
INTRODUCTION
Smoking is a serious hazard to global communal health, and according to present trends, 500 million of the world’s 1 billion smokers will die early as a result of smoking-related diseases [1]. Smoking cigarettes has been widely acknowledged as a conventional and important hazard aspect for the progression of cardiovascular disease and atherosclerosis [2]. It has recently been realized that coronary heart disease (CHD) encompasses an inflammatory module and has been considered an inflammatory disorder [3]. Cigarette smoking causes CHD in a variety of ways, including through the progression of atherosclerosis and inflammation, altering the blood’s balance of high-and low-density lipoproteins (HDL and LDL), raising blood pressure and heart rate, and encouraging clot formation. Toxic byproducts of cigarette smoke circulate in the bloodstream, including nicotine, carbon monoxide, oxidant gases, and acrolein. They disrupt endothelial function, causing blood fat anomalies and affecting glucose regulation. Additionally, smoking directly affects platelets, increasing activation and stickiness. As a result, there is a higher risk of thrombosis or the onset of atherosclerosis [4].
Human apolipoprotein-E (APO-E), a glycosylated protein of 34 kDa, has been associated with the aetiology of atherosclerosis [5]. The APO-E gene consists of 299 amino acids and is found on chromosome 19 at location q13.2, which is made up of three introns and four exons. The APO-E gene variation at both single sequences (rs429358 and rs7412) produces three unique alleles, E2, E3, and E4, resulting in six different genotypes, encompassing E2/E2, E3/E3, E4/E4, E2/E3, E2/E4, and E3/E4. Arginine or cysteine can be found in amino acid sequences 112 and 158, which differentiate the three APO-E isoforms: E2 (cysteine 112, cysteine 158), E3 (cysteine 112, arginine 158), and E4 (arginine 112, arginine 158) [6]. The structural and functional variations between the three APO-E variants can be sufficient to raise the risk of disease even if they only differ by one or two amino acids. The three APO-E isoforms differ by a single amino acid, which affects the protein’s structure, lipid association, and receptor binding. As a result, APO-E influences cholesterol homeostasis in an isoform-dependent way [7].
Inflammation-related pathways might be one of the probable mechanisms linking the APO-E gene to cardiovascular disease [8,9]. Moreover, APO-E has proinflammatory qualities and facilitates the delivery of lipid antigens to the immune system, which can contribute to chronic inflammation. Numerous studies have shown that the APO-E gene regulates metabolic processes. The crucial biomarkers for chronic systemic inflammation are high-sensitive C-reactive protein (hs-CRP) and matrix metalloproteinase-9 (MMP-9). The association between APO-E Single nucleotide polymorphisms (SNPs) and disease states was significantly impacted by serum C-reactive protein (CRP) levels [10]. CRP is a known inflammatory marker and a risk indicator for CHD and has been linked to the APOE gene variant [11]. The APO-E genetic makeup influences CRP production through a cytokine-independent pathway. However, it is necessary to identify the mechanism behind this relationship [12]. Several study findings indicate that APO-E4 macrophages exhibit altered inflammatory responses, which may be a factor in the increased CHD risk seen in APO-E4 carriers [13,14]. Therefore, the study’s goal is to investigate the relationship between APO-E genotype and inflammation and the risk of premature CHD in smokers versus non-smokers.
MATERIALS AND METHODS
Study design and participants
The case-control study included 300 subjects and was recruited between October 2020 and 2022. Three sections constituted the research investigation. Group 1 is made up of 100 healthy nonsmokers; Group 2 is made up of 100 young active smokers who have CHD, and Group 3 is made up of 100 young active smokers who have both CHD and diabetes mellitus (DM) and are undergoing cardiology and medicine Outpatient. Participants in the study were all men between the ages of 20 and 55. Smoking habits were identified based on self-reporting. Calculations were made for smoking status, smoking load (duration of smoking), and smoking intensity (number of cigarettes smoked each day). Those who regularly smoked >5 cigarettes every day over the last 12 months were considered smokers [15]. The inclusion criteria were patients with a CHD diagnosis who smoked often. The characteristic chest discomfort raised the ST segment on the electrocardiogram, abnormal lab results for creatine phosphokinase, creatine kinase-myocardial band, and troponin I, and abnormal coronary angiography with a stenosis of more than 50% in one of the major coronary arteries were used to make the diagnosis of CHD. The control participants are all non-smokers and did not have evidence of cardiovascular disease. Patients with cardiomyopathy, chronic conditions like hepatic failure, malignancy patients, people who have had heart surgery, people who have had cardiovascular accidents, autoimmune diseases, endocrine diseases, serious systemic illnesses, and systemic inflammatory diseases have been excluded from the study. To determine whether patients were fit for the study, a standard questionnaire was provided. A signed informed consent form was obtained from each study participant.
