
INTRODUCTION
Genistein, a natural isoflavone abundant in soybeans and other leguminous plants [1], exhibits numerous biological activities, including anti-inflammatory [2], antioxidant [3], anticancer [4], antibacterial [5], and antiviral effects [5]. These attributes suggest its potential therapeutic utility across various conditions. However, genistein is classified under class II of the Biopharmaceutics Classification System due to its poor water solubility (0.029 mg/ml) [6], which limits its oral bioavailability. In rats, the oral bioavailability of genistein at doses of 4, 20, and 40 mg/kg was found to be 38.58%, 24.34%, and 30.75%, respectively [7]. Low bioavailability can reduce drug efficacy and increase variability in patient responses.
Various methods have been employed to enhance the delivery of genistein, such as nanoparticle [8], microparticle [9], micelle [10], microemulsion [11], cocrystal [12], and solid dispersion (SD) [6]. SD-based formulations are particularly noteworthy because it has been successfully used to enhance the solubility of many drugs (such as itraconazole, tacrolimus, lopinavir/ritonavir, etravirine, and vemurafenib), resulting in products approved by the US FDA [13]. Additionally, SD is a simple and cost-effective method [14–16]. This technique generally involves dispersing one or more active substances in an inert carrier to form an amorphous solid. The amorphous form of the drug in SD enhances solubility by eliminating the need to disrupt crystalline lattices, and using hydrophilic polymers as inert carriers improves wettability [14,17].
The genistein-SD was previously developed using a single carrier, polyvinylpyrrolidone K30. The SD formulation, with a 1:7 ratio of genistein to polyvinylpyrrolidone K30, exhibited a 482-fold increase in drug release at 60 minutes compared to pure genistein [6]. Pharmacokinetic studies in rats further demonstrated that the Cmax and AUC0-24 of the SD formulation were 6.86-fold and 2.06-fold higher, respectively, than those of pure genistein [6]. In this study, a co-carrier system was used to develop genistein-SD, consisting of three carriers: polyethylene glycol (PEG 4000), poloxamer 407 (P 407), and crospovidone (XPVP). PEG 4000, a hydrophilic and biocompatible polymer [18], has been used as a carrier in SD to enhance the solubility of poorly water-soluble drugs [15,19,20]. P 407, a non-ionic surfactant, has also been used as a carrier [21,22] and can enhance the solubility of poorly water-soluble drugs by improving their wettability [14,21,23]. XPVP particles surrounding drug particles decrease aggregation, facilitating rapid contact with the dissolution medium and thereby enhancing solubility [24]. Co-carrier systems that combine polymers and surfactants can enhance drug solubility and stabilize the drug’s amorphous state [25]. Response surface methodology (RSM) was previously used to develop and optimize the genistein-SD formulation [26,27]. A well-developed predictive model from RSM can assist formulation optimization by estimating results without additional experiments. If accurately developed, such a model allows predicted values to align with experimental values closely. Within RSM, the Box-Behnken design (BBD) is widely used, as it identifies the most favorable response and optimizes the formulation by establishing the optimal relationship between influential variables. Additionally, this design enables the investigation of the effects of independent variables on the target response [28].
The main objectives of this study were to develop and characterize both genistein-SD and genistein-SD tablets. Genistein-SDs were prepared using the solvent evaporation method with three previously mentioned carriers. BBD was implemented to evaluate the effects of the amount of each carrier and their interactions on solubility to maximize genistein’s solubility. The optimal formulation was selected for further evaluation, including assessments of flowability and characterization using attenuated total reflectance-Fourier transform infrared (ATR-FTIR), differential scanning calorimetry (DSC), and powder X-ray diffraction (PXRD), followed by development into genistein-SD tablets with two different formulations. The tablets were evaluated for weight, thickness, diameter, hardness, friability, and disintegration time. Finally, in vitro dissolution and stability studies were conducted on the optimal genistein-SD formulation and two formulations of the genistein-SD tablets.
MATERIALS AND METHODS
Materials
The materials used in this study, along with their suppliers and locations, are summarized in Table 1.
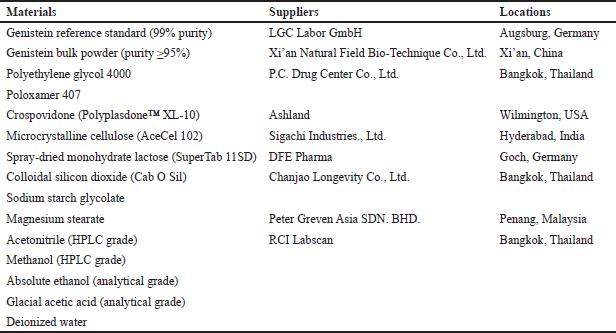 | Table 1. Materials used in this study. [Click here to view] |
Chromatographic method for quantification of the genistein and its validation
The quantification of genistein in SD and tablets was conducted using high-performance liquid chromatography (HPLC) with diode-array detection (UltiMate 3000, Thermo Scientific, MA, USA) equipped with a VWD-3400 RS detector, WPS-3000SL autosampler, LPG-34003D solvent delivery pump, and TCC-3000SD column compartment. Chromatographic separation was achieved using a VertiSep™ C18 column (250 × 4.6 mm, 5 µm) from Vertical Chromatography Co., Ltd. (Nonthaburi, Thailand), with the column temperature maintained at 27°C ± 3°C. The isocratic mobile phase system comprised solvent A (1% acetic acid) and solvent B (70% acetonitrile and 1% acetic acid), with a flow rate set at 1 ml/minute. Detection was performed at a wavelength of 260 nm. A 20 µl sample injection volume was employed for the analysis. The retention time for genistein was about 5.8–5.9 minutes (Fig. S1). The mean peak areas for each concentration were calculated from three determinations. The standard curve, generated by plotting concentrations against peak areas, demonstrated good linearity across the 3.13 to 100 µg/ml (Fig. S2), with a correlation coefficient (r²) of 1.000. The limit of detection and limit of quantification were 0.34 and 1.03 µg/ml, respectively. The method’s accuracy was assessed, and the recovery percentage for genistein ranged from 100.22% to 102.66%. The relative standard deviation for assessing repeatability ranged from 0.28% to 0.32%, and for intermediate precision, it ranged from 0.25% to 0.26%. The accuracy and precision were evaluated according to the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) guideline titled “Validation of Analytical Procedures”. The detailed evaluation protocol and results are provided in Tables S1 and S2.
