Def2-SVPD basis set [26–28] using the Q-Chem program suite [29]. This analysis was performed for mutations and residues for which the protein–ligand interaction analysis resulted in interactions above 50% (vide supra). The EDA analyses allowed us to measure the contributions of dispersion (?EDISP), polarization (?EPOL), and charge transfer (?ECT) interactions to the overall intermolecular interaction energies.
RESULTS AND DISCUSSIONS
Initial ligand conformation
The initial ligand conformation was obtained through molecular docking of the prepared OTV into NA in the WT, E119A, E119D, E119G, H274Y, I117T, I117V, I117V-E119A, K150N, N294S, R292K, V116A, and Y252H systems (Fig. 1).
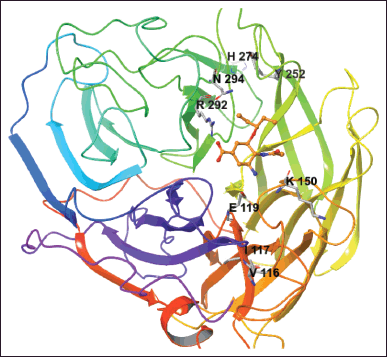 | Figure 1. The NA is complex with OTV (orange color). The mutated residues are shown. [Click here to view] |
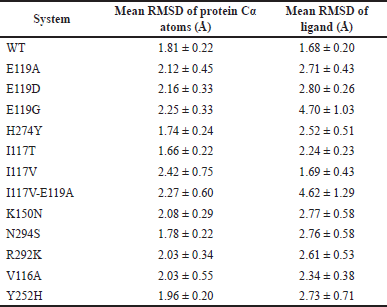 | Table 1. The mean RMSD of protein Cα atoms and mean RMSD of ligands calculated for WT and mutant systems. [Click here to view] |
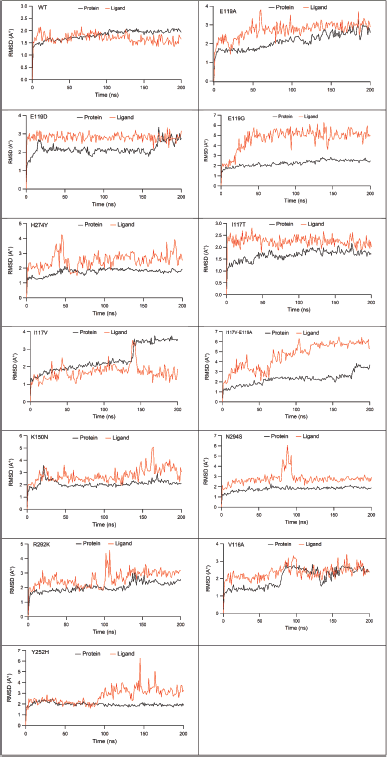 | Figure 2. The RMSD of protein Cα (black line) and ligand (red light) in WT, E119A, E119D, E119G, H274Y, I117T, I117V, I117V-E119A, K150N, N294S, R292K, V116A, and Y252H systems. [Click here to view] |
Binding affinities calculated were −12.857, −7.760, −11.683, −7.860, −10.575, −10.887, −10.898, −7.647, −10.999, −10.947, −10.534, −10.825, and −11.080 kcal/mol, for the WT, E119A, E119D, E119G, H274Y, I117T, I117V, I117V-E119A, K150N, N294S, R292K, V116A, Y252H systems, respectively (Table S1). The mutation reduced the docked binding energy of OTV to NA. The 2D interactions of OTV and WT and mutant systems are shown in Figure S1.
The root mean squared deviation (RMSD) values
Table 1 depicts the mean RMSDs of protein Cα and ligand atoms during 200 ns MDS. The protein Cα was relatively more stable in WT (mean values of 1.81 Å), compared to those in E119A (2.12 Å), E119D (2.16 Å), E119G (2.25 Å), I117V (2.42 Å), I117V-E119A (2.27 Å), K150N (2.08 Å), R292K (2.03 Å), V116A (2.03 Å), and Y252H (1.96 Å), but it was higher than that in I117T (1.66 Å), H274Y (1.74 Å), and N294S (1.78 Å).
Meanwhile, OTV also had a more stable ligand RMSD in WT (1.68 Å) compared to those in E119A (2.71 Å), E119D (2.80 Å), E119G (4.70 Å), H274Y (2.52 Å), I117T (2.24 Å), I117V (1.69 Å), I117V-E119A (4.62 Å), K150N (2.77 Å), N294S (2.76 Å), R292K (2.61 Å), V116A (2.34 Å), and Y252H (2.73 Å) systems. It was found that the OTV poses in the E11G and I117V-E119A systems were most impacted by the protein mutations (Fig. 2).