Study protocol and measures
Before laboratory measurements, participants were told to fast for at least 12 hours and rest all night without engaging in any physical activity. They were then instructed to report to the hospital between the hours of 8:00 a.m. and 9:00 a.m. 4 ml of venous fasting blood from each participant was divided into two aliquots, one of which was placed in an EDTA vial and the other in a plain vial. From the sample containing EDTA, DNA was extracted and serum will be separated from blood samples by centrifuging them at 2,000 × g for 10 minutes at 4ºC and keeping the separated samples at −20ºC until analysis.
Self-reporting is used to identify age, gender, educational background, and other sociodemographic traits, as well as prior health and medical histories. Anthropometric measurements like weight and height were taken using standard protocols. Blood pressure was assessed after 2 minutes of relaxation. A systolic blood pressure of at least 140 mmHg and a diastolic blood pressure of at least 90 mmHg are indicators of hypertension. Patients with DM were identified using the American Diabetes Association guidelines. The AU480 Beckman coulter instrument was used to evaluate fasting glucose and lipoprotein profiles. HbA1c (glycosylated hemoglobin) was determined using a high-performance liquid chromatography technique. The very high-density lipoprotein cholesterol (VLDL-C) level was determined using the formula VLDL-C = triglyceride (TGL)/5. The non-HDL level was determined by deducting the total cholesterol (TC) value from the HDL-C value.
According to the production technique, serum inflammatory marker (hs-CRP and MMP-9) concentrations were measured using an enzyme-linked immunoassay (ELISA, Abbkine, Inc., China).
DNA extraction and APO-E genotyping
Genomic DNA was extracted using a DNA extraction kit (Qiaamp DNA extraction kit) from 200 ul of whole peripheral blood mononuclear cells (Qiagen Hilden, Germany, catalogue number: 51104). A nano-drop 2000TM spectrophotometer was used to measure the quantity of DNA. The intactness of isolated DNA was confirmed by 1% agarose gel electrophoresis computed by (UV) spectroscopy, and then isolated DNA was stored at −80ºC.
The APO-E gene was genotyped utilizing an allele-specific polymerase chain reaction (PCR) method that was adapted to use with a TaqMan probe for real-time PCR [6]. The nucleotide changes observed at the two SNPs, rs429358 and rs7412, inside exon four of the APO-E gene were used to produce the initial PCR primers. Three pairs of oligonucleotide primers that specifically amplified the E2, E3, and E4 alleles were found after screening a collection of oligonucleotide primers. For each process, a single TaqMan probe with a double-dye oligonucleotide was used to track the results of real-time DNA amplification. The probe has a black hole quencher molecule and a fluorescein amide (FAM) attached to its 3′ and 5′ ends, respectively [6].
E2-Forward GCGGACATGGAGGACGTGT E2-Reverse CCTGGTACACTGCCAGGCA
E3-Forward CGGACATGGAGGACGTGT E3-Reverse CTGGTACACTGCCAGGCG
E4-Forward CGGACATGGAGGACGTGC E4-Reverse CTGGTACACTGCCAGGCG
APO-E probe: FAM-CAGCTCCTCGGTGCTCTGGC-BHQ1
Three reactions are used for utilizing real-time PCR to genotype APO-E: the E2 reaction, the E3 reaction, and the E4 reaction. The following was the PCR amplification protocol: amplitude gold DNA polymerase was activated at 95ºC for 10 minutes before being put through 40 cycles of denaturation at 95ºC for 15 seconds and extension at 64ºC for 1 minute. During the annealing/extension processes, fluorescence signals were recorded [6]. The FAM signal suggests the APOE gene. The amplification was carried out using a Quant Studio real-time PCR instrument.
Statistical analysis
The statistical analysis was carried out using the International Business Machines Corporation’s statistics software for the social sciences, version 22. The mean, as well as the SD, is used to express numerical information. One-way ANOVA has been used in descriptive analysis for continuous variables to compare means and medians. Genotype and allele risks were calculated using the odds ratio (OR). For qualitative variables, the Hardy-Weinberg equilibrium distribution was used to evaluate the allele distribution using the chi-square test. Unadjusted OR with confidence intervals (95% CL) were reported as a result of univariable logistic regression analysis conducted to assess the connection between risk indicators and CHD risk. Statistical significance was defined as p values less than 0.05.