Optimization of SD formulation using RSM
The required quantities of genistein powder (GP), PEG 4000, and P 407 were dissolved in absolute ethanol to obtain a clear solution, and the required quantity of XPVP was then dispersed into the solution. The solvent was removed by evaporation, and the resulting product was dried at 40°C for 12 hours in a vacuum oven. The dried product was sieved through mesh no.40 (sieve size opening of 0.42 mm). Each formulation of the SD was prepared with the composition ratios shown in a column of actual values in Table 2, with the genistein of 1% w/w.
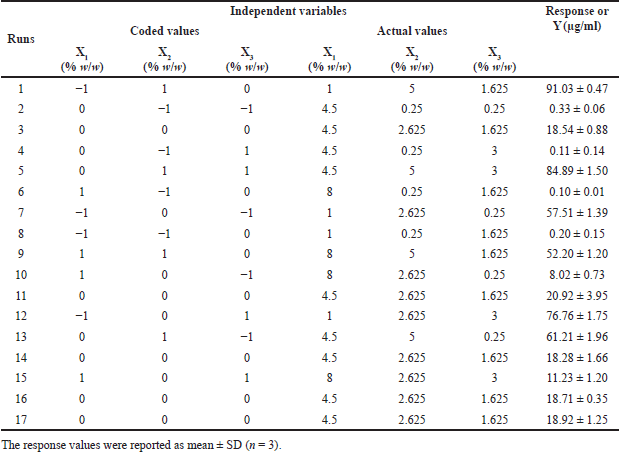 | Table 2. BBD of independent variables and response. [Click here to view] |
The study employed the BBD, a 3-level (−1, 0, +1) factorial design within RSM, to optimize the solubility of genistein in SD. The investigation focused on three independent variables: the quantity of PEG 4000 (X1), the quantity of P 407 (X2), and the quantity of XPVP (X3). Specific levels were set for each variable: X1 at 1, 4.5, and 8; X2 at 0.25, 2.625, and 5; and X3 at 0.25, 1.625, and 3. The dependent variable was genistein’s solubility (Y) (Table S3). The BBD was selected for its efficiency and suitability, as it enables the identification of the most favorable response while reducing the number of experimental runs compared to a full factorial design. In optimizing genistein-SD formulations, BBD facilitates the identification of optimal amounts of carriers (PEG 4000, P 407, and XPVP) to achieve maximum solubility. Additionally, it allows for examining both individual and combined effects of carriers on solubility.
Using a BBD, 17 experimental runs (formulations) were conducted to assess genistein’s solubility across various formulations (Table 2). Data analysis utilized Design-Expert® software version 13.0.5.0, which fitted a quadratic model and employed analysis of variance (ANOVA) to determine the significance of each factor (p ≤ 0.05). Response surface plots were generated to visually represent the relationships between the independent variables and the response.
The genistein-SD formulation was optimized to maximize Y using numerical and graphical techniques from the software. Optimum values for X1, X2, and X3 were identified, and the maximum Y was predicted. Validation involved preparing SD with the selected X1, X2, and X3 values, followed by testing the genistein’s solubility. The theoretical prediction from the software was compared to the experimental result, with prediction accuracy calculated using the following equation:
Prediction accuracy (%) = (Experimental value/ Predicted value) × 100. (1)
Determination of genistein content
The genistein content was determined to ensure the quantity of genistein in each SD (17 formulations and optimized formulation). The SD was weighed 10 mg into a volumetric flask, and absolute ethanol was added to dissolve the genistein from the SD. The solution was made up to volume with absolute ethanol and then filtered through a 0.45 µm nylon syringe filter. The assay was conducted using HPLC, as described in Section “Chromatographic method for quantification of the genistein and its validation”. Each sample was analyzed in triplicate, and the results were reported as mean ± SD.
Determination of solubility
The solubility of genistein in each SD (17 formulations and optimized formulation) was determined. An excess amount of sample was dispersed in 10 ml of 0.1 M phosphate buffer (pH 6.8) and continuously shaken for 48 hours at 30°C in a shaking incubator. The sample was centrifuged and then filtered through a 0.45 µm nylon syringe filter to keep the solution, which was analyzed using HPLC as as described in Section “Chromatographic method for quantification of the genistein and its validation”. Each sample was performed in triplicate. The results were used for formulation optimization.
Preparation of a physical mixture (PM) of genistein and carriers
The PM is a crucial control for evaluating the performance and benefits of SD formulations compared to straightforward drug-carrier mixtures. The PM was prepared by mixing the GP of 1% w/w with PEG 4000, P 407, and XPVP at an optimized ratio of 1.034, 4.992, and 3, respectively. The obtained PM was stored in a desiccator for further experiments.
Characterization of optimized SD
DSC analysis
Thermal analysis of GP, PEG 4000, P 407, XPVP, PM, and optimized SD was performed using a differential scanning calorimeter (DSC-8000, PerkinElmer, Shelton, Connecticut, USA). Approximately 5 mg of each sample was sealed in an aluminum pan, with an empty pan used as a reference. DSC thermograms were recorded at a heating rate of 10°C/minute from 25°C to 350°C under a nitrogen atmosphere with a 20 ml/minute flow rate.
ATR-FTIR spectroscopy analysis
ATR-FTIR spectroscopy analysis of GP, PEG 4000, P 407, XPVP, PM, and optimized SD was performed using an FTIR spectrometer (Tensor 27, Bruker, Hong Kong, China), coupled with ATR accessory, over a range of 4,000−400 cm−1 with a resolution of 4 cm−1 and 32 scans.
