
INTRODUCTION
Diabetes Mellitus (DM) represents a multifaceted metabolic disorder stemming from various causes, including deficiencies in insulin secretion and/or action. DM disrupts glucose homeostasis, inducing metabolic alterations and complications. Effective management strategies are vital to mitigate long-term health risks associated with glycemic dysregulation and metabolic abnormalities.[1–2] Chalcones are a family of naturally occurring 1,3-diphenyl-2-propen-1-ones Figure 1 that have fascinated researchers due to their distinctive structure and the great potential they provide across a wide range of scientific fields [3]. Chalcones exhibit promising anti-inflammatory [4–8] and anticancer activities [9–12], suggesting their potential therapeutic role in managing these severe conditions [13]. Further exploration, particularly regarding their ability to influence diabetes [14], is a crucial area of future research.
Chalcones have been widely explored for several decades and have been used for treating diabetes [15]. Their potential lies in targeting glycemic control and regulating pathways involved in carbohydrate, lipid, and protein metabolism, offering comprehensive management strategies to mitigate long-term health risks associated with diabetes [16]. PPARγ, prominently expressed in adipose tissue, orchestrates adipogenesis and insulin sensitivity, making it a key player in metabolic homeostasis. Dysregulation of PPARγ signaling has been implicated in the pathogenesis of metabolic disorders, underscoring the significance of targeting PPARγ for therapeutic purposes [17]. An important function of PPARs is to regulate the transcription of several target genes that regulate adipocyte differentiation, glucose, and lipid metabolism, as well as insulin sensitivity and inflammation. Moreover, beyond their metabolic regulatory functions, PPARγ exerts pleiotropic effects encompassing cell proliferation, differentiation, angiogenesis, inflammation, and oxidative stress. [18]. Consequently, dysregulated PPARγ activity contributes to the development and progression of metabolic disorders. By targeting PPARγ, chalcones offer a comprehensive approach to addressing the multifaceted nature of metabolic dysfunction. Through their modulation of PPARγ activity, chalcones not only impact metabolic processes but also influence cellular functions crucial for overall metabolic health. Understanding the intricate mechanisms underlying chalcone-PPARγ interactions provides insights into novel therapeutic strategies for combatting metabolic disorders, including obesity, diabetes, and atherosclerosis [19,20]. In this work, we critically studied the article Chi-Ting Hsieh et.al. [21] entitled Synthesis of chalcone derivatives as potential anti-diabetic agents which were published in Bioorganic & Medicinal Chemistry Letters in 2012 [21]. This study employs a combined computational and experimental approach to screen 61 analogs of 1,3-diphenyl-2-propen-1-ones for potential anti-diabetic properties. Computational analyses predict interactions with key molecular targets involved in diabetes, while animal experiments assess direct effects on blood glucose levels. By correlating computational predictions with empirical data, researchers identify promising compounds for further development as anti-diabetic agents. This integrated strategy aims to advance diabetes treatment by identifying compounds with favorable pharmacological profiles, facilitating subsequent experimental validation and potential therapeutic advancements.
 | Figure 1. Structure of 1,3-diphenyl-2-propen-1-ones. [Click here to view] |
MATERIALS AND METHODS
Library creation
This article from Chi-Ting Hsieh et al. [21] provided the authors with information about chalcone analogs that could be used as bioactive. The chalcone analogs were set up using computational software, and they were converted into .pdb format [20] and are represented in Table 1. The initial energy associated with the molecules was minimized before to inhibit the hindrance of that with the binding energies. The .pdb files were further analyzed to determine their reactivity and stability. The results showed that the chalcone analogs had the potential to be used as bioactives. Before the binding energy was inhibited by the initial energy associated with the molecules, these energies were minimized so that the hindrance to that was prevented from occurring. This allowed the molecules to be bound together more tightly, resulting in a stronger bond. In addition, this minimized the energy required to keep the molecules together, leading to more efficient and stable bonds.