RESULTS
Population-specific demographic, clinical, and biochemical data
Table 1 shows the socioeconomic, clinical, and biological data of the research individuals. When smokers from Groups 3 and 2 were compared to Group 1, (controls), there was a statistically significant difference (p < 0.001) in their age, body mass index (BMI), waist-hip ratio (W/H ratio), blood pressure, smoking burden, and smoking intensity. Patients’ fasting blood sugar and glycated hemoglobin (HbA1c) values were significantly higher than controls (p < 0.001). Data from lipid profiles indicate a substantial link between dyslipidemia and CHD. The study demonstrates that when compared with Group 1, smokers in Groups 3 and 2 had significantly lower levels of HDL-C and higher levels of TC, TGL, LDL-C, VLDL-C, the TC/HDL-C ratio, and the LDL-C/HDL-C ratio, and non-HDL-C.
According to the findings, Groups 3 and 2 smokers showed substantially higher blood APO-E, hs-CRP, and MMP-9 levels. Subjects with both CHD and diabetes who smoke (group 3) exhibited the highest serum concentrations of APO-E, hs-CRP, and MMP-9 when compared to controls.
To determine if there was a significant difference between the groups, a Tukey’s honest significant difference post hoc analysis was performed (Table 2). A considerable difference was observed between the subject’s age, BMI, Systolic blood pressure (SBP), smoking intensity, smoking burden, lipid profile, lipid ratios, APO-E, hs-CRP, and MMP-9 in smokers with CHD and controls (p < 0.001). The highest significant difference was found among the smokers with CHD and DM with controls (p < 0.001), which was significantly greater than the smokers with CHD without DM.
The genotypes and allele frequencies of the APO-E gene in the studied sample
Tables 3 and 4 summarise the genotype and allele frequency spectrum of the APO-E gene in study participants. All groups’ genotype distributions were in a state of Hardy-Weinberg equilibrium (p < 0.05). In all three groups, the E3/E3 genotype predominated (60% of smokers with CHD and DM patients, 58% of smokers with CHD patients without diabetes, and 72% of control participants), with the E3/E4 genotype coming in second with 25% of smokers with CHD and DM patients, 23% of smokers with CHD patients without diabetes, and 11% of participants in the control group, and the E2/E3 genotype coming in third with 7% of smokers with CHD and DM patients 9% of smokers with CHD patients without diabetes and 8% of participants in the control group. The most prevalent allele was E3 (77% of smokers with CHD and DM patients, 78% of smokers with CHD patients without diabetes, and 84% of control participants), with the E4 allele coming in second with 19% of smokers with CHD and DM patients, 17% of smokers with CHD patients without diabetes, and 9% of control participants, and the E2 allele coming in third with 4% of smokers with CHD and DM patients, 5% of smokers with CHD patients without diabetes and 7% of control participants. According to the findings, the risk frequency of the E3/E4 genotype and E4 allele was considerably higher in smokers with CHD and DM. (p = 0.011 and p < 0.001, respectively). Whereas, when compared to controls, in smokers with CHD, the risk frequency of the E3/E4 genotype and E4 allele was substantially greater (p = 0.026 and p = 0.019, respectively).
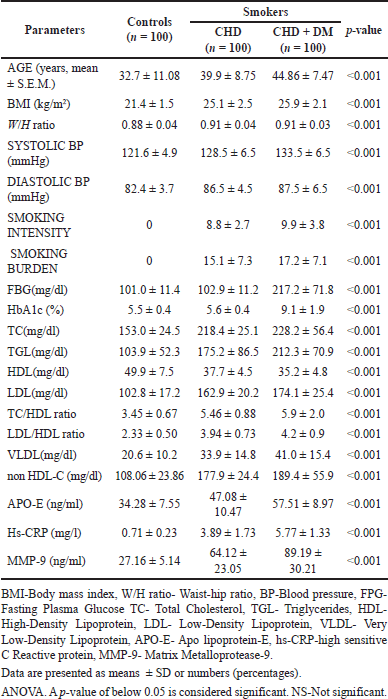 | Table 1. Demographic, clinical and biochemical data of the study population. [Click here to view] |
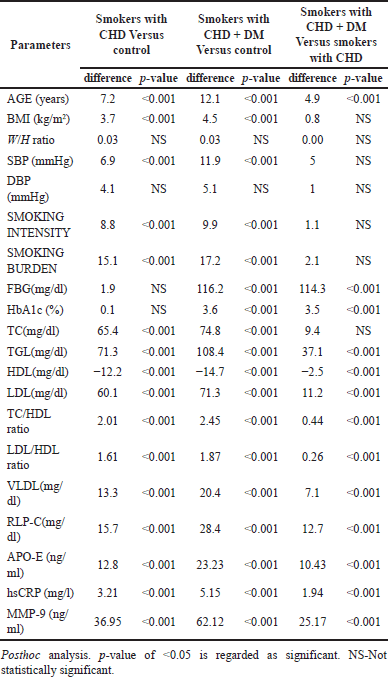 | Table 2. Posthoc analysis of demographic, clinical and biochemical data of the study population. [Click here to view] |
Distribution of lipid profile and inflammatory markers among APO-E genotypes in the study population
Tables 5 and 6 demonstrate the distribution of the lipid profile and inflammatory markers among the APO-E genotypes of the study participants. Compared with the other genotype of the APO-E gene, smokers with CHD and smokers with CHD and DM subjects with the E3/E4 genotype had lower levels of serum HDL, higher TC, TGL, LDL, APO-E, hs-CRP, and MMP-9 which were statistically significant. This demonstrates that the E3/E4 genotype is linked to an increased risk of early CHD in young smokers.
