
INTRODUCTION
Breast cancer is a complex and heterogeneous disease that affects millions of women worldwide and stands as the most prevalent form of cancer, with approximately 2.3 million new cases (11.7% of total cancer cases) reported in the year 2020 [1]. Cancer treatment typically involves surgery, radiotherapy, and chemotherapy. However, the nonselective nature of chemotherapy often leads to toxic side effects. To address this, researchers are investigating targeted therapies that focus on specific proteins, such as cyclin-dependent kinases (CDKs), crucial for cancer cell growth. These alternative approaches aim to enhance cancer drug therapy by selectively targeting key proteins involved in proliferation [2–6]. Cyclin-dependent kinase-2 (CDK-2) inhibition is a particularly attractive target to treat breast cancer for its role in cell cycle progression and its potential to prevent uncontrolled cell proliferation and promote DNA repair, making an active area of research in breast cancer treatment [7–10]. Several CDK-2 inhibitors have been developed and evaluated in preclinical and clinical studies, including flavopiridol, palbociclib, ribociclib, and abemaciclib [10–13]. However, these inhibitors have limitations such as poor selectivity, off-target effects, and dose-limiting toxicities. Especially flavopiridol (Fig. 1A), the first FDA-approved CDK-2 inhibitor, which was also later withdrawn due to these limitations [14–17]. It has been considered a privileged structure with broad medicinal utility and has shown promising activity against CDKs [18–24]. Repurposing of these drugs is, therefore, needed and one way to achieve this is by modifying its chemical structure while keeping the core scaffold 4H-chromene intact. Modifying this scaffold could lead the way for developing more potent and selective compounds with better pharmacokinetic properties. For example, 4-aryl-4H-chromene wherein modification involves the addition of an aryl group to the 4-position has attracted considerable attention as a potential source of antiproliferative agents such as compound EPC2407 (Fig. 1B) having a 4-aryl-4H-chromene scaffold, has been shown to induce apoptotic processes by hindering tubulin polymerization and is under phase II clinical trials for the treatment of anaplastic thyroid cancer [25]. Another compound (Fig. 1C) was tested using multiple human cell lines and it was discovered that it can induce nuclear fragmentation, Poly (ADP-ribose) polymerase (PARP) cleavage, and arrested cells at the G2/M stage. In addition, it was found to induce apoptosis, as confirmed by flow cytometry analysis (e.g., T47D). It also shows high growth inhibition (GI) in the 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay [26]. Similarly, another compound (Fig. 1D) shows in vitro cytotoxicity against tumor cells having IC50 values in the nM range. Also, the cytotoxic effects of this compound on normal cells were minimal [27].
Based on the above-stated facts, the study aimed to investigate the antioxidant and antiproliferative activity of 4-aryl-4H-chromene derivatives with an electron-rich substitution of a hydroxyl group at the 5th and 6th positions. Furthermore, molecular docking (XP), molecular mechanics generalized-born surface area (MM-GBSA), induced fit docking (IFD), and absorption, distribution, metabolism, excretion, and toxicity (ADMET) investigations were carried out to explore their efficiency and target interactions in the active site of the CDK-2 enzyme. This molecular approach holds promise for the development of new and effective 4-aryl-4H-chromene derivatives as a potential lead for breast cancer.
MATERIALS AND METHODS
In-silico studies
Softwares used
Commercially available software Schrödinger (Schrödinger, LLC, NY, -2021-2) was used to carry out molecular docking studies. XP docking, MM-GBSA, IFD-XP, and ADMET predictions were carried out using Glide, Prime, Induce fit, and Qikprop module incorporated in Schrödinger, LLC, NY, -2021-2 (Maestro version 11.08) [28].
Protein selection and protein preparation
Target, i.e., CDK-2 enzyme, was chosen as the protein to explore the underlying molecular mechanism of the antiproliferative effect. The structure of the target protein was retrieved from the Protein Data Bank (PDB) (www.rcsb.org accessed on 10 October 2022). Thus, the 3-D crystal structure with 2.52 Å resolution (PDB ID: 6GUB) of CDK2/Cyclin in complex with flavopiridol was used. Later, the crystal structure of PDB ID: 6GUB was optimized using the Protein Preparation Wizard in Maestro (Schrödinger,-2021-2) by performing several steps, including assigning bond orders, adding hydrogen atoms and removing water molecules beyond 5Å from the crystal structure using the OPLS3e force field. The grid box was built considering flavopiridol as the centroid of the grid.
