
INTRODUCTION
Cancer accounts for the second leading cause of global deaths after cardiovascular disease, especially in developed countries. In 2020, the projection of cancer in the United States stands at 1,806,590 new cases and 606,650 deaths. The prostate is among the four leading cancers, and the others are lung, breast, and colorectal. In a decadal trend (2008–2017), there exists a significant decline in the death rates associated with other leading cancers except for the prostate (Siegel et al., 2020). Prostate remains the most common cancer infection in males with undetectable signals in females. In 2020, for instance, estimates of 191,930 new and 33,330 death cases are projected in relevance to the prostate in the United States; all are attributed to men (Ikwu et al., 2020; Siegel et al., 2020). Despite the alarming statistics, the incessant resistance by the disease, inadequate effective prevention, treatment strategies, and accessibility remains a major stumbling block in curtailing the disease worldwide while its impact on the healthcare system lasts. Several pharmacological mechanisms across different targets are being adopted in prostate therapy while the search for effective strategy remains imperative.
The modification of the androgen receptor has been identified as an ideal therapeutic strategy toward the control of prostate cancer development (Ikwu et al., 2020). Since the pathogenesis of prostate carcinoma includes the damaging of the prostate epithelium which could be triggered by procarcinogenic inflammatory processes, the progression of which may result in the generation of proliferative inflammatory atrophy lesions. Through these important pathomechanisms, the pharmacological responses usually observed include the prevention of cell injury, clearance of necrotic cells, initiation of tissue repair, and other inflammatory processes (Fouzia and Salim, 2019). Thus, bioactive agents with anti-inflammatory and antioxidant potentials with the expression for androgen modification such as spironolactone, progestogen, and flutamide are among the common markers against the development of carcinomas including prostate (Bardia et al., 2009).
The cyclooxygenase (cox) is a multipurpose enzyme that primarily functions in the catalytic conversion of arachidonic acid into prostaglandins (PGs), physiologically active lipids present in human and other animal tissues, performing various hormone-like activities such as the control of blood flow and inflammation. They exist in dual forms as cox-1 and cox-2. While the cox-1 is usually found in tissue, majorly involved in the production of PGs, the cox-2 is physiologically undetectable in most tissues but induced by some homeostatic disorders and its inhibitors mainly the nonsteroidal anti-inflammatory drugs such as ibuprofen and meloxicam are usually applied to control neoplasmic syndromes, including oxidative stress and inflammation (Swiatek et al., 2019). Therefore, the inhibitory process against cox-2 constitutes another essential pathology in the prophylaxis and treatment of prostate cancer.
The cytochrome P450 (CYP17A1/17αhydrogenase) represents an endoplasmic reticulum membrane-bound monooxygenase with vital multifunctional roles in the biosynthesis of several human steroid hormones. Secondly, the onset initiation of prostate carcinoma in males depends greatly on the primary anabolic steroid and sex hormone, testosterone, its metabolite, 5α-dihydrotestosterone (DHT), and androgens. The 5α-reductase enzymes aid the conversion of the testosterone to DHT, which then binds to AR to trigger the transcription processes on several genes, supporting the fact that the signaling pathways of androgen play vital roles in the progression of the disease. Thus, the inhibition of CYP17A1 is also identified as an important (androgenic) antiproliferative pathway toward effective prevention and treatment of estrogen- and androgen-dependent prostate carcinoma (Brito et al., 2019; Ai et al., 2019). Abiraterone acetate and finasteride are renowned antiprostate drugs expressing these inhibitory pathways.
However, most of the orthodox medicines effectively applicable to these pathophysiological mechanisms induce incessant resistance by pathogenic targets through single-target pathways and various side effects, and often they are not easily accessible. The application of precision and/or immunotherapy enhances the survival rates in high-income countries, whereas affordability also becomes a major challenge in low-income counterparts, which makes the prevention and treatment strategies remain inadequate (Nagai and Kim, 2017). These phenomena make imperial the continuous search for alternative candidates with global accessibility, enhanced pharmacological potentials for prostate carcinoma and lesser aftereffect.
Multitarget therapeutic approaches are oftentimes employed to mitigate the redundant cellular pathways, development of pathogenic defense, and compensatory mechanisms usually in association with single-target treatment. The strategy has become attractive to basic and clinical scientists in tackling some complex diseases such as thrombotic and psychiatric disorders, inflammation, and cancers. Other advantages of this modern pharmacological innovation include the additive and synergistic effects to overcome resistance (Ayipo et al., 2021; Skok et al., 2020). Although combination therapy is usually applied in orthodox medicines to achieve this promising aim, it poses the risks of cumulative side effects. Thus, single-therapeutic candidates with enhanced potential for this important strategy especially from vast and safer natural resources could offer a breakthrough against incessant drug-resistant cancers such as prostate carcinomas through a “one-drug-multiple-target” approach.
The medicinal plant Aframomum melegueta has been reported with interesting pharmacological potentials against inflammation, nociception, Alzheimer disease, Parkinson’s disease, and some other complex disorders which threaten global healthcare, with constituents mostly studied via single-treatment mechanisms (Dzoyem et al., 2017; Umukoro and Aladeokin, 2011). Few in vivo studies have also proposed the physiological expression of its extracts for prostate dysfunction and influence on cytochrome P45001bi in animal models (Adefegha et al., 2017; Akpanabiatu et al., 2013; Biobaku et al., 2020). However, the pharmacology–target relationships for these activities are inadequately reported, especially the propensity of its phytochemicals for a “one-drug-multiple-target” therapeutic potentials for prostate carcinoma.
More so, the application of in silico computational models offers a cheap, environmentally friendly, and fast evaluation of potential drug-like candidates to develop good confidence required further translational study. Notably among the tools are molecular ligand–receptor docking simulations, predictions of bioactivity, physicochemical, pharmacodynamics, pharmacokinetics, and toxicological parameters including absorption, distribution, metabolism, excretion, and toxicity (ADMET). The structure–activity relationship (SAR) of >250,000 bioactive compounds forms building predicting parameters of the Prediction of Activity Spectra for Substances (PASS) online server-based computational tools applicable to drug candidates, lead-likeness study, and toxicological profiling. The accuracy of the prediction expressed in terms of leave-one-out cross-validation is reported to be >95% with over 4,000 biological activities including mechanisms of action, pharmacological effects, interactions with metabolic enzymes, gene expression, and toxicological and adverse effects. The developmental application of the network of these computational tools is becoming essential in the field of medicinal and pharmaceutical sciences and is documented with many success stories (Filimonov and Poroikov, 2009; Poroikov et al., 2019).