Logistic regression model of CHD risk in smokers
To investigate the predictability of several factors and their interactions in the progression of CHD, a logistic regression model has been applied. After correcting for confounding variables such as age, BMI, lipid profile, and biochemical parameters in regression analysis as independent variables, the E3/E4 genotype is associated with age, lipoprotein profile, serum APO-E hs-CRP, and MMP-9 and were statistically significant predictors for CHD (all p < 0.05), with OR shown in Tables 7 and 8.
DISCUSSION
There is no about that the adverse impacts of smoking on the cardiovascular system. The exact facts, however, remain contentious and unclear. Young cigarette smokers are more likely to develop cardiovascular disease than healthy adults. According to one study, 4,326 instances of smokers with CHD were recorded during follow-up. The study shows CHD risk among current smokers was highest among the youngest and lowest among the oldest individuals when compared to never-smokers. The study shows that among subjects aged 40 to 49 years, the risk ratio was 8.5 (95% CI = 5.0–14) and 3.1 (95% CI = 2.0–4.9) among those aged 70 years or older [16]. This can be explained by several connections, such as changes in serum lipid and lipoprotein concentrations [17]. Smoking causes cardiovascular events by a variety of mechanisms, including the development of atherosclerotic alterations with narrowing of the arterial lumen and the generation of a hypercoagulable condition, which increases the risk of acute thrombosis [18]. In this investigation, smokers’ serum APO-E concentrations were substantially higher compared to healthy controls (<0.0001). Acrolein, a cigarette smoke component, causes oxidative alteration of APO-E, reducing its ability to bind with LDL receptors. Furthermore, the acrolein-modified APO-E had a decreased ability to interact with lipid surfaces. As a result, it disrupts the control of plasma cholesterol homeostasis by interfering with APO-E’s functional activity [19,20]. One of the hypotheses states that an increase in APO-E concentration might cause CHD. High APO-E levels may reflect a determinant lipid profile. The study suggested that an increase in APO-E concentration reflects an increase in lipoprotein levels such as LDL-C and VLDL-C levels, which are pathogenic [21].
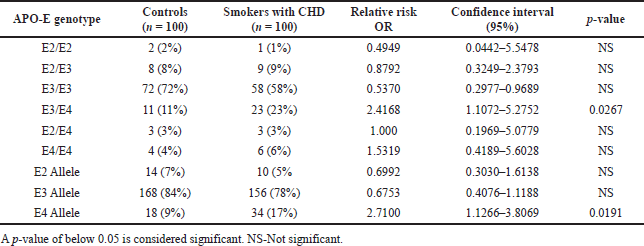 | Table 3. The genotypes and allele frequency distribution of the APO-E gene in smokers with CHD subjects. [Click here to view] |
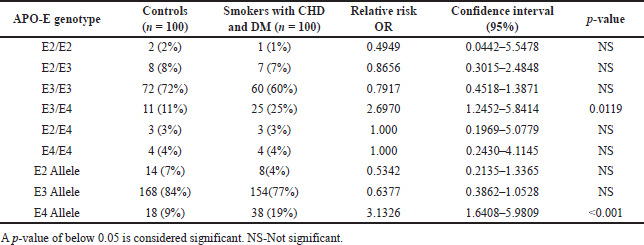 | Table 4. The genotypes and allele frequency distribution of the APO-E gene in smokers with CHD and DM subjects. [Click here to view] |
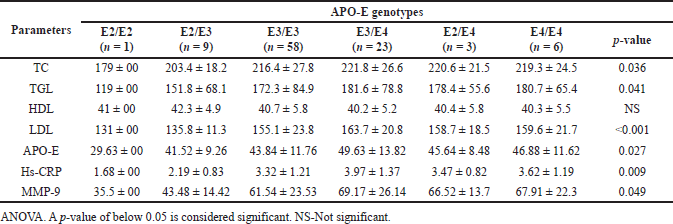 | Table 5. Distribution of lipid profile and inflammatory markers among APO-E genotypes in smokers with CHD subjects. [Click here to view] |
In recent years, a large amount of research has been conducted, on the alleged link between smoking and the activation of inflammatory pathways. A few of them are contradictory, in which serum CRP concentrations have been assessed in connection to the smoking category [22]. Smokers have more white blood cells, which is mostly due to an increase in polymorphonuclear neutrophils, which are produced in the bone marrow and attracted to inflamed tissue [23]. In one of the earliest studies of CRP levels in smokers, before the advent of assays with increased sensitivity, CRP levels were shown to be considerably elevated in male and female smokers compared to non-smokers [24]. This study highlights the complexities of cytokine-mediated inflammation by revealing that smoking status has a significant association with inflammatory markers.