Molecular docking
The virtual screening of the bioactive molecules was carried out using molecular docking studies using the referenced software under standard protocol. The autodock software [21] under the Vina module was used for molecular docking studies. A crystal structure of human PPAR-gamma was obtained from the protein database (PDB ID: 2P4Y) (https://www.rcsb.org/) [22] as a .pdb format from the protein databank. Discovery Studio was used to prepare the protein and remove the pre-associated ligand from the protein during its preparation. In Discovery Studio, the active sites of the protein were defined using the define sites module.
It was necessary to construct the ligands as 2D structures using chemsketch software and then convert them to SMILES using Avogadro software. The SMILES were then optimized for the geometry using the Avogadro software. UFF force fields were used to minimize the energy of the molecules in Avogadro software’s energy minimization module, and the data were then saved in a file format known as .pdb [23]. Analysis of the results was done using the Maestro suite of Schrodinger software [24] and Discovery Studio [25].
Structural property calculation
The structural property calculation was carried out using DruLiTo software [26]. A QSAR model was developed using the information obtained from the property calculations to predict the best antidiabetic molecules in the library. It was developed in the context of converting the molecules into .sdf files, loading them in the program, and calculating their properties. The model was then used to predict the inhibitory activity of antidiabetic molecules. The model was validated by comparing the predicted values with the experimentally obtained values from molecular docking studies.
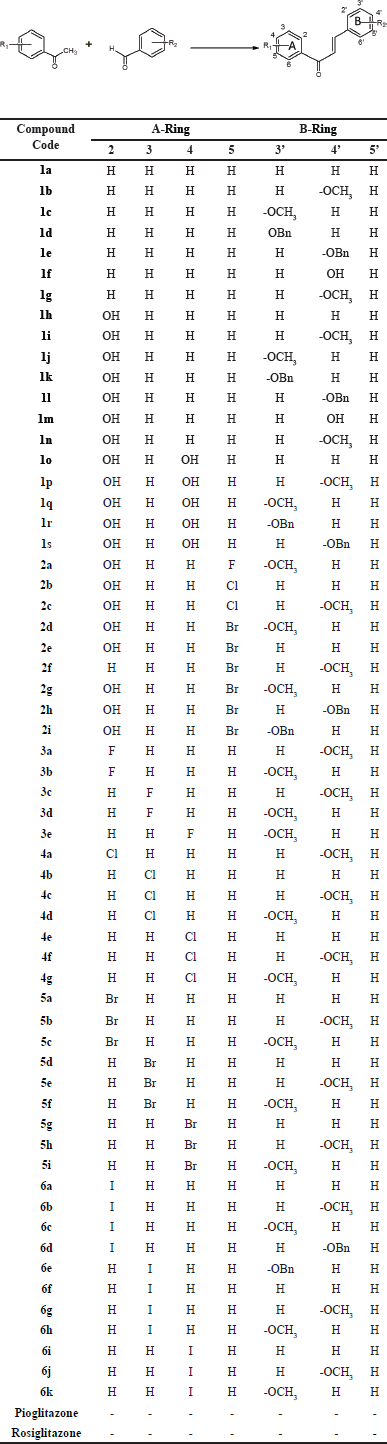 | Table 1. Analogs of 1,3-diphenyl-propen-1-ones. [Click here to view] |
QSAR studies
BuildQSAR® open-source software was used to study the correlation of the structural features with the binding affinities obtained by the screening studies [27]. All 60 molecules, along with pioglitazone and rosiglitazone, were included in the study. 10 descriptors were used for the study, and correlational studies were performed with binding energy (Kcal/mol) as activity.
RESULTS AND DISCUSSION
Library creation
A library of 60 analogs and two marketed drugs was created, and the energy of all the molecules was minimized using UFF force fields. All the molecules were saved in the required format in the working directory of the system.
