
INTRODUCTION
The pulmonary route of administration is an approach for both local and systemic delivery of drugs, due to the large surface area and direct connection between systemic circulation and pulmonary. The limitations of pulmonary delivery include drug irritation to the lung, limited drug dissolution, and high drug clearance. Devices for delivering medications by inhalation include inhalers, nebulizers, and dry powder inhalers. Particle size distribution plays an important role in penetration and deposition in the airways. Particles ranging from 5 µm to 0.5 µm are deposited in the alveoli, particles >10 µm impact the walls of the throat and do not reach the lungs, and particles size <0.5 µm are exhaled [1].
Favipiravir was initially approved in Japan as an anti-influenza agent and later during the COVID-19 pandemic crisis, it was approved by DCGI (Drug Controller General of India) as an antiviral agent against SARs-CoV-2 [2]. Favipiravir functions as a prodrug and undergoes ribosylation and phosphorylation intracellularly to become the active favipiravir-RTP. Favipiravir-RTP binds to and inhibits RNA-dependent RNA polymerase, which ultimately prevents viral transcription and replication. Favipiravir-RTP is incorporated into a nascent RNA strand, it prevents RNA strand elongation and viral proliferation [3]. The daily dose for favipiravir is 1,600 mg, and the pulmonary dose is 5% of the oral dose (5% of 1,600 mg is 80 mg) taken as a dry powder inhaler [4]. Favipiravir is available in the market as tablets, the downside is the high dose. Research has been done to formulate favipiravir with less dose but there is no evident solution to date. To target favipiravir in the lungs, the main challenge is particle size. Carriers available for pulmonary drug delivery are microparticles, microspheres, proliposomes, microsponges, and so on.
Microsponges, polymeric porous highly cross-linked microparticulate systems, imbibe water and swell, thereby having slow release at the target site achieving novel drug delivery [5].
The main objective of the present work is to reduce the dose and target the drug to pulmonary to treat the infection caused by COVID-19. Administered by inhalers, which is a pressurized container delivering a fixed amount by the actuation of a valve. In the present study, favipiravir microsponges are optimized using Box Behnken Design (BBD). In-vitro parameters, lung deposition studies were performed using twin-stage impinger.
MATERIAL AND METHODS
Materials
Favipiravir was purchased from Hubert Drugs Pvt.Ltd, ethyl cellulose, disodium hydrogen phosphate, dichloromethane, and PVA were purchased from SD Fine-Chem Limited, dimethyl sulfoxide was procured from Sisco Research Laboratories Pvt. Ltd, and potassium dihydrogen phosphate was purchased from Molychem.
Methods
Preparation of favipiravir microsponges
The emulsion solvent diffusion approach was used to prepare the favipiravir microsponges. To produce the internal phase, 15 ml of dichloromethane is used to dissolve ethyl cellulose. Next, 1 ml of dimethyl sulfoxide is used to dissolve the drug favipiravir, which is then added to the ethyl cellulose solution and mixed thoroughly using a magnetic stirrer. To prepare the external phase, dissolve the weighed amount of PVA in 100 ml of distilled water. The emulsion is prepared by adding the internal phase to the external phase drop by drop at a rate of 10 ml/minute while stirring continuously. Subsequently, a high-speed homogenizer is used to agitate the emulsion for approximately 3 hours at speeds between 1,000 and 1,400 rpm to evaporate the solvent. The prepared microsponges were dried in an oven at 40°C for 24 hours after being filtered through Whatman filter paper [5].
Optimization using BBD
For the optimization process to investigate quadratic response surfaces and create second-order polynomial models using Design Expert 11 (Version 11; Stat-Ease Inc, Minneapolis, MN), a three-factor, three-level BBD was chosen [6]. This resulted in the selection of three independent variables at three different levels: X1, ethyl cellulose quantity, X2, PVA quantity, and X3, speed (RPM) (low, medium, and high). Three criteria were chosen as dependents: R1, percent production yield; R2, entrapment efficiency; and R3, in-vitro drug release (Table 1). According to the design used, 15 formulations were created and assessed (Table 2) [7].