PXRD analysis
The crystalline or amorphous state of GP, PEG 4000, P 407, XPVP, PM, and optimized SD was analyzed by a powder X-ray diffractometer (SmartLab, Rigaku, Tokyo, Japan) using a Cu Kα radiation at a voltage of 40 kV and current of 40 mA. The diffractograms were measured over a 2θ range of 5° to 60° with a scanning speed of 2°/minute and a step size of 0.02°.
Flowability study of optimized SD
Bulk density and tapped density
The bulk and tapped densities of optimized SD were determined according to the method in a general chapter 616 of United States Pharmacopeia (USP). The SD of mass (m) 100 g was filled into a 250 ml graduated cylinder without compacting. The untapped volume (V0) was measured for bulk density, and the bulk density was calculated from the formula m/V0. After that, to obtain the tapped density, the cylinder with 100 g sample was placed on the tapped density testers (PT-TD300, Pharma Test, Hainburg, Germany). Mechanically tap 500 times first and read the volume (V500), then tap 750 times and read the volume (V1250). If the difference between V500 and V1250 is ≤ 2 ml, V1250 is the final tapped volume (VF). If the difference between V500 and V1250 is > 2 ml, repeat the tapping in increments of 1,250 taps until the difference between succeeding measurements is ≤2 ml; the latest volume is the VF. The tapped density was calculated using the formula m/VF. The measurements of each density were done in triplicate.
Compressibility index (CI) and Hausner ratio (HR)
CI and HR were calculated using the bulk density (ρbulk) and tapped density (ρtapped) by the following equations:
CI = [(ρtapped - ρbulk)/ ρtapped] × 100 (2)
HR = ρtapped/ ρbulk. (3)
Angle of repose
The angle of repose was measured by the fixed cone diameter method. The optimized SD was poured through a fixed funnel with a 10 mm orifice to form a cone until the diameter (d) of the cone reached 7.5 cm. The height (h) of the cone was measured. The measurement was done in triplicate, and the angle of repose was calculated using the following equation:
Angle of repose = tan−1 (2h/d) (4)
Formulations of genistein-SD tablets and characterization
Formulations of genistein-SD tablets were developed based on the flowability results of the genistein-SD powder, shown in Table 3. Spray-dried monohydrate lactose and colloidal silicon dioxide were incorporated to enhance the powder mixture flowability. Formulations F1 and F2 were designed with different disintegrants (XPVP and sodium starch glycolate) to investigate their disintegration abilities.
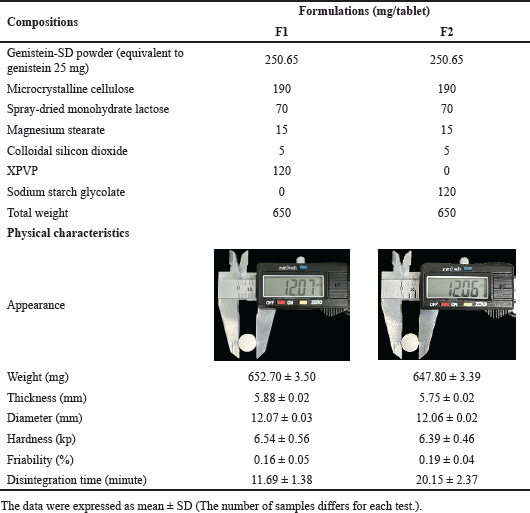 | Table 3. Composition and physical characteristics of 25 mg genistein SD tablet formulations. [Click here to view] |
Weighed quantities of genistein-SD powder and all ingredients, except magnesium stearate, were individually sieved through mesh no.18 (sieve size opening of 1.04 mm) to break powder aggregates. All materials were then mixed geometrically for 10 minutes. Subsequently, the weighed quantity of the lubricant, magnesium stearate, was sieved through a mesh no.40 (sieve size opening of 0.42 mm) and mixed with the previous materials for 3 minutes to prepare the mixture for compression. Finally, the obtained mixture was compressed into 650 mg biconvex tablets of 12 mm diameter using an electric single punch tableting machine (CMT12, Charatchai Machinery, Bangkok, Thailand).
The genistein-SD tablets characterization includes weight, thickness, diameter, hardness, friability, and disintegration time. Twenty tablets of each formulation were randomly taken at intervals from the beginning to the end of the compression process and then weighed individually. The tablet’s thickness, diameter, and hardness were evaluated after weighing, which was carried out using a 3-in-1 hardness, diameter, and thickness tester (PTB 311E, Pharma Test, Hainburg, Germany). The data were presented as mean ± SD (n = 20).
The tablet friability was determined using a tablet friability tester (RT-125, Rocknet Siam, Bangkok, Thailand). Whole tablets weighing close to 6.5 g were randomly selected from each formulation, dusted off, and placed in a drum that rotated at a fixed speed (25 rpm) for 4 minutes. Loose dust was then removed from the tablets and accurately weighed again. Tablet friability was computed as the weight loss percentage, which should not exceed 1.0%.
The disintegration test was conducted using the disintegration tester (DIST-3, Pharma Test, Hainburg, Germany). Six tablets of each formulation were placed separately into separate tubes of the basket-rack assembly, and then the disintegration tester’s disks were put. The medium tested was 900 ml of 0.1 M phosphate buffer (pH 6.8) with maintaining temperature at 37°C ± 2°C. The disintegration time of each tablet was recorded and reported as mean ± SD (n = 6).
Dissolution studies of optimized SD and genistein-SD tablets
The dissolution testing of GP, PM, optimized SD, and SD tablets (F1 and F2) was performed strictly according to the procedures outlined in the general chapter 711 of USP. This testing used a dissolution tester (Varian VK-7010, Agilent Technologies, Ontario, Canada) equipped with a paddle apparatus. All samples contained genistein equivalent to 25 mg. Each sample was immersed in 900 ml of 0.1 M phosphate buffer (pH 6.8) containing 1% w/v sodium lauryl sulfate, maintained at 37°C ± 0.5°C, with a stirring speed set at 50 rpm. A 1-ml sample solution was withdrawn at a predetermined time (0, 5, 10, 15, 30, 60, 120, and 180 minutes). After each withdrawal, an equal volume of preheated fresh medium was added to the dissolution vessels to maintain a constant volume. The withdrawn sample solutions were filtered through a 0.45 µm nylon syringe filter, and the concentration of genistein was determined using HPLC as described in Section “Chromatographic method for quantification of the genistein and its validation”. Dissolution experiments for each sample were performed in triplicate.