The protein–ligand interaction analysis
Figure 3 depicts the interaction fraction sustaining more than 20% of MDS. In the E119A system (Fig. 3B), H-bond interactions were observed with Asp151 (91% occupancy), Arg292 (81% and 72% occupancy), and Arg152 (67% occupancy). The high occupancy of H-bonds with Asp151 (99% occupancy), Arg156 (99% occupancy), Tyr406 (79% occupancy), and Asp119 (72% occupancy) was observed in E119D, but the H-bond occupancies with Arg292 in E119D (34% and 31%) and E119G (53% and 48% occupancy) were much lower than those in the E119A system (Fig. 3C). The mutation to glycine in the E119G system caused a reduction in H-bond interactions while forming modest new interactions with residues Arg371 (64% and 44% occupancy) and Glu277 (36% occupancy) (Fig. 3D).
High occupancy of H-bonds with Glu119 and Asp151 was also observed in I117V (Fig. 3G) (90% and 88% occupancies), K150N (Fig. 3I) (89% and 87% occupancies), and Y252H (Fig. 3M) (84% and 88%, respectively). The number of H-bonds with Glu119 was also high in H247Y (Fig. 3E) (86% occupancy) and N294S (Fig. 3J) (81% occupancy). However, they were significantly lower in the R292K system (Fig. 3K) (38% with Glu119 and 54% with Asp151). In the I117T and V116A systems (Fig. 3F and L), the interaction of H-Bond with Glu119 had occupancies of 86% and 39%, respectively, and the interaction with Asp151 disappeared. New bonds were formed with Arg118 (41% and 33% occupancy) and (35% occupancy), respectively. On the contrary, the H-bonds with key residues, including Glu119 and Asp151, completely disappeared in the E119G and I117V-E119A (Fig. 3D and H) systems.
The interactions with Arg292 were observed with relatively low occupancies in almost all mutant systems (56% and 43% in I117V, 34% in I117V-E119A, 54% and 38% in K150N, 58% and 54% in H294S, 59% in V116A, and 37% in Y252H); however, they were lost in the H274Y, I117T, and R292K systems, respectively. The occupancy of H-bonds with Arg371 was also observed in the E119G, H274Y, H294S, and V116A systems (64% and 44%, 96% and 92%, 92% and 90%, and 64% and 56%, respectively), but they became low in I117V (33% and 39% occupancies) and Y252H (38% occupancy).
The water-mediated H-bond varies among mutant systems, for example, E119A and E119D with Trp178 (33% and 31% occupancy) and (45% occupancy), H274Y systems with Asn294 (37% occupancy) and Lys350 (48% occupancy), and I117V, R292K, and V116A with Arg152 (52%, 40%, and 76% occupancies, respectively). Meanwhile, water-mediated H-bonds with Glu277 were also frequently present in mutants such as E119G, H274Y, I117V-E119A, H294S, R292K, V116A, and Y252H, with occupancies of 36%, 42%, 33%, 44%, 85%, 43%, and 51%, respectively. It was found that while most mutant systems preserved interactions with key residues with high occupancies, the interactions were significantly reduced or lost in the I117V-E119A system (Fig. 3).