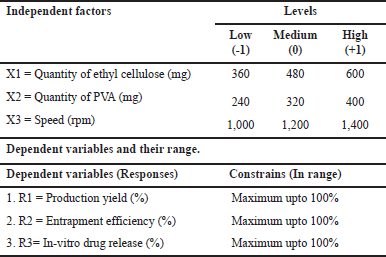 | Table 1. Box-Behnken independent and dependent variables. [Click here to view] |
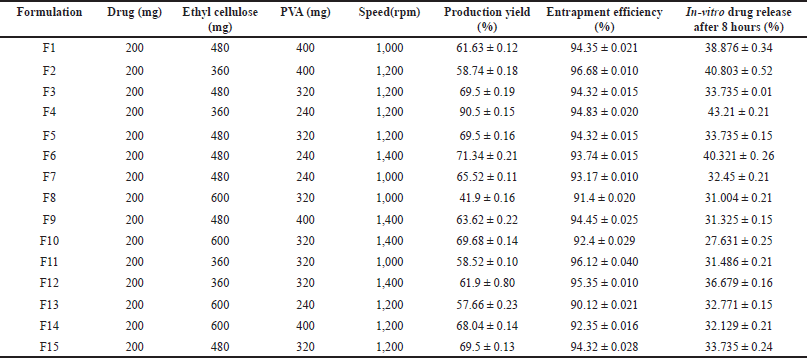 | Table 2. Formulations according to Box-Behnken design with observed values. [Click here to view] |
Evaluation
Percent production yield (%)
The following formula is used to get the percent production yield [8].
Assay
5 mg of equivalent favipiravir microsponges are taken and calculated for the concentration of the drug in comparison with placebo using the UV spectrophotometric method at 238 nm [9].
Flow properties
Angle of repose
The following formula was used to calculate the angle of repose [10], Tan θ = h/r, where θ = angle of repose, h = height of the heap (in cm), and r = radius of the base (in cm).
Compressibility index
The following equation was used to calculate Carr’s index using the bulk density (ρb) and tapped density (ρt) values from the earlier experiments [10].
Hausner’s ratio
Hausner’s ratio is an indirect index of ease of powder flow. It is calculated by the following formula, Hausner’s
ratio = ρt/ρb, where ρt is tapped density and ρb is bulk density.
Lower Hausner’s ratio (<1.25) indicated better flow properties than higher ones [10].
Particle size measurement
Determined by the microscopic technique using micrometry.
Entrapment efficiency
Powder equivalent to 5 mg of the drug is dissolved in 10 ml phosphate buffer 6.8 pH. The solution is then centrifuged at 6,000 RPM for 1 hour. The supernatant is collected and filtered, 1 ml is taken and diluted to 10 ml with phosphate buffer 6.8 pH, and then examined using the UV spectrophotometer set to λmax 238 nm. The formula is
In-vitro diffusions studies
Studies for 15 formulations were carried out in an open-end tube using a dialysis bag diffusion technique, in phosphate buffer pH 6.8. For activation, the dialysis bag was immersed in phosphate buffer solution for 12 hours to saturate. The formulation is equivalent to 5 mg of the drug and is dispersed in a phosphate buffer solution of 1 ml and put into the dialysis bag with one end sealed of the open-end tube and immersed in 200 ml of the release medium. The temperature was maintained at 37°C ± 0.5°C with continuous magnetic stirring at 100– 200 rpm. Aliquots up to 1 ml are withdrawn at predetermined intervals up to 8 hours, and sink conditions are maintained. The samples are analyzed by UV spectrophotometry at 238 nm after suitable dilutions. Percentage drug release is calculated accordingly [11].
Lung deposition studies
Twin stage impinger was employed for the determination of an In-vitro lung model for aerosolization using a nebulizer. The twin-stage impinger comprises of mouthpiece to which the nebulizer is attached, an upper stage (stage 1), and a lower stage (stage 2). Stage 1 and stage 2 represent the upper and lower airways of the respiratory tract, respectively. The deposition eligibility is based on the assumption of the application of 60 l/minute negative pressure by a vacuum pump. During assembly, the upper and lower stages are filled with phosphate buffer pH 6.8 of 7 ml and 30 ml, respectively. Powder equivalent to 5 mg of the drug is taken in the nebulizer and dissolved by adding 5 ml of phosphate buffer solution attached to the mouthpiece of the twin-stage impinger, post nebulization the solutions from stages 1 and 2 are collected and diluted with phosphate buffer. Absorbance is determined spectrophotometrically at 238 nm, and furthermore, the parameters such as mass output, respirable fraction, aerosol output rate, nebulization time, and so on, are determined [12].