Molecular docking
The results of molecular docking studies are tabulated in Table 2. The studies represent the compounds as 1a to 6k, Pioglitazone and Rosiglitazone, with their binding affinities. Total energy, internal energy, Van der Waal energy, and electrostatic energy were also calculated simultaneously and are represented in Table 2.
 | Table 2. Results of molecular docking studies. [Click here to view] |
The 2i molecule’s binding affinity was much higher than the other drugs, meaning it was more likely to bind to its target tissue. This made it a promising candidate to be developed into a drug. Figure 2 shows the results of the docking analysis carried out by maestro suite. Figure 2a represents the interacting amino acids in the periphery of 5.0 Å. The results show that glutamic acid 62 (GLU62) is firmly interacting with the hydroxyl group and glutamic acid 110 (GLU110) is interacting with bromine at the c-2 position. Other close interactions were observed with arginine 55 (ARG55), glutamine 53 (GLN53), isoleucine 108,63 (ILE108,63), serine 109,56 (SER109,56), leucine 107 (LEU107), and glycine 111,51 (GLY111,51). Figure 2b, 2c, and 2d represents the interaction at the active catalytic site. The Ramchandran plot of the interactions is observed in Figure 3.
1d molecule showed the second-best binding affinity towards the catalytic site of the receptor. Firm interactions with histidine 216 (HIS 216, 90), serine 56 (SER56), isoleucine 63, 92, 93 (ILE63, 92, 93), tyrosine 94 (TYR94), methionine 96 (MET96), and leucine 97,100 (leucine97,100). The interactions of the molecule on the catalytic side are shown in Figure 4a.2h, 6d, and 6e molecules showed significant bioactivities in comparison to the other 62 molecules in the library. The interactions are represented in Figure 4b, Figure 4c, and Figure 4d.