Thus, this study is aimed at the in silico profiling of phytochemicals in the ethanolic extract of A. melegueta as potential multimechanistic antiprostate therapeutic candidates using computational tools of molecular docking, bioactivity, ADMET, and mutagenicity predictions.
MATERIALS AND METHODS
Collection and authentication of plant part
Dried A. melegueta seeds were collected from herbs seller in Ilorin metropolis. The seeds were identified and authenticated at the Herbarium Unit of the Department of Plant Biology, University of Ilorin, Nigeria, and a voucher number UILH/001/2019/1166 was assigned and deposited at the University Herbarium. The seeds were ground into a powder and stored in airtight containers at 4°C for further use.
Extract preparation
The powdered seeds of A. melegueta were subjected to ethanol extraction adopting the existing protocols (Abubakar and Haque, 2020). Exactly 100 g of the powdered A. melegueta seeds was placed in a stoppered container and 500 ml absolute ethanol was poured to completely cover the sample material. The concoction was kept at room temperature for 3 days under periodical stirring for homogeneity and optimum yield. At the end of the extraction, the filtrate was decanted using Whatman No. 1 filter paper (125 mm) and then concentrated using a rotary evaporator. The dried extract was stored in a desiccator until needed where it was dissolved in an appropriate volume of solvents to make every desired concentration.
Phytochemical screening
Phytochemical screening was carried out to identify the secondary metabolites present in the ethanolic extract of A. melegueta seeds according to documented literature through qualitative tests: Dragendorff’s, ferric chloride, Benedict’s, modified Bontrager, lead acetate, foam and Libermann Burchard for alkaloids, flavonoids, and tannins, reducing sugars, glycosides, phenols, saponins, and steroids and terpenoids, respectively (Harborne, 1984; Trease and Evans, 2009).
Gas chromatography-mass spectroscopy (GC-MS) analysis
GC-MS analysis was carried out using a GC-MS (model: QP 2010SE, Shimadzu, Tokyo, Japan). The sample for GC-MS was prepared by dissolving 3 g of the extract powder in an aqueous solvent. To analyze the sample, the column oven temperature and the injector temperature were set at 60°C and 250°C, respectively. The flow control mode was maintained in linear velocity with a split injection mode split ratio of 1:1. The column flow was 3.22 ml/minute with a helium carrier gas of 99.99% purity. The temperature was set at 60°C with 1-minute hold time, 120°C with 2 minutes hold time by the rate of 15, and then 300°C with 3 minutes hold time by the rate of 15. The column at 5 minutes was used with a length of 30 mm and a diameter of 0.25 mm and its film thickness was 0.25 μm. The ion source temperature for MS condition was 200°C and interface temperature was 240°C. The starting m/z (mass to charge) ratio was 45 and the ending m/z ratio was 700 (45–700 m/z).
Ligand preparation
The SMILE formats of the detected phytochemicals were derived using ChemDraw 19.1 suite and imported into the workspace of Maestro 12.12 (LigPrep, Schrödinger, LCC, New York, NY, 2019). They were prepared by energy minimization, the addition of hydrogen, and conversion of 2D–3D structures. The geometry and partial atomic charges of the 2D and 3D molecular structures were calculated using Optimized Potentials for Liquid Simulations (OPLS-3e) force field (Harder et al., 2016) and the minimized ligands are saved in standard database format into the LigPrep.out folder.
Protein preparation and receptor grid generation
The 3D crystallographic structures of the human androgen receptor synthesized in complex with 2-chloro-4-[(2S,3S)-3-hydroxy-2-methylpyrrolidin-1-yl]-3-methylbenzonitrile (77U) with an X-ray resolution of 2.71 Å protein data bank (PDB 5T8E), cox-2 in complex with ibuprofen and X-ray resolution of 1.81 Å protein data bank (PDB 4PH9), and human cytochrome P450 (CYP) 17A1 in complex with abiraterone and X-ray resolution of 2.60 Å (PDB 3RUK) were retrieved from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB). The protein receptor targets were preprocessed, optimized, and refined by depleting water and covalently linked molecules, moderating the bond order, and removing other chains using Protein Preparation Wizard (Madhavi Sastry et al., 2013). Then the assignment of charges and protonation state was followed by energy minimization using the OPLS-3e force field available in Maestro 12.2 (Harder et al., 2016). The active site x, y, and z coordinates of their respective centroid co-crystallized ligands were used to generate docking grid boxes saved in gridbox.zip files.
Molecular docking
The ligand–receptor docking simulations were carried out to estimate the theoretical interaction of the prepared screened phytochemicals including the positive controls against the residues within the generated grid boxes of the protein receptor targets using Maestro 12.2 Glide (Halgren et al., 2004). The docking functions were used to virtually evaluate the interactions between the phytochemicals and amino acids in the active sites of the receptor based on the binding energy, docking score, glide standard precision score, and binding poses. The phytochemicals in complex with the receptors, having good scoring functions in comparison with the co-crystallized ligands, and the references were selected for further studies.
PASS bioactivity prediction
To further assess the proposed multitarget antiprostate mechanisms, the bioactivity of the selected phytochemicals was predicted using online PASS (http://www.pharmaexpert.ru/passonline/index.php) by input strings of SMILE formats of the selected phytochemicals. The numerically predicted results were in pa/pi representing probability to be active/inactive, respectively, in a SAR with the mechanisms of action and enzymatic pathways such as antineoplastic, apoptosis agonist, CYP2J substrate, testosterone 17beta-dehydrogenase (NADP+) inhibitor, prostate disorder treatment, and anti-inflammation. Renowned antiprostate drugs, abiraterone and bicalutamide, were used as references.
ADMET pharmacokinetics and toxicological predictions
The physicochemical, pharmacokinetics, and toxicological parameters of the selected phytochemicals were predicted using ADMET server (http://www.swissadme.ch/index.php) (Daina et al., 2017). The server-based tools provide key information such as lipophilicity, molecular weight, flexibility, solubility, polarity, topological polar surface area (TPSA), and the number of hydrogen bond acceptors/donors to probe the ideal of the selected phytochemicals for applicability within a biological system.
Drug-likeness prediction
The drug-likeness profile of each selected phytochemical was predicted as an additional screening protocol using the web-server SwissADME. The properties such as blood–brain barrier (BBB), human gastrointestinal (GI) absorption, p-glycoprotein (pg) substrate, inhibitory activity against important cytochromes were also probed using BDDCS, the rule of 5 and drugability (Benet et al., 2016).