Ligand preparation
A total of thirty-eight 4-aryl-4H-chromene derivatives were drawn using the 2-D sketcher in Maestro (Schrödinger, LLC, NY,-2021-2), and the co-crystallized ligand flavopiridol was separated from the protein structure. These designed 2-D compounds and flavopiridol were then converted into 3-D dorms using the LigPrep module under the OPLS3e force field. This step is important to generate 3-D conformation of the ligand in the lowest energy state to generate correct chirality. It also predicts the protonation state of the ligand and the presence of ionizable groups and can optimize the ionization state of the ligand to better match the conditions of the target protein [28].
Molecular docking and MM-GBSA analysis
The molecular docking was performed using Glide module in extra precision (XP) with the OPLS3e force field into the generated grid, which was centered on the active site of the protein (PDB ID: 6GUB). Molecular docking is a hierarchy system that generates the most likely binding poses of the ligand on the protein. Later, the output of molecular docking was used to calculate the ?G values of flavopiridol and the designed ligands using the Prime MM-GBSA module (Prime, Schrödinger, LLC, New York, NY,-2021-2). It determined the energy difference between the bound complex and the separate energy values of the protein and ligand when unbound [29].
IFD—XP
Induced-fit docking was later performed using the Maestro application’s induced-fit docking module. Prime side-chain prediction and minimization were executed to refine residues within 5Å of the ligand pose and side chains, enabling nearby reorienting side chains to adjust to the ligand structure and conformation. The receptor was then marked from the workspace ligand’s centroid, and a maximum of 20 poses were retained for each docked ligand to be further docked at XP mode using the standard protocol [30].
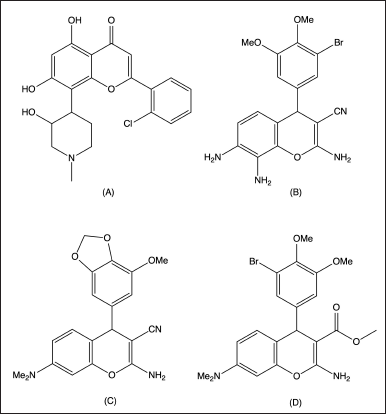 | Figure 1. Recent development and rationale for designing target 4-aryl-4H-chromene derivatives. [Click here to view] |
ADMET analysis
The drug-like activity of the selected ligand molecule was analyzed for ADMET properties using the QikProp module of the Maestro application of Schrödinger Suite-2021-2. This was used to calculate the molecular descriptor and to predict the pharmacokinetics and pharmacodynamics properties of small molecules [31].
Chemistry
General experimental materials and methods
Resorcinol and hydroquinone were purchased from (Sisco Research Laboratories Pvt. Ltd., New Delhi, India). The substituted benzaldehydes were purchased from BLD pharma (BLD Pharmatech (India) Pvt Ltd., Hyderabad, India), and all the solvents used were procured from Sigma-Aldrich Chemical Co. (Sigma-Aldrich Corp., St. Louis, MO). Melting points were recorded using the melting point apparatus by Shital Scientific Industries. IR spectra were documented on a Shimadzu FTIR 8310 spectrometer with KBr pallets. 1H NMR spectra were recorded on Bruker Ascend TM 400Hz NMR spectrophotometer using Dimethyl sulfoxide (DMSO)-d6 as a solvent and tetramethylsilane as the internal standard. The mass spectra were run on a Thermo Scientific LT-QXL spectrometer at 70 eV with the APCI method of ionization. Thin-layer chromatography on Merck silica gel 60 F254 sheets to assess the pureness of the synthesized compounds using solvent mixtures with different polarities, e.g., Hexane: Ethyl Acetate (3:2) and Ethyl Acetate: Hexane (7:3). The spots were visualized using UV light at 200 and 400 nm.
General procedure for the synthesis of 4-aryl-4H-chromene derivatives
An efficient one-pot synthesis was used with different substituted benzaldehyde (5 mmol), malononitrile (5 mmol), and resorcinol or hydroquinone (5 mmol) was taken together with piperidine as base catalyst (10 mmol) and ethanol as a solvent (20 ml). The admixture was stirred at 35°C for 6–7 hours (Fig. 2). The product was then filtered, washed with water, and dried. Later, the dried product was recrystallized with methanol to get a yield of 50%–80% [26,32].