Stability studies of optimized SD and genistein-SD tablets
The stability studies were evaluated according to the ASEAN Guideline on Stability Study of Drug Product [29]. The optimized SD and genistein-SD tablets (F1 and F2) were stored in tight, light-resistant containers under long-term conditions [30°C ± 2°C and 75% ± 5% relative humidity (RH)] and accelerated conditions (40°C ± 2°C and 75% ± 5% RH) for 3 months. Genistein content was measured monthly over this period. Each sample was analyzed in triplicate, and the results were reported as mean ± SD.
All the results obtained were compared using a one-way ANOVA followed by the least significant difference test. The p-values lower than 0.05 indicated a statistically significant difference between groups.
RESULT AND DISCUSSION
Optimized formulation of SD using RSM
This study aimed to enhance the solubility of genistein by optimizing the SD formulation. To achieve this, a BBD was utilized with 17 experimental runs. The independent variables included the amount of PEG 4000 (X1), the amount of P 407 (X2), and the amount of XPVP (X3), and the results of the dependent variable are shown in Table 2. The ANOVA demonstrated a significant model with an F-value of 146.05 and a p-value of less than 0.0001. This result indicates a high probability that the investigated independent variables influenced the observed solubility (Table 4). X1, X2, and the quadratic term for X2 (X22) were identified as significant model terms, whereas X3 and the interaction between the amount of P 407 and the amount of XPVP (X2X3) were not significant (p-value = 0.9089 and 0.0743, respectively). Despite the significant lack of fit (F-value = 67.38, p-value = 0.0005), indicating some discrepancies between the model and certain data points, the overall fit statistics were strong. The experimental data exhibited an outstanding fit, as evidenced by the model R2 value of 0.9852 and the adjusted R2 value of 0.9784. Moreover, the predicted R2 value of 0.9560 was in reasonable agreement with the adjusted R2 value.
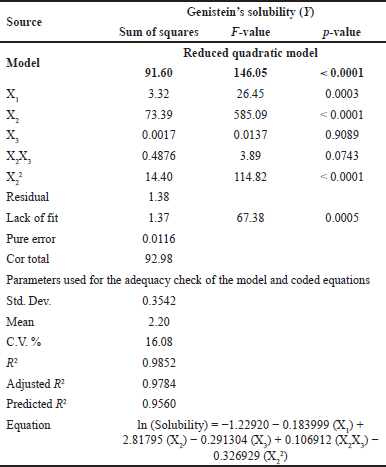 | Table 4. Analysis of RSM model variance (ANOVA) for genistein’s solubility. [Click here to view] |
Based on the regression equation in Table 4 and Figure S3, the coefficient for P 407 (X2) is positive, indicating that increasing its concentration significantly enhances genistein’s solubility. Conversely, PEG 4000 (X1) exerts a negative effect, though it is less pronounced than other factors. The impact of XPVP (X3) on solubility was found to be insignificant. According to the model, the optimal SD formulation of genistein contains 1.034% w/w of PEG 4000, 4.992% w/w of P 407, 3% w/w of XPVP, and 1% w/w of GP. The experimental validation of the predicted optimal conditions was carried out, resulting in a predicted solubility of 186.39 µg/ml and an experimental solubility of 181.12 µg/ml. The prediction accuracy between the predicted and experimental solubility was determined to be 97.17%, thereby validating the reliability of the model predictions.
The interactions between factors are shown in the 3D response surface plots (Fig. 1). The PEG 4000 and P 407 interaction (X1X2), as shown in Figure 1A, shows that as the amount of P 407 increases, the solubility appears to increase but can be influenced by the amount of PEG 4000; solubility seems to decrease when PEG 4000 increases. Interestingly, an increase for P 407 above a certain point does not result in any further increase in solubility. The PEG 4000 and XPVP interaction (X1X3), as shown in Figure 1B, shows that increasing XPVP tends to increase solubility. However, it can be interfered with the amount of PEG 4000, as higher levels of PEG 4000 are associated with reduced solubility. The P 407 and XPVP interaction (X2X3), as shown in Figure 1C, shows that as the amount of P 407 increases, the solubility appears to increase but can be influenced by the amount of XPVP, with the solubility being highest when the amount of XPVP is the highest. However, these interactions did not significantly affect the solubility. Therefore, the co-carrier system used in this study did not synergize to improve solubility significantly.
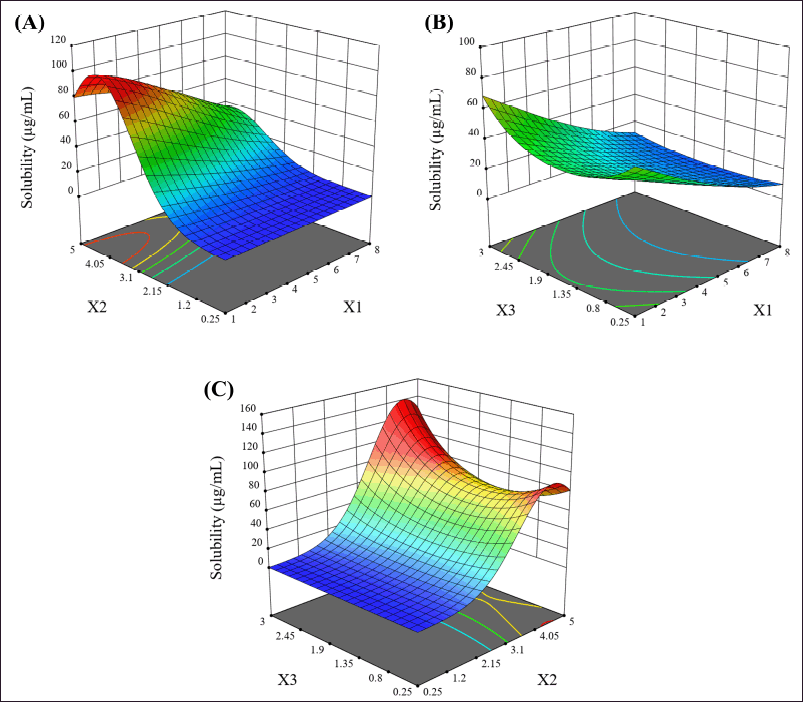 | Figure 1. The 3D response surface plots of the relationship between (A) the amount of PEG 4000 and the amount of P 407 (X1X2), (B) the amount of PEG 4000 and the amount of XPVP (X1X3), and (C) the amount of P 407 and the amount of XPVP (X2X3). [Click here to view] |
Determination of genistein content
As shown in Table S4, the genistein content in each SD formulation ranged from 98.09% ± 0.87% to 100.89% ± 0.36%. The results indicate that all SD formulations contain nearly 100% of the genistein.