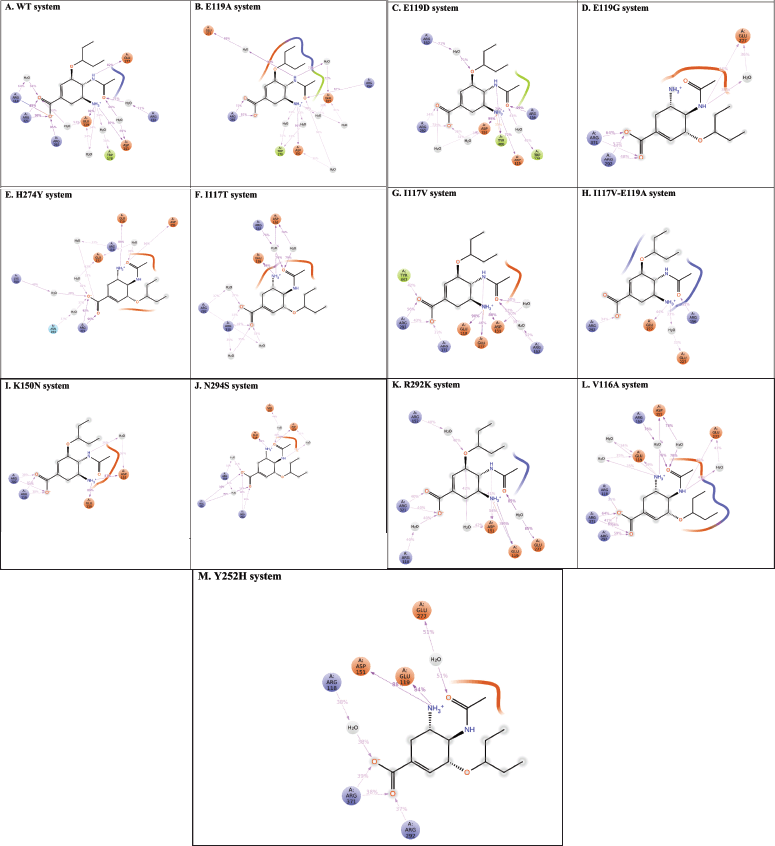 | Figure 3. The 2D protein–ligand interaction diagrams lasting more than 20% of the MDS (A) WT-OTV; (B) E119A-OTV; (C) E119D-OTV; (D) E119G-OTV; (E) H274Y-OTV; (F) I117T-OTV; (G) I117V-OTV; (H) I117V-E119A-OTV; (I) K150N-OTV; (J) N294S-OTV; (K) R292K-OTV; (L) V116A-OTV; (M) Y252H-OTV) recorded during MDSs.. [Click here to view] |
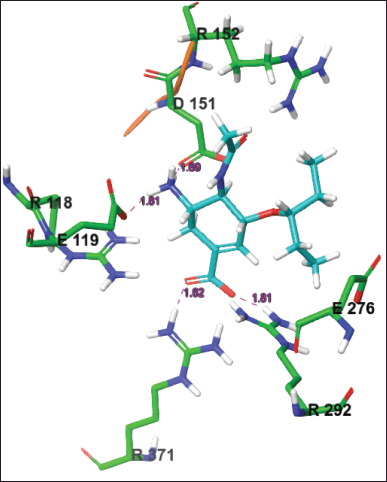 | Figure 4. The active site conformation and 2D interactions of the WT-OTV system taken from the most populated cluster. H-bonds were formed between the drug and Glu119, Asp151, Arg292, and Arg371. [Click here to view] |
Cluster analysis
H-bonds with Glu119 and Asp151 were observed in almost all mutant systems except for E119A, E119G, E117V-E119A, R292K, and V116A, whereas H-bonds with Glu277 were observed in E119A, E119G, H274Y, and E117V-E119A systems. H-bonds with Tyr406, Tyr347, and Lys292 were E119D, I117V, and R292K, respectively. The pi-pi interaction was not recorded in the WT or all mutation systems, but a salt bridge interaction with Arg292 was formed in the I117V-E119A, H294S, and V116A systems. Other interactions with Arg118 and Arg371 were noted in the K150N and V116A systems, respectively. Figure 4 shows the 2D and 3D interactions of WT OTV taken from the most populated cluster, while those of mutants systems were described in Figure S2.
Binding energy prediction
The total binding energy was −36.1 kcal/mol for the WT system, which was lower than that in the E119A (−22.6 kcal/mol), E119D (−22.3 kcal/mol), E119G (−20.9 kcal/mol), H274Y (−17.0 kcal/mol), I117T (−29.2 kcal/mol), I117V (−29.2 kcal/mol), I117V-E119A (−15.0 kcal/mol), K150N (−14.8 kcal/mol), N294S (−24.2 kcal/mol), R292K (−13.5 kcal/mol), V116A (−27.0 kcal/mol, and Y252H (−13.2 kcal/mol) systems.
The binding energy changes (ΔΔGbind) were 13.5, 13.8, 15.2, 19.1, 6.9, 6.9, 21.1, 21.3, 11.9, 22.6, 9.1, and 22.9 kcal/mol, respectively, for those between the WT system and those in the E119A, E119D, E119G, H274Y, I117T, I117V, I117V-E119A, K150N, N294S, R292K, V116A, and Y252H systems. Among these mutations, Y252H, R292K, K150N, I117V-E119A, H274Y, and E119G mutations resulted in much lower affinities for OTV. Table 2 lists the binding energies calculated for the last 100 ns of each system.
The van der Waals interactions (ΔEvdw) is the dominating favorable energy terms, each with −19.4, −22.4, −23.0, −18.4, −15.2, −23.8, −18.0, −21.0, −9.7, −16.2, −17.0, −20.5, and −13.7 kcal/mol in WT, E119A, E119D, E119G, H274Y, I117T, I117V, I117V-E119A, K150N, N294S, R292K, V116A, and Y252H, respectively. The differences of van der Waals interactions (ΔΔEvdw) were 3, 3.6, 1, 4.2, 4.4, 1.4, 1.6, 9.7, 3.2, 2.4, 1.1, and 5.7 kcal/mol in E119A, E119D, E119G, H274Y, I117T, I117V, I117V-E119A, K150N, N294S, R292K, V116A, and Y252H systems, each compared to the WT system.