Mutagenicity
To further estimate the toxicological indices of the selected phytochemicals, mutagenicity was predicted for each of them using the modeled read-across SAR available in VEGA ToxRead 0.23Beta java-based software. The similarity and fragment search from the series of compounds with known properties were used to predict the mutagenicity of the selected phytochemicals using the strings of their SMILE formats. The accuracy and reproducibility of the results are in strong agreement with the Ames in vitro experiments (Gini et al., 2014), validating the applicability of the tool.
RESULTS AND DISCUSSION
Extraction
The aqueous and ethanol extract elicited varying solubility capacities of plant phytochemicals. The aqueous extract of A. melegueta seeds has a lower yield (6.05%) of soluble phytochemicals compared to ethanol extract yield (6.16%) (Table 1). Despite being unexpectedly insignificant, the variation in the yield could be traced to the solubility property of the phytochemicals in water and ethanol since solvents are believed to dissolve more solutes of similar polarity. Thus, the phytochemicals seem more soluble in less polar ethanol than water possibly due to their organic nature. Thus, the ethanolic extract was therefore adopted for further study.
Phytochemical screening
The phytochemical screening results (Table 2) reveal that the aqueous and ethanolic extracts of A. melegueta seeds containing alkaloids, flavonoids, glycosides, phenols, saponins, steroids, tannins, and terpenoids. Alkaloid, flavonoid, phenol, saponin, and tannin were found to be more in the ethanol extract than aqueous extract, glycosides, steroids, and terpenoids which were detected in trace amount in both extracts, while reducing sugar and volatile oils was only shown to be present in ethanolic extract. Interestingly, some previous studies have also reported the presence of phytochemicals in the plant (Emeribe, 2018; Toh et al., 2019), supporting the results obtained.
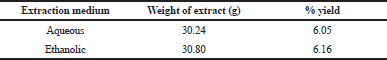 | Table 1. Weight and percentage yield of aqueous and ethanol extracts of ethanol extract of A. melegueta seeds. [Click here to view] |
GC-MS analysis
The phytochemical composition of the ethanolic extract of A. melegueta seeds detected using the GC-MS method of analysis is shown in Figure 1. From the results, 44 stable, nonfragmented compounds were detected in the ethanolic extract of the seeds. These compounds account for various properties of the plant seed extract and also provide some biochemical evidence for its ethnopharmacological use in the management of some diseases, especially the compounds 16, 17, 18, and 44. Compound 16 otherwise known as 17 is a volatile bicyclic sesquiterpene lactone derivative that is abundantly present in the essential oils of many dietary and edible plants. Supportively, some of the detected phytochemicals have been documented to be present in A. melegueta (Agim et al., 2017; Hassan et al., 2019), although isolation and more robust characterization are required to further confirm the compounds in a future study. Detailed result spectra are presented in Supplementary File 1.
Receptor grid boxes
The grid box surfaces of the active sites are presented as 3D solid mesh within the ribbon structures of the selected receptors (Fig. 2). The phytochemicals were docked within a length of 20 Å in the respective active surfaces occupied by the co-crystallized ligands. The x, y, and z coordinates of the sites generated by picking the centroid of the co-crystallized ligands are 26.62, 2.59, and 4.14 Å; 13.01, 23.49, and 25.26 Å of chain A; and −6.44, 9.30, and 62.09 Å of chain A for PDB 5T8E, PDB 4PH9, and PDB 3RUK, respectively.
Molecular docking
The result (Table 3) contains the molecular docking scores of the prepared respective phytochemicals detected in the ethanolic extract of A. melegueta in complex with the crystal structure of the human androgen receptor in complex with 77U (PDB 5T8E). The molecular docking procedure was validated by re-docking a co-crystallized ligand within the active pocket of the receptor, the complex of which gives Root mean square deviation (RMSD) of 0.073 Å in comparison with the retrieved crystal complex from RCSB PDB. Other ligands are mostly docked within the RMSD range of 0.196–2.000 Å, while only a few are within 2.000–3.991 Å, validating the protocol as good (Castro-Alvarez et al., 2017; Ramírez and Caballero, 2018). The interactive potentials of the phytochemicals are demonstrated in order of increasing docking scores and comparison with co-crystallized ligand (77U) and standard drugs. The higher the negative value, the stronger the binding interaction. As expected, the co-crystallized ligand, 77U, interacted most strongly with the receptor as shown by the lowest docking score. However, six phytochemicals, 16, 17, 23, 28, 34, and 44, also display strong binding to the active site of the receptor in strong competition with the co-crystallized ligand and higher terms than flutamide, a nonsteroidal antiandrogen drug primarily applied to treat prostate cancer. This signals a higher agonistic propensity of the phytochemicals for the human androgen receptor than the orthodox drug. From the binding poses of the ligand–receptor complexes (Fig. 3), it could be observed that the selected phytochemicals occupy similar volumes and subcavities of the active binding pocket of the receptor in comparison with the reference drugs. Again, most of the selected phytochemicals and the reference drug interacted with the amino acids within the active cavities of the receptor through nonbonding van der Waals forces, accounting for their high binding affinity (Ikwu et al., 2020). Additionally, the reference drug, flutamide, whose docking score is −7.691 kcal/mol interacted with Phe 764 through π – π stacking using its aromatic group, while compound 34 with −8.787 kcal/mol exhibited the same interactions twice through its extended aromatic π-system within bond lengths of 2.13 and 2.40 Å, respectively. The phytochemicals mostly possess ≤7 number of rotatable bonds, indicating their ideal flexibility for biological conformity (Khanna and Ranganathan, 2009). Since testosterone, DHT, and androgens play important roles in the onset of prostate carcinoma development, the inhibition of the androgenic pathway has been implicated in effective prevention and treatment of prostate carcinoma and other related prostate disorders (Brito et al., 2019). Therefore, with these interesting potentials, the ethanolic extract of A. melegueta could synergistically modulate the androgen receptor in stronger terms than some currently available orthodox medicines, representing model candidates of complementary and traditional medicine. Individually, the selected phytochemicals, 16, 17, 23, 28, 34, and 44, were further studied for wider pharmacological and toxicological profiles.