Nebulization time
The time required for the aerosol to become erratic and intermittent was determined as nebulization time.
Mass output
After nebulization, i.e., after achieving complete dryness the mass output is determined by calculating the difference in the mass (weight) of the microsponges formulation before and after the nebulization. The percentage mass output is determined by using the following formulae.
Aerosol output rate (Mg/Min)
Respirable fraction
The respirable fraction is determined by calculating the drug mass in stage 2 to the drug mass loaded in nebulizer prior to the nebulization. The respirable fraction is calculated by using the following formulae:
Evaluation of the optimized formulation
Furthermore, the optimized formulation (PMS) and the solutions from the twin-stage impinger stage 1 and stage 2 are evaluated for the SEM, zeta potential, particle size, and Thermogravimetric analysis (TGA) [13].
SEM
Scanning electron microscopy was used to analyze the surface morphology of the optimized favipiravir microsponge formulation as well as the solutions obtained from stage 1 and stage 2 of the twin-stage impinger.
Zeta potential and particle size
Using a zetasizer Malvern analyzer, Zeta potential, and particle size for the optimized favipiravir microsponges were carried out at the temperature of 25°C, conductivity of mS/cm at 39% and 100% intensity given in size (d. nm).
DSC
3– 5 mg samples were weighed and placed in closed, hermetic sample pans with pinhole. Samples were heated from 0°C to 210.0°C. The disappearance of the crystalline sharp peak of the drug and the appearance of any new peak and peak shape were noted. The thermogram of the optimized favipiravir microsponge formulation was superimposed with that of a pure drug [14].
XRD
The material under analysis is homogenized, and finely powdered, and its average bulk composition is determined. An X-ray beam is pointed at the sample in this test procedure, and the dispersed intensity is measured as a function of the outgoing direction. The scatter, also known as a diffraction pattern, shows the crystalline structure of the material once the beam has been separated [15].
Thermal analysis
TGA and differential thermal analysis (DTA) are used to determine the thermal stability of the material. An amount of 3.202 mg of drug-loaded microsponges was placed in platinum pans and sealed before the test. All samples were run at a heating rate of 10ºC/minute over a temperature range of 10°C– 1,000°C in an atmosphere of nitrogen [16].
Stability studies
Optimized formulation was stored at accelerated stability conditions (40°C ± 2°C/75% ± 5% RH) as per ICH guidelines throughout 1 month and evaluated for physical appearance, entrapment efficiency, assay, drug content, and in-vitro drug release for every week [17].
RESULTS AND DISCUSSION
Evaluation
15 formulations were evaluated for the percent production yield (Table 2), assay (90.24% ± 0.05% to 96.479% ± 0.01%), flow properties, Carr’s index within 9.09 ± 0.15–20% ± 0.25% range, Hauser’s ratio within 1.1 ± 0.19–1.25 ± 0.22 range, and angle of repose within 31.45 ± 0.16°–39.69 ± 0.10° range indicating that they have good-fair flow properties. The mean particle size of 15 formulations was in the 10– 45 μm range. Out of these formulations, F4 showed a high percentage yield, high drug content, and excellent flow properties. F2 formulation showed the highest entrapment efficiency (Table 2).
In-vitro diffusions studies
In-vitro diffusion studies were performed for all formulations for 8 hours, the release of the drug (3 hours) is constant throughout the time, formulation F4 showed maximum drug release at the end of 8 hours indicating the longer retention of the favipiravir in the pulmonary region. The dissolution profile of all formulations is represented in Figure 1. Model-dependent kinetics were performed for the formulations and they follow first-order kinetics and the Higuchi drug release mechanism [18].
 | Figure 1. In-vitro drug diffusion of favipiravir microsponges (a) F1 – F5 (b) F6 – F10 (c) F11– F15. [Click here to view] |
Lung deposition studies
Lung deposition studies were performed; maximum nebulization time was shown by F2. The mass output evaluation shows that no optimum or 100% mass output is reached after nebulization. F4 formulation shows maximum aerosol output rate. F4 formulation shows a maximum value of respirable fraction (Table 3) indicating the lower value of output rate, mass output, and respirable fraction hence the drug delivery through microsponges via nebulization is difficult therefore the alternate procedure such as the dry powder inhalers such as rota haler can be used as alternate method to deliver the complete drug.