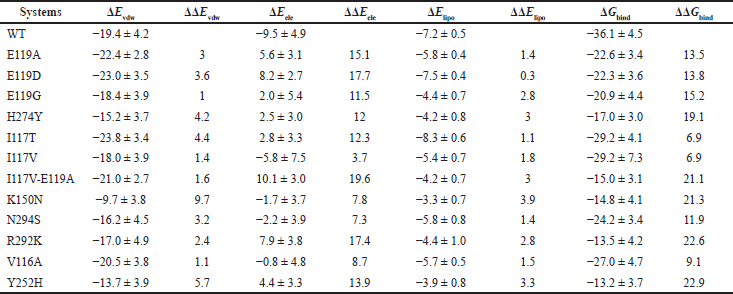 | Table 2. The binding energies calculated for the last 100 ns of each trajectory. [Click here to view] |
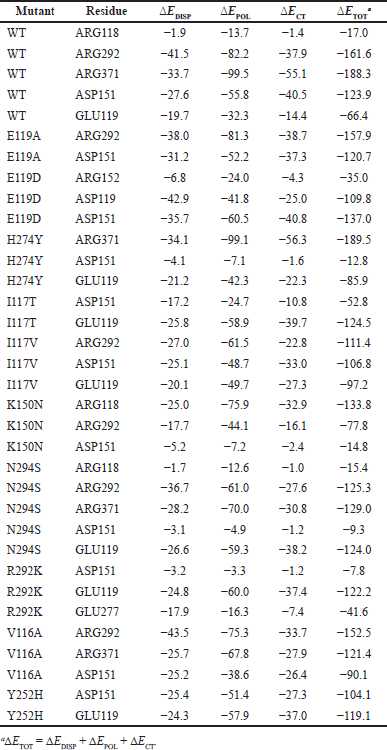 | Table 3. Breakdowns of the dispersion (?EDISP), polarization (?EPOL), and charge transfer (?ECT) interaction energies between OTV and key residues in the active site obtained from second-generation ALMO-EDA at the _B97M-V/Def2-SVPD level of theory (in kJ/mol). [Click here to view] |
Meanwhile, ΔElipo were also more negative in the E119D and I117T systems (−7.5 and −8.3 kcal/mol) than in other systems each with −7.2, −5.8, −4.4, −4.2, −5.4, −4.2, −3.3, −5.8, and −4.4 kcal/mol in WT, E119A, E119G, H274Y, I117V, I117V-E119A, K150N, N294S, and R292K, respectively. The mutations reduced the electrostatic and lipophilic interactions in the I117V-E119A system with 19.6 and 3 kcal/mol changes, respectively, compared to the WT system.
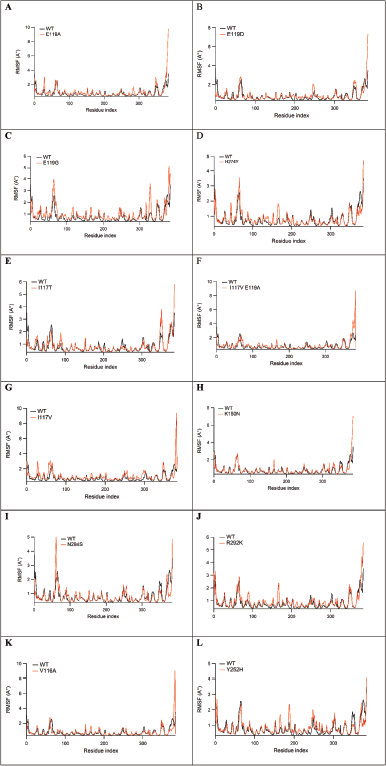 | Figure 5. The RMSF values of protein Cα atoms comparison between WT and E119A (A), WT and E119D (B), WT and E119G (C), WT and H274Y (D), WT and I117T (E), WT and I117V (F), WT and I117V-E119A (G), WT and K150N (H), WT and N294S (I), WT and R292K (J), WT and V116A (K), and WT and Y252H (L), with the mutant positions are noted by yellow asterisks. [Click here to view] |
The electrostatic energy (ΔEele) is favorable binding contributions to WT with −9.5 kcal/mol; however, it became unfavorable in E119A, E119D, E119G, H274Y, I117T, I117V-E119A, R292K, and Y252H systems each with 5.6, 8.2, 2.0, 2.5, 2.8, 10.1, 7.9, and 4.4 kcal/mol, respectively. The differences in electrostatic interactions (ΔΔEele) were 15.1, 17.7, 11.5, 12, 12.3, 19.6, 17.4, and 13.9 kcal/mol in E119A, E119D, E119G, H274Y, I117T, I117V-E119A, R292K, and Y252H systems, respectively, each compared to the WT system. However, it becomes less favorable in I117V (−5.8 kcal/mol), K150N (−1.7 kcal/mol), N294S (−2.2 kcal/mol), and V116A (−0.8 kcal/mol).