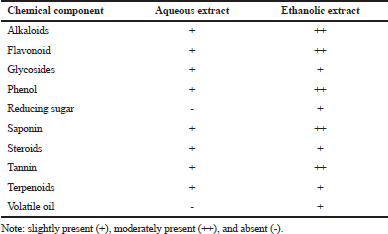 | Table 2. Qualitative phytochemical analysis of aqueous and ethanol extracts of A. melegueta seeds. [Click here to view] |
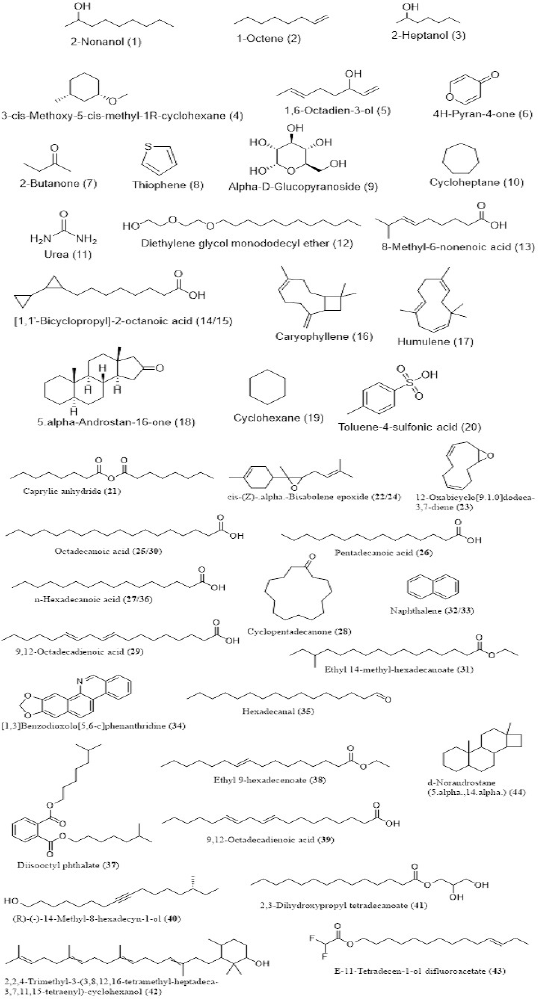 | Figure 1. Chemical structures of the phytochemicals detected in the ethanolic extract of A. melegueta using GC-MS. [Click here to view] |
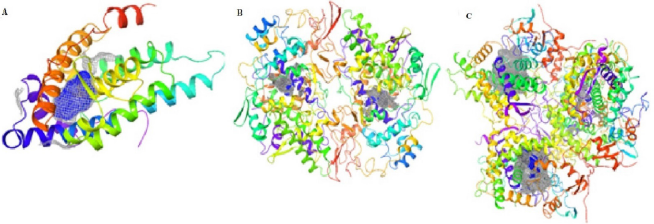 | Figure 2. Surfaces of the active binding sites of the selected receptors in solid meshes. (A) PDB 5T8E; (B) PDB 4PH9; and (C) PDB 3RUK. [Click here to view] |
The inhibitory potentials of the phytochemicals were further demonstrated against an anti-inflammatory enzyme target, cyclooxygenase-2 (cox-2) as presented (Table 4). The phytochemicals were docked within the active pocket of the crystal structure of the receptor (PDB 4PH9) in complex with a strong nonsteroidal anti-inflammatory drug, ibuprofen. The docking protocol was proved valid by the RMSD values which are mostly ≤2 Å; only a few are outside the range. From the ligand interactions of the crystal structure profiles as retrieved from the RCSB PDB, ibuprofen exhibited H-bonding interactions through the carboxyl group with Arg 120 and Tyr 355. These amino acid residues are shown in the binding poses (Fig. 4) as Arg 121 and Tyr 355 with similar interactions to further validate the docking procedure in the study. The drug also displays strong nonbonding interactions with other amino acid residues such as Val 350, Ser 354, and Met 523 in the α-helix region and Leu 385 and Trp 388 reportedly occupying the β-helix sheet of the receptor active cavity (Bittencourt et al., 2019). The phytochemicals under focus exhibit similar interactions to ibuprofen and the second nonsteroidal anti-inflammatory drug reference, meloxicam. From another report, the active amino acid residues Tyr 385 and Ser 530 form important interactions with ibuprofen in subpocket A, Arg 120 in B, and Val 523 and Ser 353 in D (Fouzia and Salim, 2019). Similar interactions are also observed with the phytochemicals here within the same subpockets but slight flip in residue numbers as Tyr 386, Ser 531, Arg 121, Val 524, and Ser 354, respectively. These favor similar potential biosystemic interactions and functionality between the selected phytochemicals and the reference drugs. Interestingly, phytochemicals 18 and 44 demonstrate stronger inhibitory potentials against the cox-2 than a renowned nonsteroidal anti-inflammatory cox-2 inhibitor, ibuprofen, as indicated by higher docking scores. Others such as 16, 22, and 34 also compete favorably with the co-crystallized ibuprofen. These indicate a more promising anti-inflammatory potentials of the phytochemicals compared to ibuprofen and meloxicam. Most of the phytochemicals possess number of rotatable bonds <10, supporting a good flexibility ideal for biological systems, and thus deserve further study.
The inhibitory potentials of the screened phytochemicals against the androgenic pathways implicated in prostate carcinoma initiation were further evaluated against the crystal structure of the human cytochrome P450 (PDB 3RUK) in complex with a renowned antiprostate, abiraterone. The molecular docking simulation was validated well with the RMSD scores mostly in the range of 0–3 Å; only a few are higher. From the docking results (Table 5), the co-crystallized abiraterone and reference drug bicalutamide show binding scores of −9.297 and −7.141, respectively. The most interacted phytochemical, 42, shows a close score of −8.524 while the compounds 18, 34, and 44 display −7.659, −7.179, and −7.722, respectively. The values are consistent with the glide energy scores in kcal/mol, with both representing binding affinity. The amino acid residues Phe 114, Ile 206, Leu 209, Arg 239, Gly 301, Ala 302, Glu 305, Ile 371, and Val 483 reportedly constitute the active site of the receptor essentially for hydrophobic interactions (Ai et al., 2019). From the binding poses of the ligand–receptor complex conformations (Fig. 5), the reference drug, abiraterone, interacted by H-bonding with Asn 202 through its OH group while similar bonding interaction is observed in bicalutamide to Asp 298. Both the references and selected phytochemicals mostly exhibit nonbonding interactions with the reported amino acid residues within the active pocket of the receptor in similar modes. These indicate potential similarity in bioactivities of the selected phytochemicals about abiraterone and a higher propensity compared to bicalutamide.