Characterization of SD
DSC analysis
The DSC thermal behaviors of the GP, PEG 4000, P 407, XPVP, PM, and optimized SD are shown in Figure 2A. The GP, PEG 4000, and P 407 exhibited a sharp endothermic peak at 304°C, 65°C, and 60°C, respectively, corresponding to their melting point [6,30–32]. XPVP has not exhibited any sharp peak due to its amorphous nature. The PM and SD exhibited similar thermograms, showing a broad endothermic peak at 54°C and 55°C, respectively, which indicate the melting of both PEG 4000 and P 407. The melting occurred at a temperature lower than their typical melting temperature, suggesting the possible formation of a eutectic mixture. Importantly, the endothermic peak of GP disappears in both the PM and SD. The absence of the GP peak in the PM is likely due to the carrier dilution effect [6,27]. In contrast, in the SD, it is attributed to the transition of genistein from its crystalline to amorphous form, as confirmed by the PXRD results.
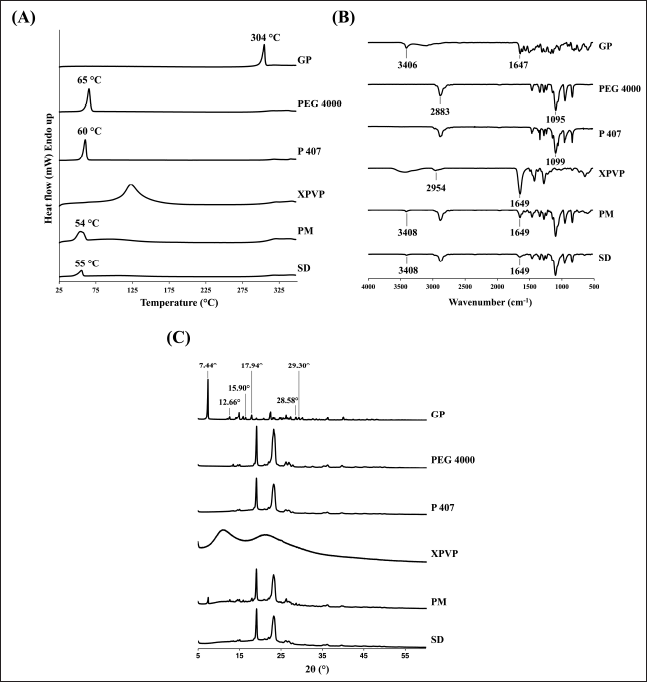 | Figure 2. Characterization results of GP, PEG 4000, P 407, XPVP, PM, and optimized SD; (A) DSC thermograms, (B) FTIR spectra, and (C) X-ray diffractograms. [Click here to view] |
ATR-FTIR spectroscopy analysis
As shown in Figure 2B, the spectrum of GP displays characteristic peaks at 3,406 and 1,647 cm−1, corresponding to the stretching of O-H and C=O, respectively [6,12,33,34]. PEG 4000 and P 407 exhibited similar spectra due to the identical functional groups in their structures. The characteristic peaks at 2,883 and 2,881 cm−1 correspond to the stretching of C-H, and at 1,095 and 1,099 cm−1 correspond to the C-O stretching for PEG 4000 and P 407, respectively [35,36]. XPVP exhibited a spectrum with characteristic peaks at 2,954 and 1,649 cm−1, corresponding to the stretching of C-H and C=O, respectively [24].
The spectra of PM and optimized SD appear to be a sum of all components in the formulation, with no significant shifting of any peaks. The presence of the peaks at 3,408 and 1,649 cm−1, which are the characteristic peaks of the GP, indicated no molecular interaction between genistein and carriers.
PXRD analysis
The diffractograms of GP, PEG 4000, P 407, XPVP, PM, and optimized SD are shown in Figure 2C. GP exhibited many sharp peaks, reflecting its crystalline nature. The peaks at 2θ values of 7.44°, 12.66°, 15.90°, 17.94°, 28.58°, and 29.30° appeared in the diffractogram of PM with lower intensity, indicating that there was no alteration of genistein’s crystallinity in the PM. The decrease in peak intensity is due to the high proportion of carriers in the formulation [27,37,38]. However, these peaks are absent in the diffractogram of optimized SD. These results indicate that the preparation of SD in this study can alter the crystallinity of genistein from the crystalline to amorphous form, thereby justifying the increased solubility and improved dissolution profile.
Flowability study of optimized SD
As shown in Table 5, the CI, HR, and angle of repose values of each batch are approximately the same, with each value consistently being classified as fair level following a general chapter 1174 of USP. Consequently, the SD powder should be slightly improved flowability to efficiently flow from the hopper into the die and prevent tablet weight variation during the tableting step. This is the reason for adding free-flowing diluent and glidant in tablet formulations.