Docking studies are vital in drug discovery, predicting how potential drugs interact with target proteins. Validating these studies is crucial for reliability, comparing predicted binding affinities with experimental data. When consistent amino acid patterns are seen in binding across different molecules, it suggests key interactions at the catalytic site, aiding drug design optimization. Identifying these key residues helps modify lead compounds for better binding affinity and specificity. Overall, validation and interpretation of docking results guide rational drug design, facilitating the discovery of new therapies for diseases like diabetes.
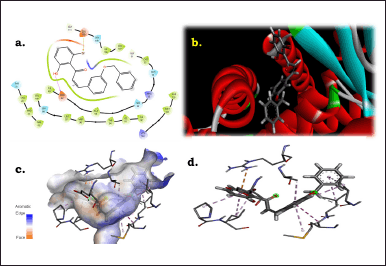 | Figure 2. Interactions of 2i molecule with amino acids. [Click here to view] |
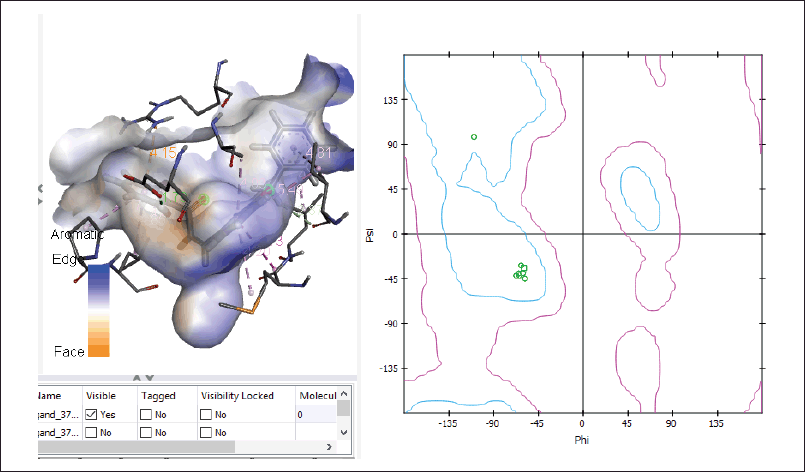 | Figure 3. Ramachandran plot of interactions. [Click here to view] |
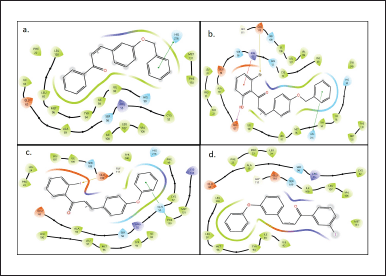 | Figure 4. a. Interactions of 1d molecule with amino acid, b. Interactions of 2h molecule with amino acids, c. Interactions of 6d molecule with amino acids, and d. Interactions of 6e molecule with amino acids. [Click here to view] |
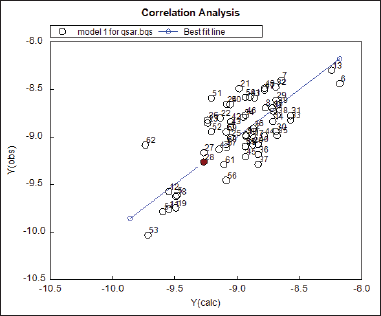 | Figure 5. Results of correlational studies. [Click here to view] |
Structural property calculation and QSAR studies
The results of the structural property calculation are represented in Table 3. The properties like molecular weight, logP, hydrogen bond acceptors (HBAs), hydrogen bond donors (HBDs), total polar surface area (TPSA), number of rotatable bonds (nRBs), number of atoms (nAtoms), number of rigid bonds (nRigid Bs), number of aromatic ring (nArom Ring), and number of hydrogen bonds (nHBs) were calculated by using DruLiTo® software. These calculated properties were truly considered as the descriptors and the binding affinities as the activity. The following structural information was used to generate the 2D QSAR model. The QSAR equation generated by the software is as follows.