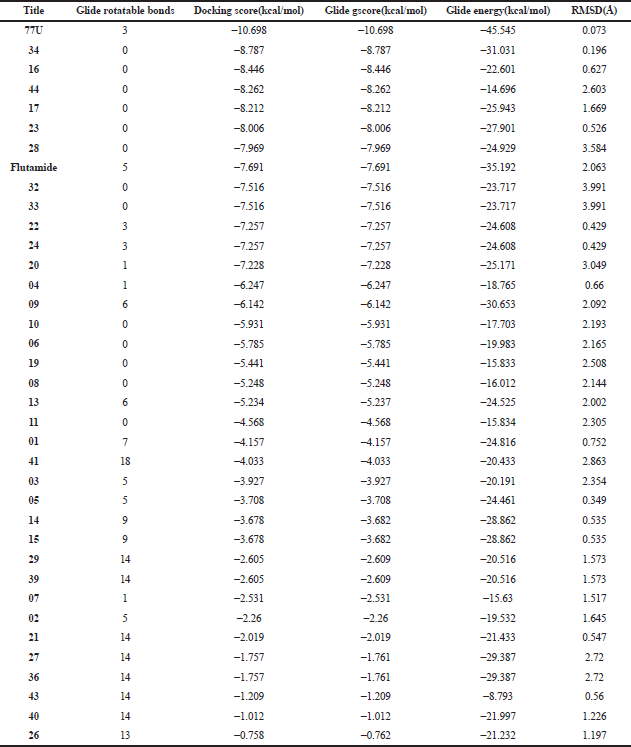 | Table 3. Molecular docking interaction of phytoconstituents against the human androgen receptor (PDB 5T8E). [Click here to view] |
The cumulative results from the docking simulations support the potentials of the selected phytochemicals 16, 17, 18, 34, and 44 to act in a “one-drug-multiple-target” approach, an important therapeutic strategy for overcoming resistance in oncology. The compounds demonstrated inhibitory activity against the protein/enzyme targets implicated in cancer therapy such as the human androgen receptor, cox-2, and CYP17A1, indicating multimechanistic pharmacology worthy of further evaluation. These are consistent with some previously reported physiological effects of the phytoconstituents in vivo (Akpanabiatu et al., 2013; Biobaku et al., 2020) and succinctly suggest the pharmacological targets for the expressions.
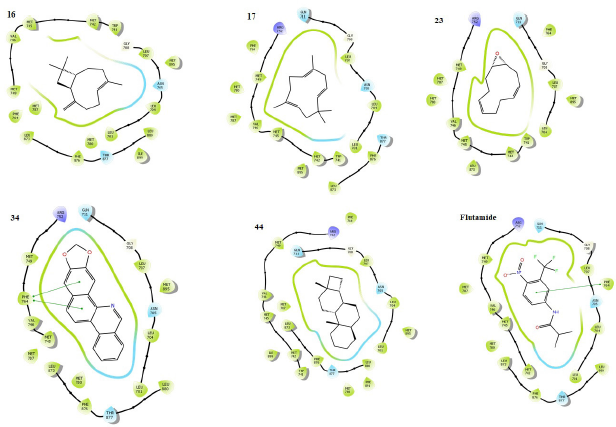 | Figure 3. Binding poses showing the interactions of selected phytochemicals as agonists with amino acids in the active cavity of human androgen receptor (PDB 5T8E). The interactions are illustrated as hydrogen bonding (magenta arrow), π – cation (blue line), salt bridge (red line), π – π stacking (green line), and van der Waals forces. [Click here to view] |
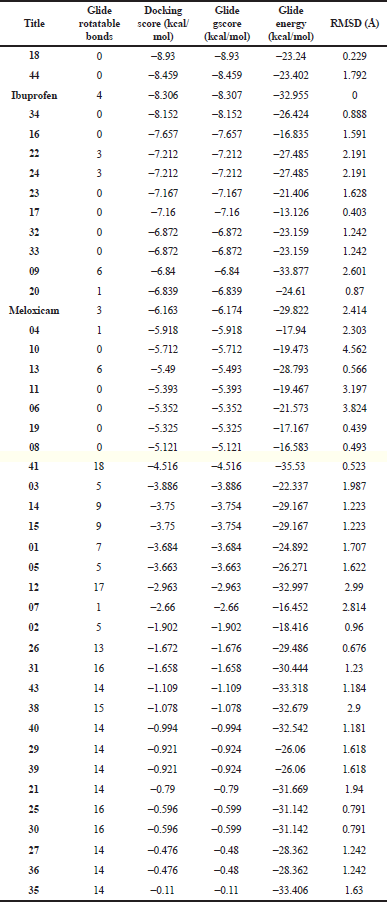 | Table 4. Molecular docking screening of phytoconstituents against the human cox-2 receptor (PDB 4PH9). [Click here to view] |
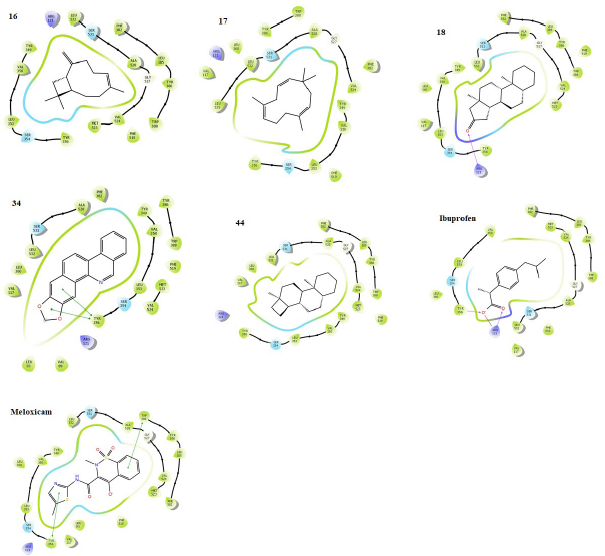 | Figure 4. Binding poses showing the interactions of selected phytochemicals with amino acids in the active cavity of human cox-2 (PDB 4PH9). The interactions are illustrated as hydrogen bonding (magenta arrow), π – cation (blue line), salt bridge (red line), π – π stacking (green line), and van der Waals forces. [Click here to view] |
PASS bioactivity prediction
Further predictions of bioactivity profiles and pharmacological potentials of the phytoconstituents, 16, 17, 18, 34, and 44, selected from molecular docking screening were carried out using PASS online tools. From the results (Table 6), the selected drugs are predictably more active as antineoplastic, apoptotic agonists, CYP2J substrates, testosterone 17beta-dehydrogenase (NADP+) inhibitors, agents for prostate disorder treatment, and anti-inflammation in stronger terms than the references, abiraterone, and bicalutamide, indicated by the probability of activity/inactivity (Pa/Pi) values. From a relevant study (Grienke et al., 2015), the tool has been used to predict the in silico pharmacological profile of some secondary metabolites with the result validated upon further biochemical experiments. The interesting prediction results here support the potentials of the selected phytoconstituents of the plant as promising agents for prostate carcinoma prevention and treatment through multitarget pathomechanisms, thus deserving further translational evaluation into therapeutics.