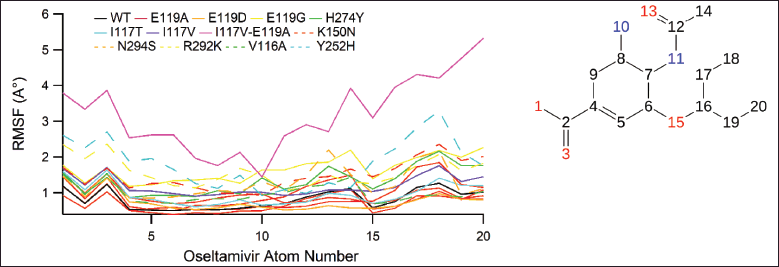 | Figure 6. The RMSF values for atoms of OTV. [Click here to view] |
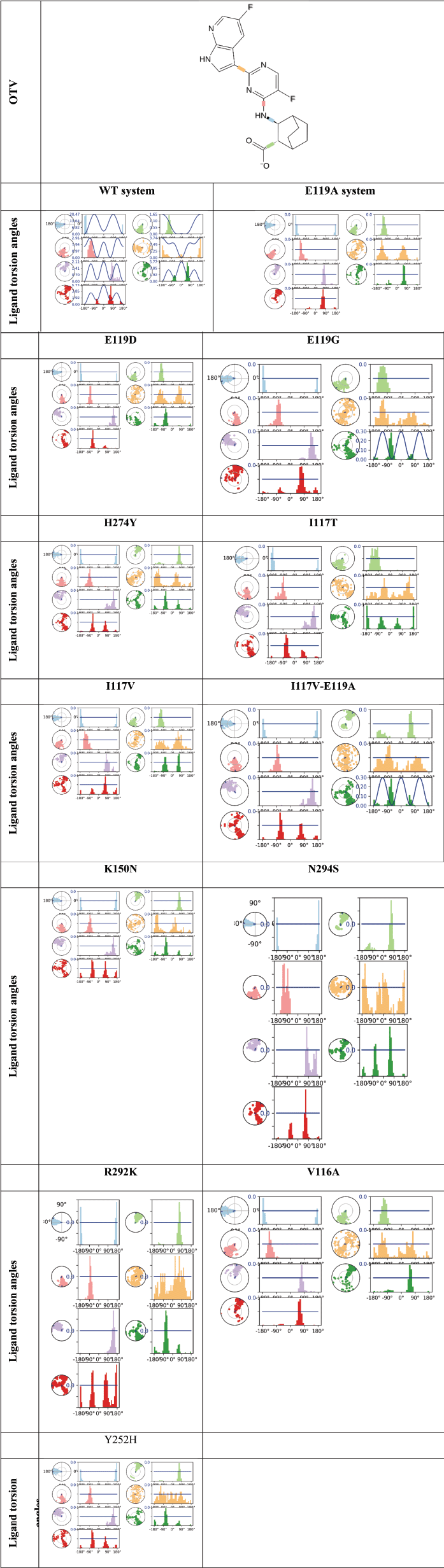 | Figure 7. The dihedral angle of OTV profiles during 200 ns MDS which is the conformational progression of the nine rotatable bonds of OTV. The dial plots describe the conformation of the torsion throughout the course of the simulation. The beginning of the simulation is in the center of the radial plot and the time evolution is plotted radially outward. The bar plots summarize the data on the dial plots, by showing the probability density of the torsion. In addition, the 2D structure of OTV in the top panel is for reference. [Click here to view] |
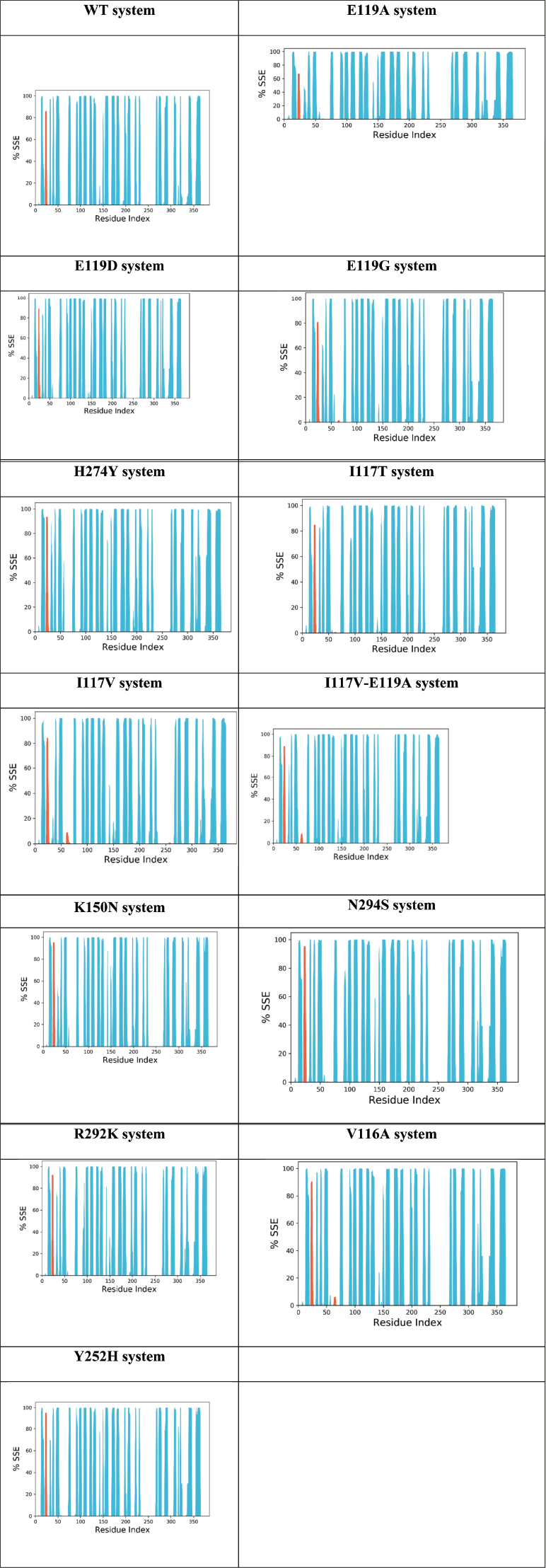 | Figure 8. Protein secondary structure element (SSE) for the WT, E119A, E119D, E119G, H274Y, I117T, I117V, I117V-E119A, K150N, N294S, R292K, V116A, and Y252H systems during MDS. The alpha helices, beta sheets, and random coil were represented by red, blue, and white spaces. [Click here to view] |
Energy decomposition analysis
EDA results
The EDA calculation revealed the nature of the H-bonding and dispersion interactions between OTV and the active site residues. Each of the small models comprises a drug in addition to a key residue of the active site. The geometries of the dimers were obtained from converged MD trajectories. Table 3 lists the resulting breakdowns of the intermolecular interaction energies between OTV and key residues in the active site.