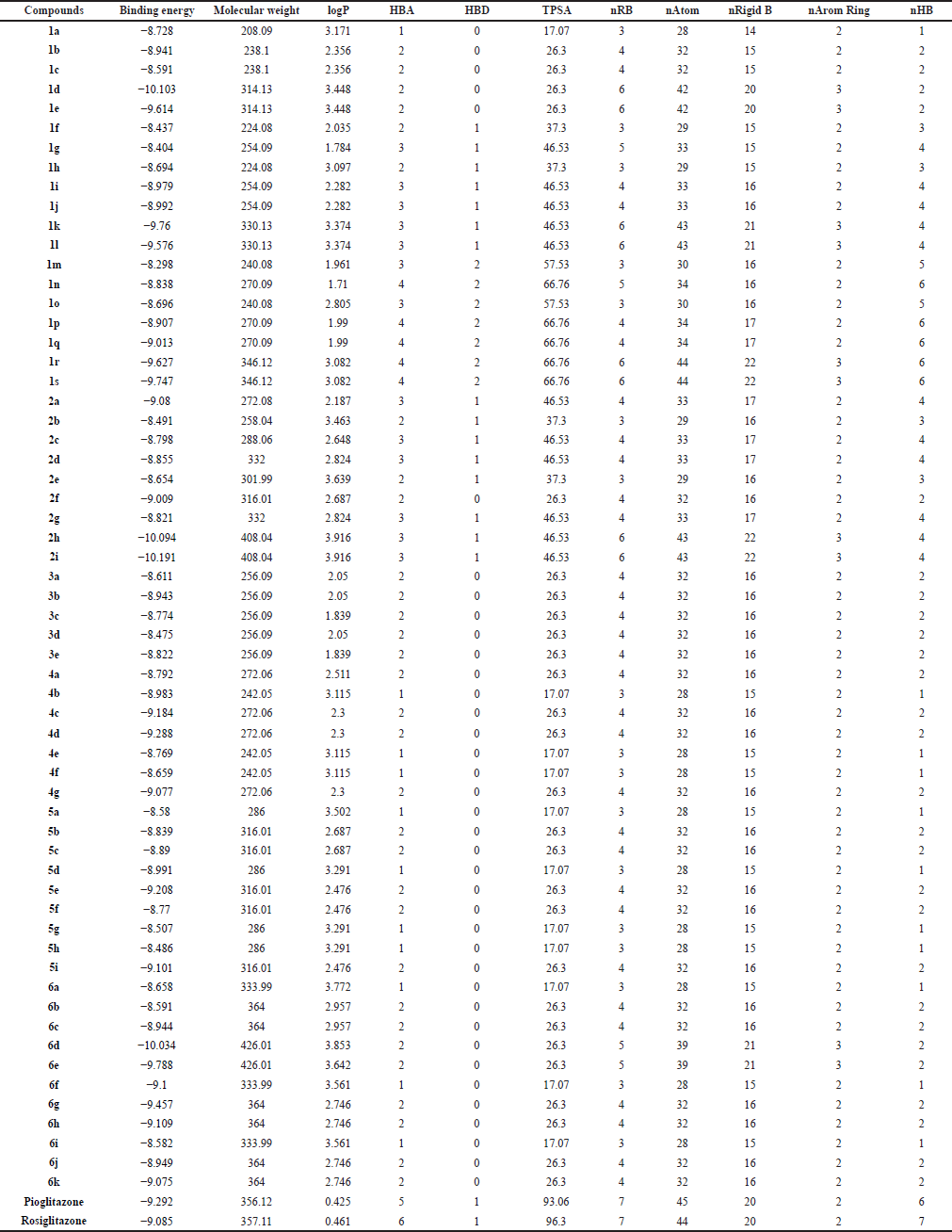 | Table 3. Results of structural property calculation. [Click here to view] |
Binding Energy = - 0.0001 (± 0.0025) MW–0.5643 (± 0.2177) LogP–0.6106 (± 0.2386) HBA + 0.5035 (± 0.2568) HBD–6.2982 (± 0.5709) (n = 62 ; R = 0.790 ; s = 0.285 ; F = 23.722 ; p < 0.0001 ; Q2 = 0.507 ; SPress = 0.327 ; SDEP = 0.316).
The results QSAR studies are as follows:
 | [Click here to view] |
The plot of the correlational matrix is represented in Figure 5 and shows that serial number 28, i.e., 2i is the best-fit molecule.
CONCLUSION
It was possible to effectively carry out molecular docking investigations in the library. The computation of the structural properties of the whole library was carried out, and the findings were computed with the assistance of software that is open source. In the course of the docking study, the compound with the code 2i was determined to be the most effective molecule. The QSAR investigations were used to carry out the correlational examinations of the structural attributes with the binding affinities that were ultimately achieved. It was determined that the 2i molecule, which had a serial number of 28, was the most suitable for the whole research and the most effective peroxisome proliferator-activated receptor-γ agonists when compared to medications that are already on the market. Studies on the link between structural activity and bioactivity revealed that the presence of a hydroxy group in the ortho position of the acetophenone analog is necessary for bioactivity. A large amount of activity is shown by the electron-withdrawing groups that have been replaced on the benzaldehyde derivatives
ACKNOWLEDGMENT
The authors are thankful to Shri Vile Parle Kelavani Mandal’s Institute of Pharmacy, Dhule.