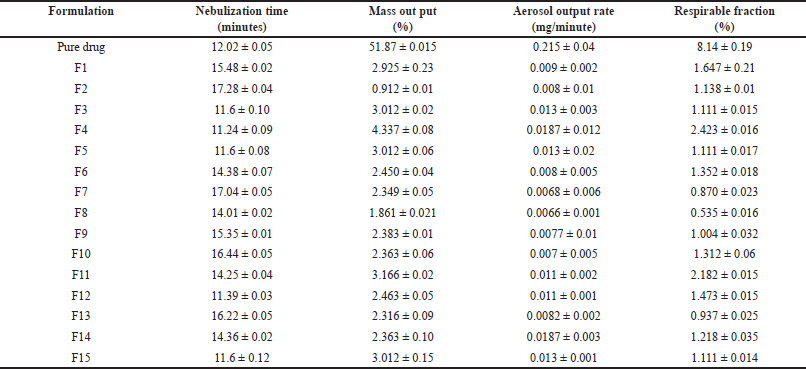 | Table 3. Results of lung deposition studies. [Click here to view] |
Optimization by BBD
Mathematical relationship between the independent factor and dependent factor are statistically related to the model considering the following parameters F value greater than 1, p value less than 0.05, difference between adjusted R2 and predicted R2 should not be more than 0.2, adequate precision should be greater than 4, lack of fit should be insignificant. Based on these the design space can be navigated using the model. These responses are given in Table 4, where the quadratic model is significant for production yield and in-vitro drug release, and the linear model for entrapment efficiency.
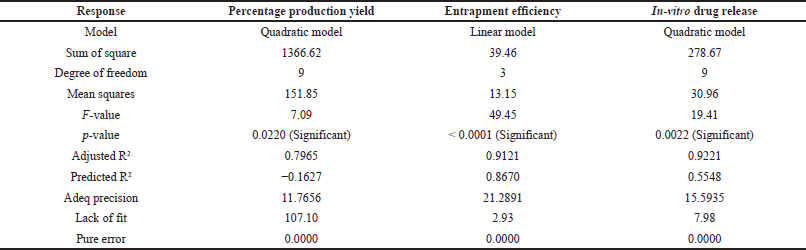 | Table 4. Fit statistics of observed responses. [Click here to view] |
From the 2D contour plots and 3D response surface plots (Fig. 2), it is observed with an increase in the quantity of ethyl cellulose there is a decrease in the percentage production yield, entrapment efficiency (%), and in-vitro drug release. Similarly, with an increase in the quantity of PVA, there is an increase in percentage production yield initially and a further increase in PVA resulted in a decrease in percentage production yield, increase in entrapment efficiency (%), and in vitro drug release. The design facilitates simultaneous assessment of the impact of independent variables and their actual significance on selected responses, where the quantity of ethyl cellulose was significant on entrapment efficiency (%) (p-value < 0.0001) and in-vitro drug release (p-value 0.0012), the quantity of PVA was significant on entrapment efficiency (%) (p-value 0.0018), and speed was significant on production yield (p-value 0.0309). The interaction effect of quantity of EC and PVA was significant on production yield (p-value 0.0061), quantity of EC and speed was significant on production yield (p-value 0.0462), and in-vitro drug release (p-value 0.0388). Quantity of PVA and speed was significant on in-vitro drug release (p-value 0.0041). The actual responses and predicted responses had in high degree of correlation (Fig. 2), with R2 of 0.9273 for production yield, R2 of 0.931 for entrapment efficiency (%), and R2 of 0.9357 for in-vitro drug release.