Inspection of Table 3 reveals that, with a few exceptions, the polarization interaction is the dominant interaction, followed by charge-transfer and dispersion interactions. It is instructive to begin with a discussion on the WT system. The strongest overall interaction was obtained for the ARG371 residue, where the sum of the ?EDISP, ?EPOL, and ?ECT components (?ETOT) amounted to as much as −188.3 kJ/mol. We note that polarization interactions account for −99.5 kJ/mol of this total interaction. The ARG292 residue also interacts strongly with the drug, with an overall interaction energy of −161.6 kJ/mol. For both ARG371 and ARG292 residues, we obtained high percentage interactions of 95%–98% from the protein–ligand interaction analysis. We also obtain a strong overall interaction of −123.9 kJ/mol with the ASP151 residue, where the smaller overall interaction energy is mainly due to a reduction in the polarization component to merely −55.8 kJ/mol (Table 1). Accordingly, the protein–ligand interaction analysis resulted in a smaller value of 87%. Finally, for the ARG118 residue, we obtained a low overall interaction of −17.0 kJ/mol, consistent with an even smaller percentage interaction of 62% obtained from the protein–ligand interaction analysis.
Let us now discuss the E119A mutant. In this mutation, we observed a strong interaction with the ARG292 residue, where the overall interaction amounts to −157.9 kJ/mol. Polarization interaction accounts for approximately half of the total interaction (kJ/mol). We also obtained a strong overall interaction with the ASP151 residue at −120.7 kJ/mol. However, dispersion interactions play a lesser role. These results are consistent with the protein–ligand interaction analysis, where both residues resulted in percentage interactions between 81% and 91%. For the related E119D mutation, we obtained a significant overall interaction of −137.0 kJ/mol with the ASP151 residue. Again, the polarization component accounted for nearly 50% of the overall interaction energy. It should be noted that the protein–ligand interaction analysis resulted in 99% interaction for this residue.
For the H274Y mutation, we obtained a very large overall interaction of −189.5 kJ/mol with the ARG371 residue, where the polarization component accounts for over 50% of the overall interaction energy, namely −99.1 kJ/mol. Protein–ligand interaction analysis resulted in 96% interaction for this residue. For the GLU119 residue, we obtain a smaller overall interaction of −85.9 kJ/mol, consistent with a percent interaction of 86%. For the ASP151 residue, we obtained a low overall interaction of merely −12.8 kJ/mol, consistent with an interaction of only 50%.
For the I117T mutation, we obtained a significant interaction of −124.5 kJ/mol with the GLU119 residue. The interaction with the ASP151 residue was significantly smaller and amounted to −52.8 kJ/mol. Both interactions are dominated by the polarization component, and both are consistent with the percent interactions of 86% and 79% obtained from the protein–ligand interaction analysis. For the I117V mutation, we obtained similar interaction energies ranging between −97.2 and −111.4 for the three residues GLU119, ASP151, and ARG292.
For the K150N mutation, the interaction energies of the three residues varied widely. For example, we obtain the following interaction energies −133.8 (ARG118), −77.8 (ARG292), and −14.8 (ASP151). Therefore, ARG118 plays a significantly more important role in drug binding. We note, however, that in contrast to the other mutations, for the K150N mutation, there was no agreement with the protein–ligand interaction analysis. For example, we obtained percent interactions of 51% (ARG118), 54% (ARG292), and 87% (ASP151).
In the case of the N294S mutation, three residues interacted strongly with the drug. ARG292, ARG371, and GLU119 resulted in binding energies ranging between −124.0 and −129.0 kJ/mol. For the R292K mutation, we obtain a similarly strong interaction of −122.2 kJ/mol with the GLU119 residue. For the V116A mutation, both arginine residues ARG292 and ARG371 interact strongly with the drug, with interaction energies of −152.5 and −121.4 kJ/mol, respectively. Finally, for the Y252H mutation, the ASP151 and GLU119 residues result in appreciable interaction energies of −104.1 and −119.1 kJ/mol, respectively.
The root mean square fluctuation (RMSF) values
Figure 5 depicts the RMSF of the protein Cα atoms in the WT and mutant strains. The highest peaks were for residues of the carboxyl and amino ends of the protein. The high peaks around 29, 60, 167, 188, 329, and 351 were protein loops, while the key residues interacting with the protein were observed to be stable.