AUTHOR CONTRIBUTION
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
Shri Vile Parle Kelavani Mandal’s Institute of Pharmacy, Dhule provided necessary support for the completion of work.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
CONSENT TO PARTICIPATE
All authors agree to publish the article.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Singh P, Anand A, Kumar V. Recent developments in biological activities of chalcones: a mini review. Eur J Med Chem[Internet]. 2014 Oct [cited 2024 Jan 18];85:758–77. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0223523414007685
2. Mahapatra DK, Bharti SK, Asati V. Chalcone derivatives: anti-inflammatory potential and molecular targets perspectives. CTMC [Internet]. 2017 Nov 20 [cited 2024 Jan 27];17(28):3146–69. Available from: http://www.eurekaselect.com/155594/article
3. Wu J, Li J, Cai Y, Pan Y, Ye F, Zhang Y, et al. Evaluation and discovery of novel synthetic chalcone derivatives as anti-inflammatory agents. J Med Chem [Internet]. 2011 Dec 8 [cited 2024 Jan 27];54(23):8110–23. Available from: https://pubs.acs.org/doi/10.1021/jm200946h
4. Zhang XW, Zhao DH, Quan YC, Sun LP, Yin XM, Guan LP. Synthesis and evaluation of antiinflammatory activity of substituted chalcone derivatives. Med Chem Res [Internet]. 2010 May [cited 2024 Jan 27];19(4):403–12. Available from: http://link.springer.com/10.1007/s00044-009-9202-z
5. Hsieh HK, Tsao LT, Wang JP, Lin CN. Synthesis and anti-inflammatory effect of chalcones. J Pharm Pharmacol [Internet]. 2010 Feb 18 [cited 2024 Jan 27];52(2):163–71. Available from: https://academic.oup.com/jpp/article/52/2/163/6157559
6. Won SJ, Liu CT, Tsao LT, Weng JR, Ko HH, Wang JP, et al. Synthetic chalcones as potential anti-inflammatory and cancer chemopreventive agents. Eur J Med Chem [Internet]. 2005 Jan [cited 2024 Jan 27];40(1):103–12. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0223523404001904
7. Ouyang Y, Li J, Chen X, Fu X, Sun S, Wu Q. Chalcone derivatives: role in anticancer therapy. Biomolecules [Internet]. 2021 Jun 16 [cited 2024 Jan 27];11(6):894. Available from: https://www.mdpi.com/2218-273X/11/6/894
8. Modzelewska A, Pettit C, Achanta G, Davidson NE, Huang P, Khan SR. Anticancer activities of novel chalcone and bis-chalcone derivatives. Bioorg Med Chem [Internet]. 2006 May [cited 2024 Jan 27];14(10):3491–5. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0968089606000253
9. Farooq S, Ngaini Z. One-pot and two-pot synthesis of chalcone based mono and bis-pyrazolines. Tetrahedron Lett [Internet]. 2020 Jan [cited 2024 Jan 27];61(4):151416. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0040403919312079
10. Uniyal A, Thakur BK, Singh S, Gairolla J, Anthwal A. Synthesis, in-silico antiviral screening and in-vitro antibacterial screening of a novel chalcone derivatives. J Indian Chem Soc [Internet]. 2023 Jul [cited 2024 Jan 27];100(7):101038. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0019452223001619
11. Gaonkar SL, Vignesh UN. Synthesis and pharmacological properties of chalcones: a review. Res Chem Intermed [Internet]. 2017 Nov [cited 2024 Jan 18];43(11):6043–77. Available from: http://link.springer.com/10.1007/s11164-017-2977-5
12. Blair M. Diabetes mellitus review. UNJ [Internet]. 2016 [cited 2024 Jan 27];36(1):27. Available from: https://library.suna.org/suna/articles/883/view
13. Kamya G, Rajwinder K, Anju G, Rajendra A. Chalcones: a review on synthesis and pharmacological activities. J Appl Pharm Sci [Internet]. 2021 Mar 5 [cited 2024 Jan 18];11. Available from: https://japsonline.com/abstract.php?article_id=3324&sts=2
14. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care [Internet]. 2010 Jan 1 [cited 2024 Jan 27];33(Supplement_1):S62–9. Available from: https://diabetesjournals.org/care/article/33/Supplement_1/S62/25777/Diagnosis-and-Classification-of-Diabetes-Mellitus