Biological evaluation
In vitro evaluation, such as antioxidant and anticancer activities, is crucial for assessing the potential therapeutic efficacy of the synthesized compounds, particularly in the context of breast cancer treatment. Hence, the 2,2-Diphenyl-1-picrylhydrazyl (DPPH) and 2,2′-azino-bis (3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) assays were employed to evaluate the antioxidant potential of the compounds to measure the ability of compounds to scavenge free radicals, simulating the oxidative stress environment often encountered in cancer cells.
Antioxidant assay
The reagent DPPH and ABTS were procured from Sigma-Aldrich Chemical Co. (Sigma-Aldrich Corp., St. Louis, MO) and the radical scavenging assays have been performed with slight modification [33].
DPPH free radical scavenging: A solution is prepared by dissolving 3.94 mg DPPH in 50 ml methanol and keeping it in the dark for 2 hours. Subsequently, the test solutions were prepared with different concentrations ranging from 500 to 62.5 μg/ml, and in each well of 96-well plates, 100 μl of the test compound of different concentrations was mixed with 0.1 ml of the DPPH solution (0.2 mg/ml in ethanol). The plate is then incubated in a dark room at 37°C for 30 minutes. Later, the absorbance of the reaction mixture was measured at 517 nm. The absorbance of the methanol was also measured as blank. The capability to scavenge the DPPH radical was calculated using the following equation:
DPPH scavenged (%) = {(AB–AA)/AB} ×100
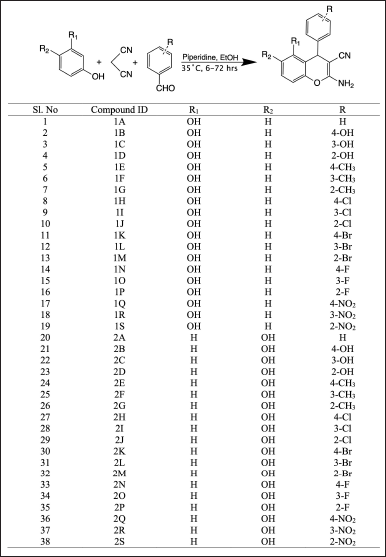 | Figure 2. One-pot synthesis of 4-aryl-4H-chromene derivatives in the presence of piperidine. [Click here to view] |
where AB is the absorbance of the DPPH solution and AA is the absorbance of the DPPH solution containing the test solution after 30 minutes of incubation.
For DPPH assay, after the addition of the reagent, the plate was incubated in a dark room at 25°C for 30 minutes. After incubation, the absorbance of the resulting solution was measured at 517 nm.
For the ABTS assay, ABTS radical scavenging was performed as follows.
ABTS radical scavenging: ABTS + cation radical was produced by the reaction between 7 mM ABTS in ultrapure water and in that 6.6 mg of potassium persulphate is added and stored in the dark at room temperature for 12–16 hours before use. ABTS·+ solution was then diluted with methanol to obtain an absorbance of 0.700 at 734 nm. The samples were prepared in different concentrations ranging from 500 to 62.5 μg/ml. Subsequently, 10 μl of different concentrations were mixed with 190 μl of the ABTS radical solution, and the absorbance was measured in each cell of 96-well plates and then mixed with ABTS solutions. The plate is then incubated in the dark at 25°C for 15 minutes, and the absorbance was measured at 734 nm.
ABTS+ scavenging effect (%) = {(AB–AA)/ AB} ×100
Here, AB is the absorbance of the ABTS solution and AA is the absorbance of the ABTS solution containing the test solution after 15 minutes of incubation.
Cell proliferation assay
The formed compounds were dissolved in DMSO and subsequently diluted with sterile deionized water for primary screening. For IC50 determination, positive controls were prepared by making six concentrations of the compound from 100 to 3.175 μM using a twofold serial dilution in 10% DMSO. The MCF-7 cell line was obtained from the National Centre for Cell Science, Maharastra, India.
Later, the cytotoxic activity of these compounds was assessed against the MCF-7 cell line using the sulforhodamine B (SRB) colorimetric assay as previously reported [34]. The assay uses the bright-pink amino xanthene dye, which binds stoichiometrically to cell protein components in proportion to cell mass. To prepare the cells, phosphate buffer was used to wash the cell monolayers, followed by the addition of trypsin to dissociate the cells. Cell concentration was determined with a haematocytometer chamber and adjusted to a seeding density of 1.9 × 10^4 cells per well. To expose the cells to the compound, 190 μl of the cell suspension was added to each well and incubated for 72 hours. A no-growth control is also incubated, which includes only cell suspension. Later, the cells were fixed with Trichloroacetic acid (TCA) and stained with SRB dye, and absorbance was measured at 540 nm and % cell growth was calculated. The formulas were as follows:
% Cell growth = Absorbance sample/Absorbance negative control or untreated × 100.