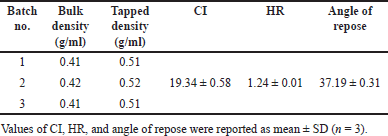 | Table 5. Results of flowability assessment of optimized SD. [Click here to view] |
Characterization of Genistein-SD tablets
Genistein-SD tablets were investigated using the direct compression process, and their physical characteristics are shown in Table 3. Two formulations, F1 and F2, were evaluated, with the difference being the disintegrant used—XPVP in F1 and sodium starch glycolate in F2. Both formulations produced white to off-white tablets, round-shape, and biconvex with beveled edges. They exhibited similar physical characteristics, including weight, thickness, diameter, hardness, and friability. The tablets weighed approximately 650 mg, with a diameter of about 12 mm and a thickness of approximately 5.82 mm. The hardness was around 6.46 kp, and the weight loss percentage in the friability test was less than 0.18% for all formulations. These results indicate that the tablets possess adequate mechanical strength and low friability, meeting the official limits. Notably, the disintegrant significantly affected the disintegration time, with XPVP in F1 resulting in faster disintegration than sodium starch glycolate in F2. XPVP in the tablet markedly decreases the interfacial tension between genistein SD and the medium, leading to rapid water absorption and swelling, creating a wicking action that quickly breaks apart the tablet [39].
Dissolution studies
The dissolution profiles of GP, PM, and optimized SD were investigated using a dissolution apparatus type 2 with 0.1 M phosphate buffer (pH 6.8) containing 1% w/v sodium lauryl sulfate as the dissolution medium. As shown in Figure 3A, GP exhibited a poor dissolution rate, with only 25.56% and 41.39% of the drug released after 60 and 180 minutes, respectively. This poor solubility in aqueous solution can be attributed to the naturally low solubility of genistein [6].
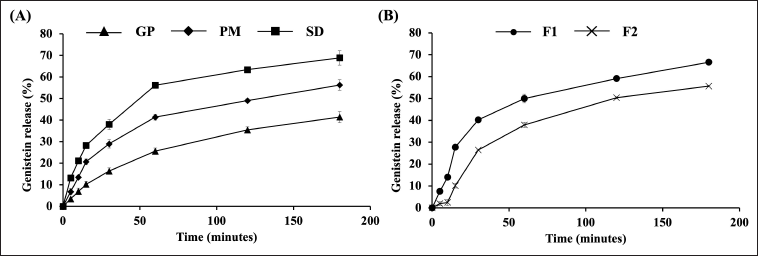 | Figure 3. Genistein release profiles from (A) GP, PM, and optimized SD and (B) genistein-SD tablets. The data were reported as mean ± SD (n = 3). [Click here to view] |
In contrast, the SD exhibited significantly higher drug release than the GP at all time points, with 56.16% and 68.87% of genistein released from the SD after 60 and 180 minutes, respectively. Notably, the amount of genistein released from the SD after 60 minutes was approximately 2.2 times greater than that of the GP. In this study, genistein SDs were prepared using PEG 4000, P 407, and XPVP. The dispersion of genistein within the hydrophilic matrix of PEG 4000 allows for rapid contact with the dissolution medium, thereby enhancing genistein’s solubility and dissolution rate [15,19]. P 407 acts as a wetting agent and a solubilizer, reducing surface tension and increasing the solubility and dissolution rate [14,21,23]. XPVP surrounds the drug particles, decreasing aggregation, which results in rapid contact with the dissolution medium and better wetting of their surface [24]. These factors collectively promote a more efficient dissolution process for genistein-SD.
The dissolution behavior of PM was slightly better than that of GP, with 41.34% of genistein released from the PM within 60 minutes. The PM was prepared by simple mixing, so genistein remained crystalline. In contrast, in the SD, the drug was converted into an amorphous state, as confirmed by PXRD observations. These results demonstrate that the SD approach is necessary for enhancing the dissolution rate and extent of genistein.
The release profiles of genistein from SD tablets are shown in Figure 3B. Genistein was released slower than SD because the tablets needed to disintegrate into smaller fragments before dissolving. However, the amount of genistein released from tablet formulation F1 was nearly identical to that from the SD, with 66.56% released from F1 and 68.87% from the SD after 180 minutes. Tablets are commonly used as oral dosage forms due to their convenience in handling, storage, and transport. Their simple design, easy swallowing, and potential for less frequent dosing enhance patient compliance with prescribed treatments.
As expected, F1, which contains XPVP, exhibited a faster drug release rate and extent than F2, which contains sodium starch glycolate. After 15 minutes, the drug release from F1 was 27.68%, compared to 10.09% from F2. By the end of the test, drug release from F1 reached 66.56%, while F2 achieved 55.65%. These results are consistent with the disintegration test, which showed that F1 had a faster disintegration time. XPVP’s high-wicking capacity quickly pulls water into the tablet structure, causing rapid disintegration. It swells extensively upon contact with water, aiding in the quicker breakup of the tablet into smaller fragments and facilitating faster drug release [40]. Additionally, its highly porous structure increases surface area and enhances water absorption, contributing to faster disintegration and dissolution [41]. The superior properties of XPVP significantly enhance the disintegration and drug release rates, making it a preferable choice over sodium starch glycolate for this formulation.
Stability studies
The data of the stability studies is summarized in Table 6. The optimized SD and genistein-SD tablets (F1 and F2) were stable at 30°C ± 75% RH for up to 3 months, with the genistein content remaining in the range of 99.25%–99.85%. At 40°C ± 75% RH at 3 months, the genistein content in the optimized SD, genistein-SD tablet (F1 and F2) remained at 96.61%, 97.20%, and 97.76%, respectively. Comparison of genistein content between 30°C and 40°C at 3 months found that genistein content in optimized SD and genistein-SD tablets (F1 and F2) at 40°C significantly lower than 30°C (p-values were 0.006, 0.045, and 0.042, respectively). Therefore, higher temperatures may accelerate the degradation of the genistein.