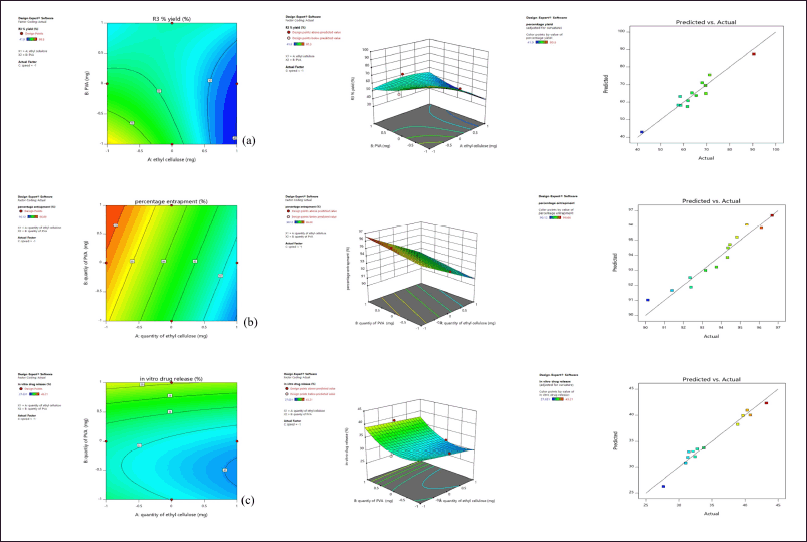 | Figure 2. (a) Effect on percentage production yield (b) Effect on Entrapment Efficiency (c) Effect on in-vitro drug release showing contour plot, response surface plot and Predicted versus Actual response. [Click here to view] |
Among the formulations, least percentage production yield (41.9% ± 016%) was by F8 containing 600 mg EC, 320 mg PVA, and 1,000 rpm, lowest EE (%) was by F13 containing 600 mg EC, 240 mg PVA, and 1,200 rpm and less in-vitro drug release (27.631% ± 0.25%) was by F10 containing 600 mg EC, 320 mg PVA, and 1,400 rpm. In F8, F10, and F13, there is an increase in drug to polymer ratio, where a similar effect was observed with curcumin microsponges [5] and ketoprofen microsponges [8]. Maximum in-vitro drug release (43.21% ± 0.21%), percentage production yield (90.5% ± 0.15%) was by F4 containing 360 mg EC, 240 mg PVA, and 1,200 rpm, and highest EE (%) by F2 containing 360 mg EC, 400 mg PVA, and 1,200 rpm where in these formulations minimum amount of EC was used.
Using a higher amount of PVA resulted in a slight increase in the dispersed phase viscosity when all the solvents diffused out, and nearly all the dispersed phases converted into solid microsponges. An increase in the polymer resulted in the thickening of the polymer matrix wall leading to the extended diffusion path and decrease in the drug release. A mathematical equation of the effect of independent variables on responses is given, where the +ve sign symbolizes the additive effect of that variable on the response and the -ve sign, and vice versa.
Quadratic equation for percent production yield (%) = +69.50 −4.05A −4.12B +4.87C +10.54AB +6.1AC - 0.9575BC −4.15A2+3.38B2 - 7.35C2.
Linear equation for Entrapment efficiency (%) = +93.86 −2.09A +0.7462B +0.1125C.
Quadratic equation for In-vitro drug release (%) =+33.74−3.96A−0.7024B +0.6425C+0.4413AB−2.89AC−3.86BC+0.1001A2+3.39B2−1.39C2.
From Table 4, for in-vitro drug release, the difference between adjusted R2 and predicted R2 is greater than 0.2 indicating model reduction. The reduced quadratic model was significant with p value < 0.0001, adjusted R2 (0.8981), and predicted R2 (0.7727), where the difference is less than 0.2 and adequate precision 16.84, which is greater than 4. Indicating it can be used to navigate the design space.
Reduced quadratic equation for In-vitro drug release (%)=+33.56−3.58A−2.14AC−3.85BC+3.79B2−1.74C2.
Similarly, for production yield, a negative value of predicted R2 is not satisfactory, so a reduced quadratic model was analyzed.
Reduced quadratic equation for production yield (%) = 69.06+4.87C+10.54AB−7.30C2.