As far as our information goes, this is the foremost study to examine the gene-environment interaction of APO-E gene variation in smokers and its association with inflammatory markers in the Indian population.
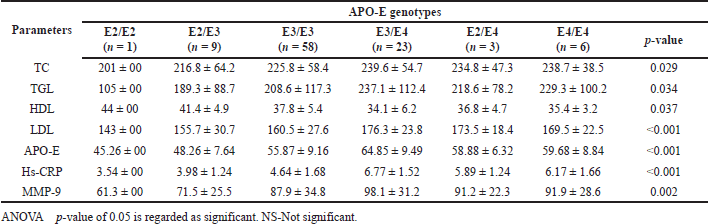 | Table 6. Distribution of lipid profile and inflammatory markers among APO-E genotypes in smokers with CHD and DM subjects. [Click here to view] |
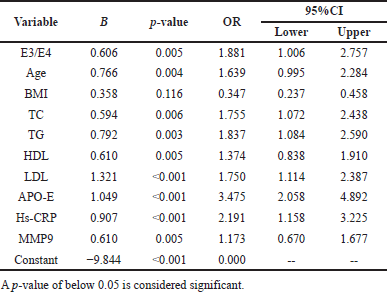 | Table 7. Logistic regression assessment of APO-E genotype, cardiovascular risk variables, in smokers with CHD subjects. [Click here to view] |
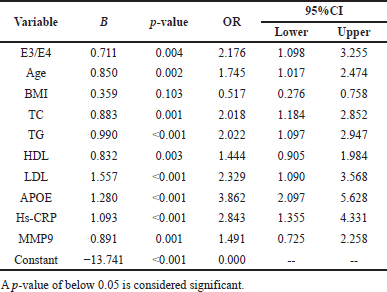 | Table 8. Logistic regression assessment of APO-E genotype, cardiovascular risk variables, in smokers with CHD and DM subjects. [Click here to view] |
The result of the present study indicates that smoking increases the risk for CHD, particularly in males with the E3/E4 genotype and the E4 allele. The study shows a strong relationship between the E3/E4 genotype and smoking and the risk of developing CHD and DM in young smokers, which is in line with earlier research [25,26]. Our findings support the smoking/genotype interaction and extend the findings to younger age groups than earlier research. Two potential explanations for the relationship between smoking and multiple APO-E genotypes with CHD are changes in lipid concentrations and an inflammatory response to different APO-E genotypes [27]. Moreover, the effect of the APO-E-smoking interaction on lipid concentrations and CHD risk has been studied [28] but the biological basis for it is still unclear.
First, while examining the connection between smoking and different APO-E variants and lipid levels, the study result shows that APO-E gene carriers had significantly different levels of TC, TGL, LDL-C, and HDL-C in current smokers. The study findings demonstrated that smokers with the E3/E4 genotype were related to greater levels of TC and TGL in all participants investigated, as well as higher LDL-C levels in smokers with CHD and T2DM, underscoring the importance of elevated LDL-C levels in CHD development. Previous research on the effects of smoking and the APO-E gene and its association with plasma lipid profiles has yielded inconclusive results across different ethnic groups [29]. Indians with the E3/E4 genotype exhibited lower HDL-C levels and higher LDL-C concentrations in patients with CHD [30], as well as increased TG concentrations in T2DM patients [31]. According to a recent survey of the Kashmiri community, CHD patients with the E4 allele exhibited significantly higher LDL and TC values [32]. Secondly, regarding the relationship between APO-E and inflammation in smokers, several studies have discovered that inflammatory biomarkers related to the progression of CHD in smokers, such as CRP, MMP-9, TNF-α, and interleukins [33–35], can enhance CHD risk classification, particularly CRP and IL-6 [36–38]. Furthermore, several studies have shown that the association between smoking, the APO-E gene, and CHD risk is partially regulated by substantial impacts on inflammatory biomarkers like CRP and IL-6 [39,40]. The result of the present study also supports previous results showing that CRP levels are associated with the APO-E genotype, indicating that smokers with APO-E4 carriers with the E3/E4 genotype have significantly higher levels of serum hs-CRP than APO-E2 carriers with the E2/E2 genotype [41,42]. A recent study has also revealed the connection between inflammation and APO-E genotype and the risk of CHD, showing that smokers with APO-E4 carriers and E3/E4 genotypes are associated with higher CHD risk in young smokers [39].