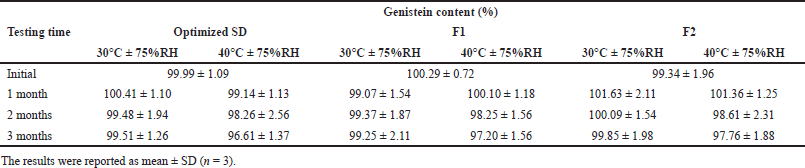 | Table 6. The genistein content of the optimized SD and genistein-SD tablet (F1 and F2). [Click here to view] |
CONCLUSION
This study demonstrated that SD is an effective strategy for enhancing genistein’s solubility and dissolution profile using the solvent evaporation method with PEG 4000, P 407, and XPVP as carriers. BBD optimization resulted in an optimized formulation comprising 1.034% w/w PEG 4000, 4.992% w/w P 407, 3% w/w XPVP, and 1% w/w genistein, achieving an experimental solubility of 181.12 µg/ml with a model’s prediction accuracy of 97.17%. The carriers that significantly affected genistein’s solubility in this study’s model were P 407 and PEG 4000. P 407 had a positive effect, while PEG 4000 had a less pronounced but negative effect. Additionally, interactions between carriers affected genistein’s solubility, though these effects were not statistically significant. Characterization of optimized SD using DSC, ATR-FTIR, and PXRD confirmed the transformation of genistein from a crystalline to an amorphous state, and in vitro dissolution studies showed superior dissolution profiles for optimized SD and genistein-SD tablets (F1) compared to the PM and GP. Stability studies further demonstrated high retention of genistein content under long-term and accelerated conditions over three months, supporting the potential for pharmaceutical development. However, in vivo bioavailability studies under physiological conditions are essential to confirm these findings. Additionally, exploring alternative carriers may provide further improvements and reveal potential synergistic interactions.
ACKNOWLEDGMENT
The authors would like to thank the Research and Innovation Institute of Excellence (RIIE), Walailak University, for the funding provided under the Individual Research Grant.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
This work was supported by Walailak University under the Individual Research Grant (Contract Number WU66235).
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
The data supporting this study’s findings are available in this article and its supplementary material. Further data are available from the corresponding author upon reasonable request.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors, and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declare that they have not used AI-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Jaiswal N, Akhtar J, Singh SP, Ahsan F. An overview on genistein and its various formulations. Drug Res. 2018;69:305–13. CrossRef
2. Goh Y, Jalil J, Lam D, Husain K, Premakumar C. Genistein: a review on its anti-inflammatory properties. Front Pharmacol. 2022;13:820969. CrossRef
3. Yoon GA, Park S. Antioxidant action of soy isoflavones on oxidative stress and antioxidant enzyme activities in exercised rats. Nutr Res Pract. 2014;8:618–24. CrossRef
4. Chen X, Wu Y, Gu J, Liang P, Shen M, Xi J, et al. Anti-invasive effect and pharmacological mechanism of genistein against colorectal cancer. BioFactors. 2020;46:620–8. CrossRef
5. Sharifi-Rad J, Quispe C, Imran M, Rauf A, Nadeem M, Gondal TA, et al. Genistein: an integrative overview of its mode of action, pharmacological properties, and health benefits. Oxid Med Cell Longev. 2021;2021(1):3268136. CrossRef
6. Qiu C, Zhang Y, Fan Y, Li S, Gao J, He X, et al. Solid dispersions of genistein via solvent rotary evaporation for improving solubility, bioavailability, and amelioration effect in HFD-induced obesity mice. Pharmaceutics. 2024;16:306. CrossRef
7. Kwon SH, Kang MJ, Huh JS, Ha KW, Lee JR, Lee SK, et al. Comparison of oral bioavailability of genistein and genistin in rats. Int J Pharm. 2007;337(1):148–54. CrossRef
8. Meteoglu I, Erdemir A. Genistein and temozolomide-loaded polymeric nanoparticles: a synergistic approach for improved anti-tumor efficacy against glioblastoma. Process Biochem. 2021;110:9–18. CrossRef
9. Zhang X, Huang Y, Zhu H, Liu Z, Zhang L, Li Z, et al. Genistein microparticles prepared by antisolvent recrystallization with low-speed homogenization process. Food Chemistry. 2022;408:135250. CrossRef
10. Cheng Q, Qin W, Yu Y, Li G, Wu J, Zhuo L. Preparation and characterization of PEG-PLA genistein micelles using a modified emulsion-evaporation method. J Nanomat. 2020;2020:1–15. CrossRef
11. Vu Q, Fang CW, Suhail M, Wu PC. Enhancement of the topical bioavailability and skin whitening effect of genistein by using microemulsions as drug delivery carriers. Pharmaceuticals. 2021;14:1233. CrossRef
12. Wang Z, Li Q, An Q, Gong L, Yang S, Zhang B, et al. Optimized solubility and bioavailability of genistein based on cocrystal engineering. Nat Prod Bioprospect. 2023;13:30. CrossRef
13. Huang Y, Dai WG. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm Sin B. 2014;4(1):18–25. CrossRef
14. Tambe S, Jain D, Meruva S, Rongala G, Juluri A, Nihalani G, et al. Recent advances in amorphous solid dispersions: preformulation, formulation strategies, technological advancements and characterization. Pharmaceutics. 2022;14:2203. CrossRef
15. Malkawi R, Malkawi W, Al-Mahmoud Y, Tawalbeh J. Current trends on solid dispersions: past, present, and future. Adv Pharmacol Pharm Sci. 2022;2022:1–17. CrossRef
16. Phuong T, Pyo YC, Kim DH, Lee SE, Kim JK, Park JS. Overview of the manufacturing methods of solid dispersion technology for improving the solubility of poorly water-soluble drugs and application to anticancer drugs. Pharmaceutics. 2019;11:132. CrossRef
17. Bhujbal S, Mitra B, Jain U, Gong Y, Agrawal A, Karki S, et al. Pharmaceutical amorphous solid dispersion: a review of manufacturing strategies. Acta Pharm Sin B. 2021;11:2505–36. CrossRef