ADMET pharmacokinetics and toxicological predictions
The physicochemical profiles are predicted for the selected phytochemicals, 16, 17, 18, 34, and 44, as presented in Table 7. All the compounds have a molecular weight of <500. They are poorly soluble in water as indicated by the Log S Estimated solubility (Log S ESOL) which falls outside the standard range of 0.25–1.00. They are mostly lipophilic as shown by their predicted partition coefficient (XLOGP3) values which are lower than 5.0. Their TPSA is all within 0–130 Å2 and all have good flexibility. The predicted parameters support the previous studies which qualify the phytochemicals as lead-like molecules worthy of more assessment.
From the pharmacological and toxicological profiles (Table 8), only two phytochemicals 18 and 34 can potentially cross the BBB; others cannot. This demonstrates their suitability for homeostatic processes in the central nervous system, determined by various factors including molecular weight and flexibility, lipophilicity, an affinity for efflux mechanisms, and systemic enzymatic stability (Meairs, 2015), although this is less significant to their pharmacological potentials as antiprostate. Similarly, only the two molecules, 18 and 34, have high GI absorptivity others are low. Only molecule 34 demonstrates virtual inhibitory expressions against various CYPs and pg substrate, such as mutagenic, while others display fair expressions and are predicted as nonmutagenic. The mutagenic profiles of the selected phytochemicals about the most structurally similar compounds or fragments with known properties suggest nonmutagenicity except for 34 (Fig. 6), although the selectivity of its cytotoxicity in favor of nontumor cells could infer its antiprostate pharmacology. The higher the negative value of Log Kp, the less the chance of the molecules to permeate the skin (Daina et al., 2017). All the selected molecules have high negative values for Log Kp, favoring a low probability for skin permeation and supporting their druggability. They have good synthetic accessibility, zero pain alert, good bioavailability scores, and insignificant violation of the BDDCS rule of 5 and druggability. These indicate the safety and potential drug-likeness of the selected phytochemicals amenable for experimental evaluation.
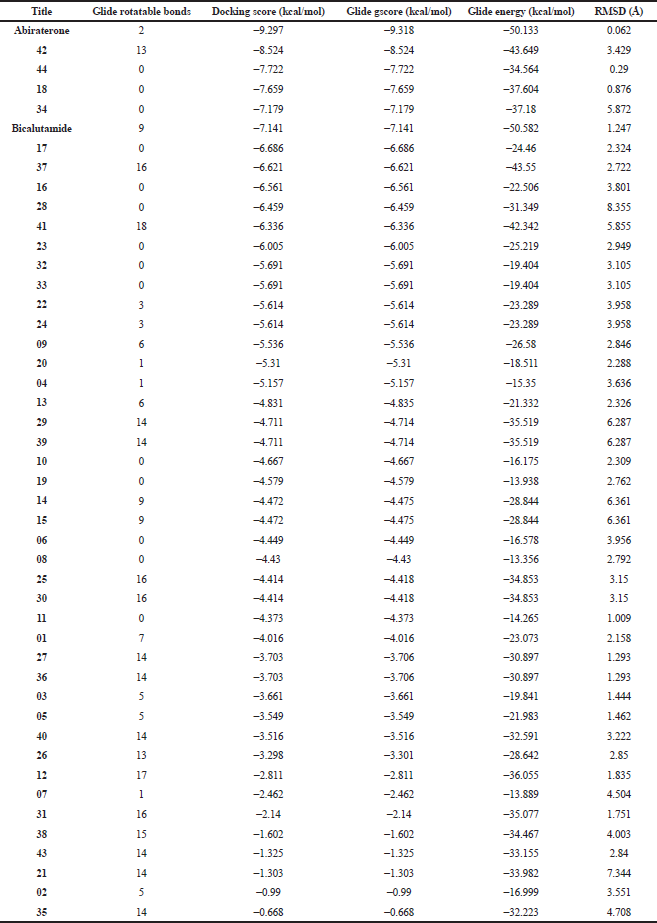 | Table 5. Molecular docking screening of phytoconstituents against CYP17A1 receptor (PDB 3RUK). [Click here to view] |
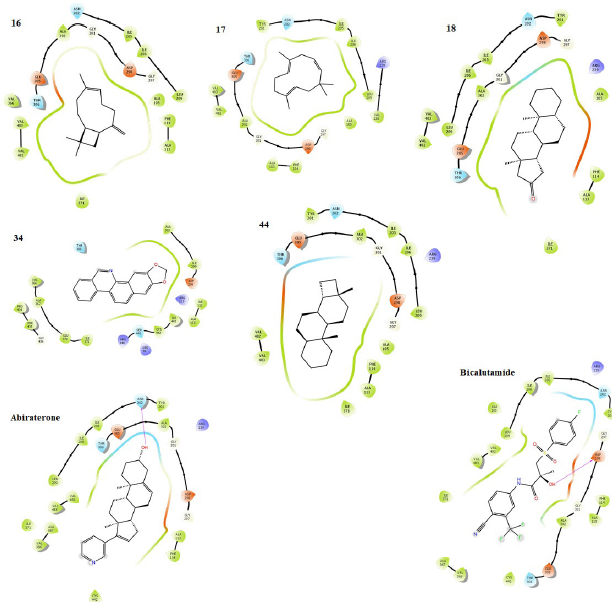 | Figure 5. Binding poses showing the interactions of selected phytochemicals with amino acids in the active cavity of CYP 17A1 (PDB 3RUK). The interactions are illustrated as hydrogen bonding (magenta arrow), π – cation (blue line), salt bridge (red line), π – π stacking (green line), and van der Waals forces. [Click here to view] |
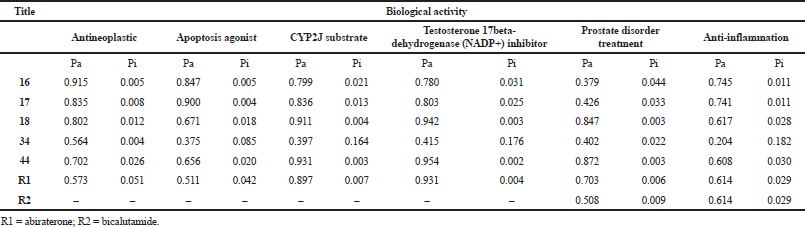 | Table 6. Predicted biological activity of selected phytoconstituents using PASS. [Click here to view] |
 | Table 7. Physicochemical parameters of selected phytoconstituents of ethanolic extract of A. melegueta seeds. [Click here to view] |
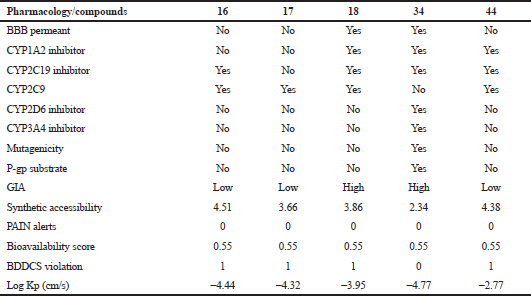 | Table 8. Pharmacological and toxicological parameters. [Click here to view] |
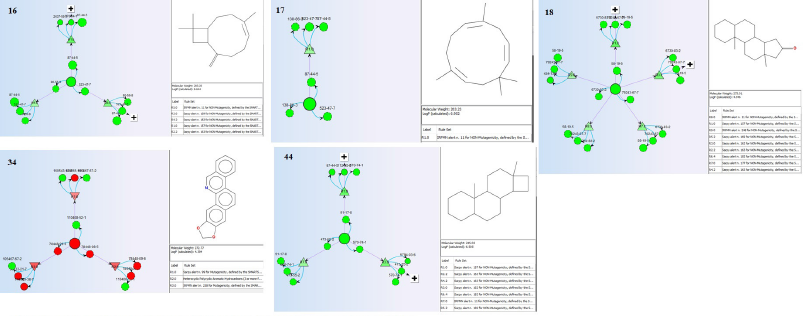 | Figure 6. Mutagenicity of the selected phytoconstituents in SAR reference to the most similar compounds/fragments with known identities. [Click here to view] |