Limitations of the study
Some potential limitations of the current study include the potential for selection bias because the population that the recruited control participants came from was undergoing a comprehensive health checkup at a hospital. Secondly, the research’s sample size was inadequate, which may have underpowered the study. Further investigations with prospective follow-up of subjects are needed to confirm our observation. Additionally, we did not evaluate some traditional cardiovascular risk factors such as sex because we were unable to add female smokers since smoking declaration for women is socially unacceptable in southern India.
CONCLUSION
In conclusion, our data revealed that smoking alters the link between Lipids and the APO-E gene. In addition, our result indicates that the levels of inflammatory markers have been robustly associated with the APO-E genotype, with APO-E4 individuals having higher CRP levels than E3 individuals, whereas E2 carriers had lower CRP levels. The study established that APO-E4 carriers are strongly associated with inflammation in young smokers, which is expected to play a role in mediating the influence of genotype on CHD risk in young smokers.
ACKNOWLEDGMENT
The authors would like to gratefully acknowledge the Departments of Cardiology, Translation Medicine, and General Medicine for their authorization and assistance.
AUTHOR CONTRIBUTIONS
All authors contributed significantly to the study. Author 1 contributed to the collection of data, data analysis, interpretation, statistical analysis, and writing the manuscript. Author 2 contributed to the inception, design, writing, review, and final authorization for publication of the manuscript. Author 3 contributed to the review and final authorization for the publication of the manuscript. All writers are eligible to be authors in accordance with the prerequisites and rules established by the International Committee of Medical Journal Editors (ICMJE).
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
The study was authorized by the SRM Medical College Hospital and Research Centre Human Research Ethics Committee (IEC No: 1763).
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Kaleta D, Kozie? A, Mi?kiewicz P. Mpower--a strategy for fighting the global tobacco epidemic. Med Pr. 2009;60(2):145–9.
2. Smith SC, Milani RV, Arnett DK, Crouse JR, McDermott MM, Ridker PM, et al. American Heart Association. Atherosclerotic vascular disease conference: writing group II: risk factors. Circulation. 2004;109(21):2613–6. doi: https://doi.org/10.1161/01.CIR.0000128519.60762.84
3. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–43. doi: https://doi.org/10.1161/hc0902.104353
4. Briffa T, Winnall WR, Greenhalgh EM, Winstanley MH. 3.1 Smoking and cardiovascular disease. In: Greenhalgh EM, Scollo MM, Winstanley MH, editors. Tobacco in Australia: facts and issues. Melbourne, Australia: Cancer council Victoria; 2021. Available from: https://www.tobaccoinaustralia.org.au/chapter-3-health-effects/3-smoking-and-cardiovascular-disease
5. Kumar P, Luthra K, Dwivedi M, Behl VK, Pandey RM, Misra A. Apolipoprotein E gene polymorphisms in patients with premature myocardial infarction: a case-controlled study in Asian Indians in North India. Ann Clin Biochem. 2003;40(4):382–7. doi: https://doi.org/10.1258/000456303766477020
6. Zhong L, Xie YZ, Cao TT, Wang Z, Wang T, Li X, et al. A rapid and cost-effective method for genotyping apolipoprotein E gene polymorphism. Mol Neurodegener. 2016;11:2. doi: https://doi.org/10.1186/s13024-016-0069-4
7. Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: insights into function. Trends Biochem Sci. 2006;31(8):445–54. doi: https://doi.org/10.1016/j.tibs.2006.06.008
8. Schönknecht YB, Crommen S, Stoffel-Wagner B, Coenen M, Fimmers R, Stehle P, et al. APOE ?4 is associated with postprandial inflammation in older adults with metabolic syndrome traits. Nutrients. 2021;13(11):3924. doi: https://doi.org/10.3390/nu13113924
9. Hasel P, Liddelow SA. Isoform-dependent APOE secretion modulates neuroinflammation. Nat Rev Neurol. 2021;17(5):265–6. doi: https://doi.org/10.1038/s41582-021-00483-y
10. Aiello AE, Nguyen HO, Haan MN. C-reactive protein mediates the effect of apolipoprotein E on cytomegalovirus infection. J Infect Dis. 2008;197(1):34–41. doi: https://doi.org/10.1086/524144
11. Chai JT, Ruparelia N, Goel A, Kyriakou T, Biasiolli L, Edgar L, et al. Differential gene expression in macrophages from human atherosclerotic plaques shows convergence on pathways implicated by genome-wide association study risk variants. Arterioscler Thromb Vasc Biol. 2018;38(11):2718–30. doi: https://doi.org/10.1161/ATVBAHA.118.311209
12. Paffen E, DeMaat MP. C-reactive protein in atherosclerosis: a causal factor? Cardiovascular Res. 2006;71(1):30–9. doi: https://doi.org/10.1016/j.cardiores.2006.03.004
13. Jofre-Monseny L, Loboda A, Wagner AE, Huebbe P, Boesch-Saadatmandi C, Jozkowicz A, et al. Effects of apoE genotype on macrophage inflammation and heme oxygenase-1 expression. Biochem Biophys Res Commun. 2007;357(1):319–24. doi: https://doi.org/10.1016/j.bbrc.2007.03.150
14. Huebbe P, Lodge JK, Rimbach G. Implications of apolipoprotein E genotype on inflammation and vitamin E status. Mol Nutr Food Res. 2010;54(5):623–30. doi: https://doi.org/10.1002/mnfr.200900398.