Ligand RMSF values
Figure 6 depicts the RMSF values of the OTV atoms, in which the RMSF of OTV in WT system was lower with mean RMSF values (0.81 Å) compared to those in E119A (0.92 Å), E119G (1.66 Å), H274Y (1.32 Å), I117T (0.95 Å), I117V (1.19 Å), I117V-E119A (3.19 Å), K150N (1.48 Å), N294S (1.26 Å), R292K (1.57 Å), V116A (0.98 Å), and Y252H (1.382 Å), but it was higher than those in E119D (0.76 Å) systems, which indicated that OTV was more stable in the WT system than in I117V-E119A but it was less stable compared to in E119D systems. The atom numbers 16-20 (ethylpropoxy group) in I117V-E119A were considered the most fluctuant atoms.
The OTV dihedral angle profiles
Figure 7 depicts the dihedral angle of OTV, in which the rotatable bonds colored light blue, light green, yellow, light purple, red, and orange show a wider distribution in mutant systems. Wider dihedral angle distributions were also observed in yellow for all mutants and light green in the E119G, I117T, I117V-E119A, and V116A systems. Briefly, the mutant systems induced a wider distribution of dihedral angles within OTV.
The secondary structure of protein
The total percentage of SSE for WT, E119A, E119D, E119G, H274Y, I117T, I117V, I117V-E119A, K150N, N294S, R292K, V116A, and Y252H was 38.86%, 37.74%, 38.75%, 38.89%, 40.07%, 38.85%, 37.72%, 39.15%, 38.48%, 39.31%, 38.39%, 39.49%, and 39.10%, respectively (Fig. 8). It was clear that the mutation in the H274Y system increased the SSE percentage in the system, whereas the mutation in other systems had minor effects on the SSE of the protein.
OTV is an FDA-approved drug that inhibits the NA activity of IVA. Previous experimental studies have shown that Arg118, Arg292, and Arg371 play vital roles in the active site of NA, whereas Glu276 interacts with acetamido from the substrate to form hydrogen bonds. Asp151 and Glu119 are responsible for cavity widening at the active site of NA [30]. These experimental observations correlated well with the results of the present study for the WT system.
Recent findings suggest that resistance to OTV arises from a NA mutation in IAV. In vitro assays revealed mutations, including E119A, E119D, E119G, H274Y, I117T, I117V, I117V-E119A, K150N, N294S, R292K, V116A, and Y252H, resulting in decreased sensitivity to OTV. Here, we show that NA mutations have reduced OTV affinity due to reduced electrostatic interactions that become more positive in all systems. In the K150N system, the mutation induced a more positive van der Waals interaction, resulting in weakened binding of OTV to IAV. Meanwhile, the I117V-E119A and R292K systems also exhibited low OTV binding energies owing to increased electrostatic values during the simulation. In addition, it may be triggered by reduced occupancy and loss of interaction with several key residues such as Arg292, Arg371, Glu119, and Asp151.
CONCLUSION
The molecular details of OTV binding to IAV NA in WT and mutant systems were explored using a computational study. The key residues that interact with OTV include Arg118, Arg292, Arg371, Arg152, Glu276, Asp151, and Glu119, which are in line with the experimental results. The binding modes of OTV changed, resulting in reduced affinity in all mutant systems. In this case, the mutation caused weakened electrostatic interactions and reduced OTV affinity, particularly in the I117V-E119A and R292K systems. The present study revealed the molecular details of how mutations in NA of IAV affect the affinities of OTV. The future work would be on designing new analogs of OTV with better affinities based on the present results.
ACKNOWLEDGMENTS
MA would like to thank the Ministry of Education, Culture, Research, and Technology, Republic of Indonesia, for supporting the research through World Class Professor Grant and Hibah Penelitian Internal UHO 2023. Wu acknowledges the New Jersey Health Foundation (PC 76-21); the US National Science Foundation under Grants NSF ACI-1429467/RUI-1904797 and XSEDE MCB 170088; and the Anton2 machine time at the Pittsburgh Supercomputing Center (PSCA170090P).
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Harrington WN, Kackos CM, Webby RJ. The evolution and future of influenza pandemic preparedness. Exp Mol Med. 2021;53(5):737–49.
2. Hanpaibool C, Leelawiwat M, Takahashi K, Rungrotmongkol T. Source of oseltamivir resistance due to single E119D and double E119D/H274Y mutations in pdm09H1N1 influenza neuraminidase. J Comput-Aided Mol Des. 2020;34(1):27–37.
3. Abed Y, Nehmé B, Baz M, Boivin G. Activity of the neuraminidase inhibitor A-315675 against oseltamivir-resistant influenza neuraminidases of N1 and N2 subtypes. Antivi Res. 2008;77(2):163–6.
4. Guo L, Garten RJ, Foust AS, Sessions WM, Okomo-Adhiambo M, Gubareva LV, et al. Rapid identification of oseltamivir-resistant influenza A(H1N1) viruses with H274Y mutation by RT-PCR/restriction fragment length polymorphism assay. Antivi Res. 2009;82(1):29–33.