The solutions for the optimized favipiravir microsponge formulation were given by design expert software 11 with the criteria for numerical optimization maximum percent production yield, maximum entrapment efficiency, and least release of the drug. This was represented in graphical optimization as an overlay plot. In the overlay plot (Fig. 3), the criteria of highest percentage production yield were taken in the probability of 74.9413%–99.655%, highest EE % (94.154%–96.2812%), and least release in the range of 39.8378%–45.826%, which are given by software on the overlay plot (representation of graphical optimization). Out of 53 solutions predicted by software, a solution having a desirability of 0.900 was selected. Which is X1, EC quantity as −0.99, X2 (PVA) as −0.99, and speed 0.122 coded values. Responses were also predicted, 87.2981% yield, 95.2176%% EE, 43.21% release. The formulation (PMS) was decoded 372 mg EC, 248 mg PVA, and a speed of 1222 rpm, prepared and evaluated. The observed responses were in correlation with predicted responses with R2=1. Among all formulations F4 and F2 formulations were better, so optimized formulation close to these was given by software.
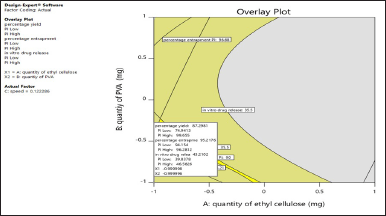 | Figure 3. Overlay plot of optimized formulation by graphical optimisation. [Click here to view] |
Evaluation of the optimized formulation
The optimized formulation PMS were evaluated for percent production yield, flow properties, lung deposition studies, and so on, results are given in Table 5.
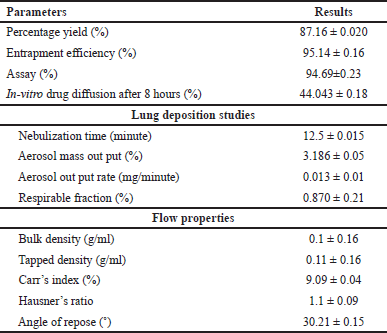 | Table 5. Evaluation parameters of optimized formulation. [Click here to view] |
SEM
The optimized favipiravir microsponges’ surface morphology was examined Figure 4 displays the microsponges’ spherical shape and nano-sized pores, as well as their homogeneous and non-segmented nature. SEM images of stage 1 and stage 2 samples of optimized formulation PMS show swelling of microsponges by imbibing solvent.
 | Figure 4. (a) SEM of PMS (b) SEM of PMS solution obtained from twin stage impinger stage1 (c) SEM of PMS solutions obtained from twin stage impinger stage2. [Click here to view] |
Zeta potential
Optimized favipiravir microsponge formulation (PMS) zeta potential was found as +1.04mv (Fig. 5) as a charge inducer was not used. A Zeta potential of ±30 mv for a system is considered to be stable.
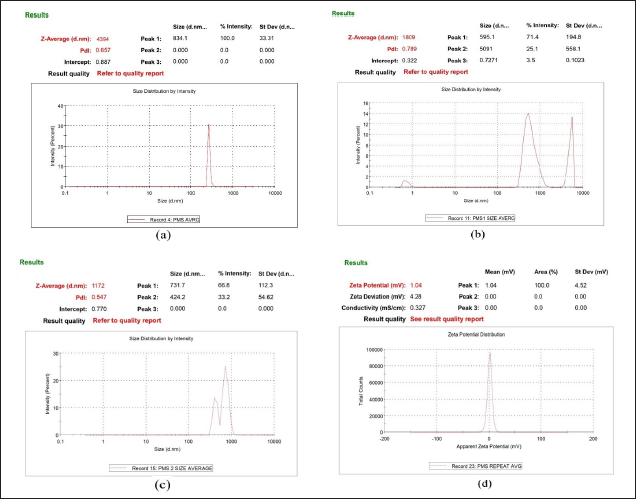 | Figure 5. (a) size of particles of PMS (b)Particle size of solution of optimized formulation at stage1 and (c) stage2 of twin stage impinger (d) Zeta potential of PMS. [Click here to view] |
Particle size
The size of the particles in the favipiravir microsponge formulation (PMS) was 4.394 ± 0.35μm (Fig. 5). The particle size of optimized formulation under twin stage impinger stage 1 and stage 2 was found 1.809 μm and 1.172 μm, respectively. Particles range 0.5μm– 5μm in size and are usually the most effective for inhalation and deposition in the alveoli of the lungs. Particles less than 1 μm are more likely to be exhaled again, while larger particles tend to deposit in the upper part of the respiratory tract [1]. Poly dispersibility index (PDI) of PMS is 0.416 indicating uniformity of particle size, whereas samples of stage 1 and stage 2 had PDI greater than 0.5 indicating non-uniformity of particles (represented as bimodal and trimodal peaks in Fig. 5), as microsponges powder sample is dispersed in phosphate buffer solvent leading to increase or decrease of the size of the particles in the process of solubilization.