15. Rees VW, Kreslake JM, O’Connor RJ, Cummings KM, Parascandola M, Hatsukami D, et al. Methods used in internal industry clinical trials to assess tobacco risk reduction. Cancer Epidemiol Biomarkers Prev. 2009;18(12):3196–208. doi: https://doi.org/10.1158/1055-9965.EPI-09-0819
16. Tolstrup JS, Hvidtfeldt UA, Flachs EM, Spiegelman D, Heitmann BL, Bälter K, et al. Smoking and risk of coronary heart disease in younger, middle-aged, and older adults. Am J Public Health. 2014;104(1):96–102. doi: https://doi.org/10.2105/AJPH.2012.301091
17. Deepa Singh. Effect of cigarette smoking on serum lipid profile in male population of Udaipur. Biochem Anal Biochem. 2016;5(3):283. doi: https://doi.org/10.4172/2161-1009.1000283
18. Office of the Surgeon General (US), Office on Smoking and Health (US). The health consequences of smoking: a report of the surgeon general. Atlanta, GA: Centers for Disease Control and Prevention (US); 2004.
19. Zak I, Niemiec P, Balcerzyk A, Krauze J. Combined “pro-atherosclerotic” variants of the ACE and APOE genes increase the risk of the coronary artery disease associated with the presence of cigarette smoking. Acta Cardiol. 2008;63(6):741–7. doi: https://doi.org/10.2143/AC.63.6.2033392
20. Tamamizu-Kato S, Wong JY, Jairam V, Uchida K, Raussens V, Kato H, et al. Modification by acrolein, a component of tobacco smoke and age-related oxidative stress, mediates functional impairment of human apolipoprotein E. Biochemistry. 2007;46(28):8392–400. doi: https://doi.org/10.1021/bi700289k
21. Freeman DJ, Packard CJ. Smoking and plasma lipoprotein metabolism. Clin Sci (Lond). 1995;89(4):333–42. doi: https://doi.org/10.1042/cs0890333
22. Yanbaeva DG, Dentener MA, Creutzberg EC, Wesseling G, Wouters EF. Systemic effects of smoking. Chest. 2007;131(5):1557–66. doi: https://doi.org/10.1378/chest.06-2179
23. Van Eeden SF, Hogg JC. The response of human bone marrow to chronic cigarette smoking. Eur Respir J. 2000;15(5):915–21. doi: https://doi.org/10.1034/j.1399-3003.2000.15e18.x
24. Tonstad S, Cowan JL. C-reactive protein as a predictor of disease in smokers and former smokers: a review. Int J Clin Pract. 2009;63(11):1634–41. doi: https://doi.org/10.1111/j.1742-1241.2009.02179.x
25. Guang-da X, You-ying L, Zhi-song C, Yu-sheng H, Xiang-jiu Y. Apolipoprotein e4 allele is predictor of coronary artery disease death in elderly patients with type 2 diabetes mellitus. Atherosclerosis. 2004;175(1):77–81. doi: https://doi.org/10.1016/j.atherosclerosis.2004.02.015
26. McGill HC Jr, McMahan CA, Zieske AW, Malcom GT, Tracy RE, Strong JP. Effects of nonlipid risk factors on atherosclerosis in youth with a favorable lipoprotein profile. Circulation. 2001;103(11):1546–50. doi: https://doi.org/10.1161/01.cir.103.11.1546
27. Corsetti JP, Bakker SJ, Sparks CE, Dullaart RP. Apolipoprotein A-II influences apolipoprotein E-linked cardiovascular disease risk in women with high levels of HDL cholesterol and C-reactive protein. PLoS One. 2012;7(6):e39110. doi: https://doi.org/10.1371/journal.pone.0039110
28. Talmud PJ. Gene-environment interaction and its impact on coronary heart disease risk. Nutr Metab Cardiovasc Dis. 2007;17(2):148–52. doi: https://doi.org/10.1016/j.numecd.2006.01.008
29. Humphries SE, Talmud PJ, Hawe E, Bolla M, Day IN, Miller GJ. Apolipoprotein E4 and coronary heart disease in middle-aged men who smoke: a prospective study. Lancet. 2001;358(9276):115–9. doi: https://doi.org/10.1016/S0140-6736(01)05330-2
30. Singh PP, Singh M, Bhatnagar DP, Kaur TP, Gaur SK. Apolipoprotein E polymorphism and its relation to plasma lipids in coronary heart disease. Indian J Med Sci. 2008;62(3):105–12.