15. Mekala KC, Bertoni AG. Epidemiology of diabetes mellitus. In: Transplantation, bioengineering, and regeneration of the endocrine pancreas [Internet]. Amsterdam, The Netherlands: Elsevier; 2020 [cited 2024 Jan 27]. pp. 49–58. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780128148334000046
16. Campos C. Chronic hyperglycemia and glucose toxicity: pathology and clinical sequelae. Postgrad Med [Internet]. 2012 Nov [cited 2024 Jan 27];124(6):90–7. Available from: http://www.tandfonline.com/doi/full/10.3810/pgm.2012.11.2615
17. Abbas A, Blandon J, Rude J, Elfar A, Mukherjee D. PPAR- γ Agonist in treatment of diabetes: cardiovascular safety considerations. CHAMC [Internet]. 2012 Apr 1 [cited 2024 Jan 18];10(2):124–34. Available from: http://www.eurekaselect.com/openurl/content.php?genre=article&issn=1871-5257&volume=10&issue=2&spage=124
18. Rocha S, Ribeiro D, Fernandes E, Freitas M. A systematic review on anti-diabetic properties of chalcones. CMC [Internet]. 2020 Apr 29 [cited 2024 Jan 18];27(14):2257–321. Available from: http://www.eurekaselect.com/165765/article
19. Hsieh CT, Hsieh TJ, El-Shazly M, Chuang DW, Tsai YH, Yen CT, et al. Synthesis of chalcone derivatives as potential anti-diabetic agents. Bioorg Med Chem Lett [Internet]. 2012 Jun [cited 2024 Jan 18];22(12):3912–5. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0960894X12005586
20. Kondapuram SK, Sarvagalla S, Coumar MS. Docking-based virtual screening using PyRx tool: autophagy target Vps34 as a case study. In: Coumar MS, Editor. Molecular docking for computer-aided drug design [Internet]. Amsterdam, The Netherlands: Elsevier; 2021 [cited 2024 Jan 18]. pp. 463–77. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780128223123000199
21. El Aissouq A, Chedadi O, Bouachrine M, Ouammou A. Identification of novel SARS-CoV-2 inhibitors: a structure-based virtual screening approach. In: Leal JOP, editor. J Chem [Internet]. 2021 Feb 8 [cited 2024 Jan 18];2021:1–7. Available from: https://www.hindawi.com/journals/jchem/2021/1901484/
22. Einstein M, Akiyama TE, Castriota GA, Wang CF, McKeever B, Mosley RT, et al. The differential interactions of peroxisome proliferator-activated receptor γ ligands with Tyr473 is a physical basis for their unique biological activities. Mol Pharmacol [Internet]. 2008 Jan [cited 2024 Jan 27];73(1):62–74. Available from: http://molpharm.aspetjournals.org/lookup/doi/10.1124/mol.107.041202
23. Anwar T, Nadeem H, Sarwar S, Naureen H, Ahmed S, Khan A, et al. Investigation of antioxidant and anti-nociceptive potential of isoxazolone, pyrazolone derivatives, and their molecular docking studies. Drug Dev Res [Internet]. 2020 Nov [cited 2024 Jan 21];81(7):893–903. Available from: https://onlinelibrary.wiley.com/doi/10.1002/ddr.21711
24. Yadav M, Dhagat S, Eswari JS. Structure based drug design and molecular docking studies of anticancer molecules paclitaxel, etoposide and topotecan using novel ligands. CDDT [Internet]. 2020 Jun 19 [cited 2024 Jan 21];17(2):183–90. Available from: https://www.eurekaselect.com/170521/article
25. Shaker B, Ahmad S, Lee J, Jung C, Na D. In silico methods and tools for drug discovery. Comput Biol Med [Internet]. 2021 Oct [cited 2024 Jan 21];137:104851. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0010482521006454
26. Pangastuti A, Amin IF, Amin AZ, Amin M. Natural bioactive compound from Moringa oleifera against cancer based on in silico screening. J Teknol [Internet]. 2016 Apr 18 [cited 2024 Jan 21];78(5). Available from: https://journals.utm.my/index.php/jurnalteknologi/article/view/8328
27. De Oliveira DB, Gaudio AC. BuildQSAR: A new computer program for QSAR analysis. Quant Struct-Act Relat [Internet]. 2000 Dec [cited 2024 Jan 21];19(6):599–601. Available from: https://onlinelibrary.wiley.com/doi/10.1002/1521-3838(200012)19:6<599::AID-QSAR599>3.0.CO;2-B