DSC
Studies showed that, when comparing the physical mixture to the pure medication, there is minimal variation in the favipiravir drug’s melting point. Since the blend was a physical mixture containing ethyl cellulose, the melting point of the drug was almost 190°?, and the polymer peaks were produced at that same temperature (Fig. 6).
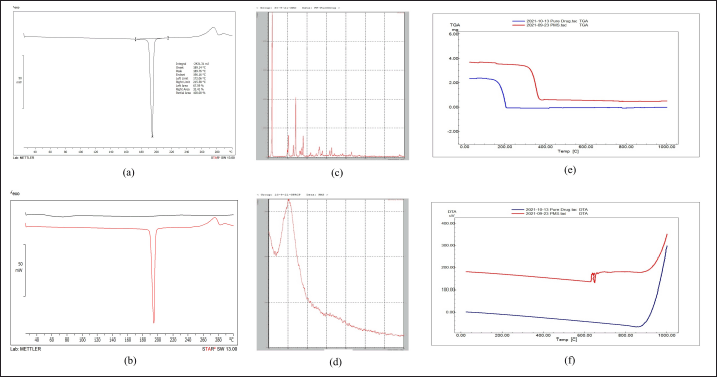 | Figure 6. (a) DSC of pure drug (b) DSC of pure drug and PMS (c) XRD of pure drug (d) XRD of PMS (e) TGA of pure drug and PMS (f) DTA of pure drug and optimized PMS. [Click here to view] |
XRD
Sharp peaks can be seen in the favipiravir spectra at 11.68?, 22.9?, 27.93?, 41.66?, and 52.75?(2θ) which indicates the drug is crystalline. The optimized favipiravir microsponges’ diffractogram displays peaks at 10.890?, 22.52?, 27.63?, 41.77?, and 51.66? (2θ) with decreased intensity which indicates a decrease in crystalline nature of the microsponges (Fig. 6). DSC and XRD studies confirm the conversion of a crystalline form of a pure drug to an amorphous form and excipient compatibility.
Thermal analysis
TGA thermograms of optimized favipiravir microsponge formulation showed a downshift which indicates the loss of mass due to evaporation of solvent, loss of moisture, and degradation. Weight loss in the region 300°C– 400°C indicates degradation of the amorphous form of the entrapped drug in the microsponges. The DTA of favipiravir pure drug is compared with the DTA of PMS. At 650°C–670°C, a DTA effect occurs but this effect not occurs on the TG curve, because this event corresponds to the recrystallization of the dehydrated material. This recrystallization process is exothermic. TG-DTA modes determine the melting points, glass transition temperatures, crystallinity, moisture volatile content, thermal and oxidative stability, purity, and transformation temperatures (Fig. 6).TGA and DTA studies prove the characteristics of microsponges wherein they remain stable upto 130°C within a pH range of 1–11 [18].
Stability studies
Accelerated stability studies were performed for 1 month as per ICH guidelines and parameters were evaluated at respective time intervals (Table 6). There was no change in the physical appearance of microsponges, whereas EE% varied within ±1%, assay within ±3%, and in-vitro drug release within ±3%, where it is less than 5%. Therefore, there is no significant change in physical appearance, EE%, assay, and in-vitro drug release studies.