31. Inamdar PA, Kelkar SM, Devasagayam TP, Bapat MM. Apolipoprotein E polymorphism in non-insulin-dependent diabetics of Mumbai, India and its effect on plasma lipids and lipoproteins. Diabetes Res Clin Pract. 2000;47(3):217–23. doi: https://doi.org/10.1016/s0168-8227(99)00126-6
32. Afroze D, Yousuf A, Tramboo NA, Shah ZA, Ahmad A. ApoE gene polymorphism and its relationship with coronary artery disease in ethnic Kashmiri population. Clin Exp Med. 2016;16(4):551–6. doi: https://doi.org/10.1007/s10238-015-0389-7
33. Li J, Lee DH, Hu J, Tabung FK, Li Y, Bhupathiraju SN, et al. Dietary inflammatory potential and risk of cardiovascular disease among men and women in the U.S. J Am Coll Cardiol. 2020;76(19):2181–93. doi: https://doi.org/10.1016/j.jacc.2020.09.535
34. Schnabel RB, Yin X, Larson MG, Yamamoto JF, Fontes JD, Kathiresan S, et al. Multiple inflammatory biomarkers in relation to cardiovascular events and mortality in the community. Arterioscler Thromb Vasc Biol. 2013;33(7):1728–33. doi: https://doi.org/10.1161/ATVBAHA.112.301174
35. Elisia I, Lam V, Cho B, Mariah Hay, Michael Yu Li, Michelle Yeung, et al. The effect of smoking on chronic inflammation, immune function and blood cell composition. Sci Rep. 2020;10:19480. doi: https://doi.org/10.1038/s41598-020-76556-7
36. McEvoy JW, Nasir K, DeFilippis AP, Lima JA, Bluemke DA, Hundley WG, et al. Relationship of cigarette smoking with inflammation and subclinical vascular disease: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35(4):1002–10. doi: https://doi.org/10.1161/ATVBAHA.114.304960
37. Woodward M, Welsh P, Rumley A, Tunstall-Pedoe H, Lowe GD. Do inflammatory biomarkers add to the discrimination of cardiovascular disease after allowing for social deprivation? Results from a 10-year cohort study in Glasgow, Scotland. Eur Heart J. 2010;31(21):2669–75. doi: https://doi.org/10.1093/eurheartj/ehp115
38. Ridker PM. Clinician’s guide to reducing inflammation to reduce atherothrombotic risk: JACC review topic of the week. J Am Coll Cardiol. 2018;72(25):3320–31. doi: https://doi.org/10.1016/j.jacc.2018.06.082
39. Grammer TB, Hoffmann MM, Scharnagl H, Kleber ME, Silbernagel G, Pilz S, et al. Smoking, apolipoprotein E genotypes, and mortality (the Ludwigshafen RIsk and Cardiovascular Health study). Eur Heart J. 2013;34(17):1298–305. doi: https://doi.org/10.1093/eurheartj/eht001
40. Kofler BM, Miles EA, Curtis P, Armah CK, Tricon S, Grew J, et al. Apolipoprotein E genotype and the cardiovascular disease risk phenotype: impact of sex and adiposity (the FINGEN study). Atherosclerosis. 2012;221(2):467–70. doi: https://doi.org/10.1016/j.atherosclerosis.2012.01.042
41. Schick UM, Auer PL, Bis JC, Lin H, Wei P, Pankratz N, et al. Association of exome sequences with plasma C-reactive protein levels in >9000 participants. Hum Mol Genet. 2015;24(2):559–71. doi: https://doi.org/10.1093/hmg/ddu450