The molecular docking simulations show that the selected phytochemicals demonstrate strong inhibitory interactions against the human androgen receptor, cox-2 enzyme, and CYP17A1 receptor. This supports their potential applicability in a “one-drug-multiple-target” which could provide a therapeutic breakthrough against the drug resistance associated with prostate cancer upon further studies. Consistently, the PASS online predicted them with good activity as antineoplastic agents, apoptotic agonists, CYP2J substrates, testosterone 17beta-dehydrogenase (NADP+) inhibitors, agents for prostate disorder treatment, and an anti-inflammatory. Their physicochemical profiles from SwissADME favor ideal characters for druggability with an insignificant violation of the BDDCS rule of 5, while the toxicological evaluation using VEGA ToxRead indicates nonmutagenicity. The parameters are favored mostly by the phytochemicals especially 16, 17, 18, and 44 in better terms than the reference drugs.
CONCLUSION
The ethanolic extract of A. melegueta seeds was shown to contain 44 stable phytochemicals belonging to various organic families as indicated by GC-MS analysis. A series of computational tools have been applied to investigate the activity of the phytoconstituents for effective antiprostate carcinomas with the potentials for mitigating the challenges of drug resistance through a multitarget mechanism, inaccessibility, and various aftereffects associated with orthodox medicines. The possibilities of these pharmaceutical breakthroughs are especially favored by some phytochemicals, 16, 17, 18, and 44 over others. The selected compounds could be deservedly isolated or synthetically derivatized for further evaluation. They also deserve more exploration for application in the field of complementary medicines against prostate cancer. Their additive/synergistic activities could be rationally tapped as an opportunity for effective combination therapy in prostate oncology. Individually, they display enhanced potentials for replacing the currently available antiprostate drugs with various shortfalls. The study requires the incorporation of more sophisticated computing algorithms to accurately assess the phytochemicals as bona fide agents. More so, basic and clinical experimental evaluations are necessarily required in vitro and in vivo for the affirmation of the interesting pharmacology and biosafety. However, it represents a promising model for future therapeutic designs for overcoming the incessant resistance, accessibility, and safety challenges associated with the currently available orthodox antiprostate drugs.
AUTHORS’ CONTRIBUTIONS
NA coordinated the extraction, phytochemical screening, GC-MS analysis, and contributed to manuscript preparation. YOA designed the project, carried out the docking simulations, PASS online activity, ADMET prediction, and prepared the manuscript. DIK, SS, and BB collected the samples and carried out the extraction. MAA, AB and ATA contributed to manuscript revision. MNM supervised the whole project and revised the final manuscript.
CONFLICT OF INTEREST
The authors have no conflict to declare in this report.
FUNDING
The authors wish to acknowledge the support of the Ministry of Higher Education of Malaysia, HICoE (Grant no. 311/CDADAH/4401009). YOA thanks the Tertiary Education Fund, Nigeria, and Universiti Sains Malaysia for PhD Scholarship and GA Scheme (Grant no. 308.AIPS.415401), respectively.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Abubakar AR, Haque M. Preparation of medicinal plants: basic extraction and fractionation procedures for experimental purposes. J Pharm Bioallied Sci, 2020; 12:1–10. CrossRef
Adefegha SA, Oboh G, Okeke BM, Oyeleye SI. Comparative effects of alkaloid extracts from Aframomum melegueta (alligator pepper) and Aframomum danielli (bastered Melegueta) on enzymes relevant to erectile dysfunction comparative effects of alkaloid extracts from Aframomum melegueta (alligator pepper) and Aframomum danielli (bastered). J Diet Suppl, 2017; 14:542–52. CrossRef
Agim C, Nosiri C, Chukwuma A, Okechukwu A. Evaluation of the phytochemical and GC-MS analysis of the aqueous seed extract of Aframomum melegueta. J Pharmacogn Phytochem 2017;6:101–104.