5. Earhart KC, Elsayed NM, Saad MD, Gubareva LV, Nayel A, Deyde VM, et al. Oseltamivir resistance mutation N294S in human influenza A(H5N1) virus in Egypt. J Infect Public Health. 2009;2(2):74–80.
6. McKimm-Breschkin JL, Selleck PW, Usman TB, Johnson MA. Reduced sensitivity of influenza A (H5N1) to oseltamivir. Emerg Infect Dis J. 2007;13(9):1354.
7. Kiso M, Mitamura K, Sakai-Tagawa Y, Shiraishi K, Kawakami C, Kimura K, et al. Resistant influenza A viruses in children treated with oseltamivir: descriptive study. Lancet. 2004;364(9436):759–65.
8. de Jong MD, Thanh TT, Khanh TH, Hien VM, Smith GJD, Chau NV, et al. Oseltamivir resistance during treatment of influenza A (H5N1) infection. N Engl J Med. 2005;353(25):2667–72.
9. Choi WS, Jeong Ju H, Kwon Jin J, Ahn Su J, Lloren Khristine Kaith S, Kwon HI, et al. Screening for neuraminidase inhibitor resistance markers among avian influenza viruses of the N4, N5, N6, and N8 neuraminidase subtypes. J Virol. 2017;92(1):e01580–17.
10. Arba M, Wahyudi ST, Brunt DJ, Paradis N, Wu C. Mechanistic insight on the remdesivir binding to RNA-dependent RNA polymerase (RdRp) of SARS-cov-2. Comput Biol Med. 2021;129:104156.
11. Mihajlovic ML, Mitrasinovic PM. Another look at the molecular mechanism of the resistance of H5N1 influenza A virus neuraminidase (NA) to oseltamivir (OTV). Biophys Chem. 2008;136(2):152–8.
12. Karthick V, Ramanathan K. Computational investigation of oseltamivir resistance in influenza A (H5N1) virus. Med Chem Res. 2013;22(12):5764–71.
13. Nguyen H, Tran T, Fukunishi Y, Higo J, Nakamura H, Le L. Computational study of drug binding affinity to influenza A neuraminidase using smooth reaction path generation (SRPG) method. J Chem Inf Model. 2015;55(9):1936–43.
14. Arba M, Wahyudi ST, Zubair MS, Brunt D, Singh M, Wu C. Binding of GS-461203 and its halogen derivatives to HCV genotype 2a RNA polymerase drug resistance mutants. Sci Pharm [Internet]. 2022;90(2):26.
15. Uba AI, Bui-Linh C, Thornton JM, Olivieri M, Wu C. Computational analysis of drug resistance of taxanes bound to human β-tubulin mutant (D26E). J Mol Graph Model. 2023;123:108503.
16. Thierry E, Guilligay D, Kosinski J, Bock T, Gaudon S, Round A, et al. Influenza polymerase can adopt an alternative configuration involving a radical repacking of PB2 domains. Mol Cell. 2016;61(1):125–37.
17. Arba M, Ningsih AS, Bande LOS, Wahyudi ST, Bui-Linh C, Wu C, et al. Computational insights into the binding of pimodivir to the mutated PB2 subunit of the influenza A virus. Mol Simul. 2023;49(10):1–13.
18. Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput-Aided Mol Des. 2013;27(3):221–34.
19. Mark P, Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J Phys Chem A. 2001;105(43):9954–60.
20. Jorgensen WL, Maxwell DS, TiradoRives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118(45):11225–36.
21. Bailey AG, Lowe CP. Milch shake: an efficient method for constraint dynamics applied to alkanes. J Comput Chem. 2009;30(15):2485–93.
22. Shan YB, Klepeis JL, Eastwood MP, Dror RO, Shaw DE. Gaussian split Ewald: a fast Ewald mesh method for molecular simulation. J Chem Phys. 2005;122(5):054101.
23. Stuart SJ, Zhou RH, Berne BJ. Molecular dynamics with multiple time scales: the selection of efficient reference system propagators. J Chem Phys. 1996;105(4):1426–36.
24. Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL; 2006. p 84.
25. Li JN, Abel R, Zhu K, Cao YX, Zhao SW, Friesner RA. The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins-Struct Funct Bioinform. 2011;79(10):2794–812.
26. Mardirossian N, Head-Gordon M. ωB97M-V: a combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation. J Chem Phys. 2016;144(21):214110.
27. Weigend F, Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys. 2005;7(18):3297–305.
28. Horn PR, Mao Y, Head-Gordon M. Probing non-covalent interactions with a second generation energy decomposition analysis using absolutely localized molecular orbitals. Phys Chem Chem Phys. 2016;18(33):23067–79.
29. Shao Y, Gan Z, Epifanovsky E, Gilbert ATB, Wormit M, Kussmann J, et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol Phys. 2015;113(2):184–215.
30. Wang NX, Zheng JJ. Computational studies of H5N1 influenza virus resistance to oseltamivir. Protein Sci. 2009;18(4):707–15.
SUPPLEMENTARY MATERIAL
Supplementary data can be downloaded from the journal’s website