 | Table 6. Stability studies for optimized favipiravir microsponge formulation (PMS). [Click here to view] |
CONCLUSION
Favipiravir microsponges for pulmonary drug delivery were optimized using the BBD in the study based on the objective of reducing the dose and targeting the lungs. The formulation prepared was evaluated for physiochemical parameters and lung deposition. From lung deposition studies it was observed the output rate, mass output, and respirable fraction were less as microsponges containing ethyl cellulose, were dispersed in 6.8 pH phosphate buffer for nebulization, and were not completely solubilized in the media. Hence dry powder inhalation using a rota haler can be suggested. SEM and particle size parameters prove they can be targeted to alveoli (target area of the symptom) and non-uniformity of particles in stage 1 and stage 2 indicates the release of drug or biodegradability of the polymer. From thermal analysis, it is proven microsponges are stable upto 130°C. Stability studies show formulation is stable. Therefore, we can conclude favipiravir microsponges at a very reduced dose when compared to oral dose can be targeted for pulmonary using formulations such as dry powder inhalers.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FUNDING
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Shah ND, Shah VV, Chivate ND. Pulmonary drug delivery: a promising approach. J Appl Pharm Sci. 2012;02(06):33–7. CrossRef
2. Agrawal U, Raju R, Udwadia ZF. Favipiravir: a new and emerging antiviral option in COVID-19. Med J Armed Forces India. 2020 Oct;76(4):370–6. CrossRef
3. Hung DT, Ghula S, Aziz JMA, Makram AM, Tawfik GM, Abozaid AA, et al. The efficacy and adverse effects of favipiravir on patients with COVID-19: a systematic review and meta-analysis of published clinical trials and observational studies. Int J Infect Dis. 2022 Jul;120:217–27. CrossRef
4. Lau M, Young PM, Traini D. A review of co-milling techniques for the production of high dose dry powder inhaler formulation. Drug Dev Ind Pharm. 2017 Aug;43(8):1229–38. CrossRef
5. Bhatia M, Saini M. Formulation and evaluation of curcumin microsponges for oral and topical drug delivery. Prog Biomatter. 2018 Sep;7(3):239–48. CrossRef
6. Gade R, Nama S, Avula PR. Formulation development and evaluation of once daily fexofenadine hydrochloride microsponge tablets. Trends Sci. 2022;20(1):4271. CrossRef
7. Moin A, Roohi NKF, Rizvi SMD, Ashraf SA, Siddiqui AJ, Patel M, et al. Design and formulation of polymeric nanosponges tablets with enhanced solubility for combination therapy. RSC Adv. 2020 Sep 21;10(57):34869–84. CrossRef
8. Como?lu T, Gönül N, Baykara T. Preparation and in-vitro evaluation of modified release ketoprofen microsponges. Farmaco. 2003 Feb;58(2):101–6. CrossRef
9. Ibrahim B. HPLC-UV method for quantification of favipiravir in pharmaceutical formulations. Acta Chromatogr. 2021;33(3)209–15. CrossRef
10. Subrahmanyam CVS. Micromeritics. Text book of Physical Pharmaceutics, 2nd ed. Delhi, India: VallabhPrakashan publishers; 2000.
11. Mali AJ, Rokade A, Kamble R, Pawar A, Bothiraja C. Resveratrol-loaded microsponge as a novel biodegradable carrier for dry powder inhaler: a new strategy in lung delivery. Bio Nanosci. 2021;11:1–12. CrossRef
12. Khan I, Lau K, Bnyan R, Houacine C, Roberts M, Isreb A, et al. A facile and novel approach to manufacture paclitaxel-loaded proliposome tablet formulations of micro or nano vesicles for nebulization. Pharm Res. 2020 Jun 2;37(6):116. CrossRef
13. Pandey P, Mishra S, Anupriya K, Sharma N, Kumari Ppa. Tolnaftate microsponges embedded biocompatible gels for controlled and effective antidermatophytic activity. Int Res J Pharm. 2018;9(6):128–33. CrossRef
14. Mahant S, Kumar S, Nanda S, Rao R. Microsponges for dermatological applications: perspectives and challenges. Asian J Pharm Sci. 2020;15(3):273–91. CrossRef
15. Suryanarayana R. X ray powder diffractometry physical characterization of pharmaceutical solids. Boca Raton, FL: CRC press; 1995. pp 187–221.
16. Giordano F, Novak C, Moyano JR. Thermal analysis of cyclodextrins and their inclusion compounds. Thermochimica Acta. 2001;380(2):123–51. CrossRef
17. Raut AV, Kathar N, Sanap G. Microsponges: the drug delivery system. Int J Pharm Sci. 2024;2(1):328–41. CrossRef
18. Vidya KP, Gopinath E, Nesalin JA, Chandy V. Unveiling the potential of microsponges: enhancing oral bioavailability. World J Biol Pharm Health Sci. 2024;17(02):405–14. CrossRef