18. Yao X, Qi C, Sun C, Huo F, Jiang X. Poly(ethylene glycol) alternatives in biomedical applications. Nano Today. 2023;48:101738. CrossRef
19. Alshehri S, Imam SS, Altamimi MA, Hussain A, Shakeel F, Elzayat E, et al. Enhanced dissolution of luteolin by solid dispersion prepared by different methods: physicochemical characterization and antioxidant activity. ACS Omega. 2020;5(12):6461–71. CrossRef
20. Fazil M, Ansari SH, Ali J. Development and evaluation of solid dispersion of spironolactone using fusion method. Int J Pharm Investig. 2016;6(1):63. CrossRef
21. Yusuf H, Meidy Nurintan Savitri O, Primaharinastiti R, Agus Syamsur Rijal M. A lyophilized surfactant-based rutin formulation with improved physical characteristics and dissolution for oral delivery. Saudi Pharm J. 2023;31(6):1077–83. CrossRef
22. Hurley D, Potter CB, Walker GM, Higginbotham CL. Investigation of ethylene oxide-co-propylene oxide for dissolution enhancement of hot-melt extruded solid dispersions. J Pharm Sci. 2018;107(5):1372–82. CrossRef
23. Zhang J, Guo M, Luo M, Cai T. Advances in the development of amorphous solid dispersions: the role of polymeric carriers. Asian J Pharm Sci. 2023;18:100834. CrossRef
24. Sermkeaw N, Plyduang T. Development of delayed-release matrix tablets comprising solid dispersion of mefenamic acid. Trends Sci. 2023;20(6):6078. CrossRef
25. Tran P, Nguyen TN, Park JS. Co-carrier-based solid dispersion of celecoxib improves dissolution rate and oral bioavailability in rats. J Drug Deliv Sci Tech. 2023;79:104073. CrossRef
26. Patel K, Patel J, Shah S. Development of delayed release oral formulation comprising esomeprazole spray dried dispersion utilizing design of experiment as an optimization strategy. AAPS PharmSciTech. 2023;24(7):186. CrossRef
27. Mohapatra D, Kumar D, Shreya S, Pandey V, Dubey P, Agrawal A, et al. Quality by design-based development and optimization of fourth-generation ternary solid dispersion of standardized Piper longum extract for melanoma therapy. Drug Deliv Transl Res. 2023;13:3094–131. CrossRef
28. Fayyaz Shahandashty B, Fallah N, Shamsi M, Nasernejad B, Afkhamipour M. Evaluation of enhanced chemical coagulation method for a case study on colloidal liquid particle in wastewater treatment: statistical optimization analysis and implementation of machine learning. J Environ Manage. 2024;370:122345. CrossRef
29. ACCSQ, PPWG. ASEAN guideline on stability study of drug product. Jakarta, Indonesia: ASEAN Secretariat; 2005.
30. Emam M, Taha N, Emara L. A novel combination of Soluplus® and Poloxamer for Meloxicam solid dispersions via hot melt extrusion for rapid onset of action—part 1: dissolution and stability studies. J Appl Pharm Sci. 2020;11:141–50. CrossRef
31. Suhail M, Chiu IH, Lin IL, Tsai MJ, Wu PC. A novel approach of polyethylene glycol-4000 hydrogels as controlled drug carriers. Micro. 2023;3:578–90. CrossRef
32. Garbiec E, Rosiak N, Zalewski P, Tajber L, Cielecka-Piontek J. Genistein co-amorphous systems with amino acids: an investigation into enhanced solubility and biological activity. Pharmaceutics. 2023;15(12):2653. CrossRef
33. Xiao Y, Ho CT, Chen Y, Wang Y, Wei Z, Dong M, et al. Synthesis, characterization, and evaluation of genistein-loaded zein/carboxymethyl chitosan nanoparticles with improved water dispersibility, enhanced antioxidant activity, and controlled release property. Foods. 2020;9(11):1604. CrossRef
34. Li X, Liu X, Song J, Wang C, Li J, Liu L, et al. Drug–drug cocrystallization simultaneously improves pharmaceutical properties of genistein and ligustrazine. Cryst Growth Des. 2021;21(6):3461–8. CrossRef
35. Amaral V, Souza J, Alves T, Batain F, Crescencio K, Komatsu D, et al. Chemical processing and physical-chemical and mechanical characterizations of poly (L-co-D, L lactic acid)/polyethylene glycol mixtures for application as a biomedical device. Adv Mater Lett. 2024;15:1–9. CrossRef
36. Lu X, Huang H, Zhang X, Lin P, Huang J, Sheng X, et al. Novel light-driven and electro-driven polyethylene glycol/two-dimensional MXene form-stable phase change material with enhanced thermal conductivity and electrical conductivity for thermal energy storage. Compos B Eng. 2019;177:107372. CrossRef
37. Slámová M, Školáková T, Školáková A, Patera J, Zamostny P. Preparation of solid dispersions with respect to the dissolution rate of active substance. J Drug Deliv Sci Technol. 2020;56:101518. CrossRef
38. Liu P, Zhou JY, Chang JH, Liu XG, Xue HF, Wang RX, et al. Soluplus-mediated diosgenin amorphous solid dispersion with high solubility and high stability: development, characterization and oral bioavailability. Drug Des Devel Ther. 2020;14:2959–75. CrossRef
39. Samanthula K, Gna L, Reddy J, Hoque N, Sen S, Bairi A, et al. Design, development and influence of super disintegrants on solid dispersion based taste masking orodispersible tablets of racecadotril. Eur Chem Bull. 2023;12:12474–500. CrossRef
40. Iffat W, Shoaib MH, Yousuf RI, Qazi F, Mahmood ZA, Muhammad IN, et al. Use of Eudragit RS PO, HPMC K100M, ethyl cellulose, and their combination for controlling nicorandil release from the Bilayer tablets with atorvastatin as an immediate-release layer. J Pharm Innov. 2022;17(2):429–48. CrossRef
41. Zhao J, Koo O, Pan D, Wu YM, Morkhade D, Rana S, et al. The impact of disintegrant type, surfactant, and API properties on the processability and performance of roller compacted formulations of acetaminophen and aspirin. AAPS J. 2017;19:1387–95. CrossRef
SUPPLEMENTARY MATERIAL
The supplementary material can be accessed from the link here: [https://japsonline.com/admin/php/uploadss/4486_pdf.pdf]