Ai CZ, Man HZ, Saeed Y, Chen DC, Wang LH, Jiang YZ. Computational insight into crucial binding features for metabolic specificity of cytochrome P450 17A1. Inform Med Unlocked, 2019; 15:100172. CrossRef
Akpanabiatu MI, Ekpo ND, Ufot UF, Udoh NM, Akpan EJ, Etuk EU. Acute toxicity, biochemical and haematological study of Aframomum melegueta seed oil in male Wistar albino rats. J Ethnopharmacol, 2013; 150:590–4. CrossRef
Ayipo YO, Osunniran WA, Mordi MN. Metal complexes of b -carboline: advances in anticancer therapeutics. Coord Chem Rev, 2021; 432:213746. CrossRef
Bardia A, Platz EA, Yegnasubramanian S, De Marzo AM, Nelson WG. Anti-inflammatory drugs, antioxidants, and prostate cancer prevention. Curr Opin Pharmacol, 2009; 9:419–26. CrossRef
Benet LZ, Hosey CM, Ursu O, Oprea TI, Sciences T, Division I. BDDCS, the rule of 5 and drugability leslie. Adv Drug Deliv Rev, 2016; 101:89–98. CrossRef
Biobaku KT, Azeez OM, Amid SA, Asogwa TN, Abdullahi AA, Raji OL, JA Abdulhamidc, et al. Thirty days oral Aframomum melegueta extract elicited analgesic effect but influenced cytochrome p4501BI, cardiac troponin T, testicular alfa-fetoprotein and other biomarkers in rats. J Ethnopharmacol, 2020; 113493. CrossRef
Bittencourt JAHM, Neto MFA, Lacerda PS, Bittencourt RCVS, Silva RC, Lobato CC, Silva LB, Leite FHA, Zuliani JP, Rosa JMC, Borges RS, Santos CBR, et al. In silico evaluation of ibuprofen and two benzoylpropionic acid derivatives with potential anti-inflammatory activity. Molecules, 2019; 24(8):1476. CrossRef
Brito V, Santos AO, Almeida P, Silvestre S. Novel 4-azaandrostenes as prostate cancer cell growth inhibitors: synthesis, antiproliferative effects, and molecular docking studies. C R Chim, 2019; 22:73–83. CrossRef
Castro-Alvarez A, Costa AM, Vilarrasa J. The performance of several docking programs at reproducing protein-macrolide-like crystal structures. Molecules, 2017; 22(1):136. CrossRef
Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep, 2017; 7:1–13. CrossRef
Dzoyem JP, McGaw LJ, Kuete V, Bakowsky U. Anti-inflammatory and anti-nociceptive activities of African medicinal spices and vegetables. Elsevier Inc, Amsterdam, Netherlands, 2017. CrossRef
Emeribe EO. Phytochemicals and organic compounds of two nigerian insecticidal plants. J Agric Food Sci, 2018; 16:1–12. CrossRef
Filimonov D, Poroikov V. Probabilistic approaches in activity prediction. In: Varnek A, Tropsha A (eds.). Chemoinformatics approaches to virtual screen, RSC Publishing, Cambridge, UK, pp 182–216, 2009. CrossRef
Fouzia M, Salim B. Virtual screening of natural and synthetic inhibitors of cyclooxygenase COX-2 enzyme using docking-scoring functions. J Appl Pharm Sci 2019; 9:20–7. CrossRef
Gini G, Franchi AM, Manganaro A, Golbamaki A, Benfenati E. ToxRead: a tool to assist in read across and its use to assess mutagenicity of chemicals. SAR QSAR Environ Res, 2014; 25:999–1011. CrossRef
Grienke U, Kaserer T, Pfluger F, Mair CE, Langer T, Schuster D, Rollinger JM. Accessing biological actions of Ganoderma secondary metabolites by in silico profiling. Phytochemistry, 2015; 114:114–24. CrossRef
Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem, 2004; 47:1750–9. CrossRef
Harborne JB. Phytochemical methods a guide to modern techniques of plant analysis. Chapman and Hall in association with Methuen, Inc, New York, NY, 1984.
Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J Chem Theory Comput, 2016; 12:281–96. CrossRef
Hassan T, Irondi A, Ajani E, Ismail O. Quantitative analysis of phytochemicals constituent of melegueta pepper seed. Int J Innov Sci Res Technol, 2019; 4:595–9.
Ikwu FA, Shallangwa GA, Mamza PA. QSAR, QSTR, and molecular docking studies of the anti-proliferative activity of phenylpiperazine derivatives against DU145 prostate cancer cell lines. Beni Suef Univ J Basic Appl Sci, 2020; 9(35): 1-12. CrossRef
Khanna V, Ranganathan S. Physiochemical property space distribution among human metabolites, drugs and toxins. BMC Bioinformatics, 2009; 10:1–18. CrossRef
Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 2013; 27:221–34. CrossRef
Meairs S. Facilitation of drug transport across the blood–brain barrier with ultrasound and microbubbles. Pharmaceutics, 2015; 7:275–93. CrossRef
Nagai H, Kim YH. Cancer prevention from the perspective of global cancer burden patterns. J Thorac Dis, 2017; 9:448–51. CrossRef
Poroikov VV, Filimonov DA, Gloriozova TA, Lagunin AA, Druzhilovskiy DS, Rudik AV, Stolbov LA, Dmitriev AV, Tarasova OA, Ivanov SM, Pogodin PV. Computer-aided prediction of biological activity spectra for organic compounds: the possibilities and limitations. Russ Chem Bull, 2019; 68:2143–54. CrossRef
Ramírez D, Caballero J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data? Molecules 2018; 23:1–17. CrossRef
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin, 2020; 70:7–30. CrossRef
Skok Ž, Zidar N, Kikelj D, Ilaš J. Dual inhibitors of human DNA topoisomerase II and other cancer-related targets. J Med Chem 2020; 63:884–904. CrossRef
Swiatek P, Gebczak K, Gebarowski T, Urniaz R. Biological evaluation and molecular docking studies of dimethylpyridine derivatives. Molecules, 2019; 24. CrossRef
Toh EYS, Lim CL, Ling APK, Chye SM, Koh RY. Overview of the pharmacological activities of Aframomum melegueta. Pertanika J Trop Agric Sci, 2019; 42:1–13.
Trease G, Evans W. Pharmacognosy. 16th edition, Saunders Publishers, London, UK, 2009.
Umukoro S, Aladeokin AC. Therapeutic effects of grains of Paradise (Aframomum melegueta) seeds. Elsevier Inc, Amsterdam, Netherlands, 2011. CrossRef