% GI was calculated is calculated from % cell growth.
% GI = 100 − % cell growth.
Next, from the % GI, the IC50 value for each compound was calculated.
RESULT AND DISCUSSION
In-silico study
Molecular docking and MM-GBSA analysis
Molecular docking and MM-GBSA studies of different compounds were performed using Schrödinger Suite’s Maestro on CDK2/Cyclin A complexed with flavopiridol. Ligands with docking scores greater than −8.000 were chosen, resulting in 13 compounds with scores ranging from −9.180 to −8.006 Kcal/mol, and MM-GBSA of these compounds confirmed their stability in the docked pose with ΔG binding energy >−30 Kcal/mol. The co-crystallized drug flavopiridol had a ΔG binding energy of −49.83 Kcal/mol (Table 1).
The standard drug, i.e., flavopiridol forms hydrogen bonds with LYS33 and GLN131, and hydrophobic interactions with VAL18, VAL64, TYR15, ILE10, PHE80, PHE82, LEU83, LEU134, ALA31, and ALA144 amino acid residues of the protein. Most of the compounds, including 2C, 2N, 1C, 2K, 2M, 2J, 2O, 2I, 2F, and 2B, showed similar interaction with the protein, such as hydrogen bonds occur primarily with residues ILE10, LYS33, LEU83, ASN132, GLN131, and ASP145. In addition, many of the compounds share hydrophobic interactions primarily with residues such as VAL18, VAL64, TYR15, PHE80, PHE82, LEU83, LEU134, ALA31, and ALA144. However, there are also notable differences in the interaction patterns; for example, 2P and 1E form hydrogen bonds with different residues than the other compounds, including ASP145 and LYS33, respectively. Furthermore, 2P lacks hydrophobic interactions with residues TYR15 and PHE82 that are present in other compounds. In contrast, 2K, 2M, and 1L form halogen bonds with GLU81, LYS33, and LEU83, respectively, which is not observed in any other compound. Lastly, 1C has a pi-pi stacking interaction with PHE80, which is not seen in any of the other compounds. The halogen bond observed in 2K, 2M, and 1L may provide additional stability and could be explored further in drug design. The pi-pi cation interaction with PHE80 is also of interest, as it may be exploited in the design of more specific and potent CDK inhibitors (Table 1).
IFD—XP
Following the molecular docking, the selected 13 compounds underwent IFD, which showed changes in protein-ligand interactions, such as H-bond, hydrophobic interactions, halogen bonding, pi-cation, and pi-pi stacking for different compounds. The 3-D interaction plot of top IFD poses for compounds 2C, 2N, 2J, 2P, 2F, and 1L are shown in Figure 3. Compound 2C exhibited a new set of interactions, involving TYR15, LEU83, GLN131, and ASP145, indicating the potential for additional stabilizing forces. Similarly, compound 2N showed alterations in its interactions, with LYS33, LEU83, and GLN131 replacing previous residues. Notably, compound 1C displayed modifications in its bonding pattern, by forming interactions with LYS33, LEU83, and ASP145, while lacking the previously observed pi-pi stacking. Compound 2K demonstrated a unique interaction profile, featuring new hydrophobic interactions with PHE146 and LEU55, and a change from halogen bonding to a pi-pi stacking interaction with PHE146. Compound 2M exhibited a switch from halogen bonding to pi-cation interaction, involving LYS33, and a new hydrophobic interaction with LEU55. Compound 2J displayed the formation of a novel halogen bond with ASP145 and the absence of hydrophobic interaction with ALA31. In compound 2P, a new hydrogen bond emerged with LYS33, and hydrophobic interactions shifted from ALA31 to ALA144, accompanied by a newly formed pi-pi stacking interaction with PHE80. Compound 2O exhibited alterations in hydrogen bonding and hydrophobic interactions, as well as the formation of a new pi-pi stacking interaction with PHE80 and a pi-cation interaction with LYS33. Compound 2I displayed new halogen bonding interactions with LYS33 and ASP145, while compound 2B featured changes in hydrogen bonding and hydrophobic interactions, including the involvement of additional residues. Compound 1E showed an additional hydrogen bond with residue ILE10, while compound 1L formed a new halogen bond with residue LEU83. These findings from IFD highlight the dynamic nature of protein-ligand interactions and the potential for additional stabilization through the formation of various bonds.
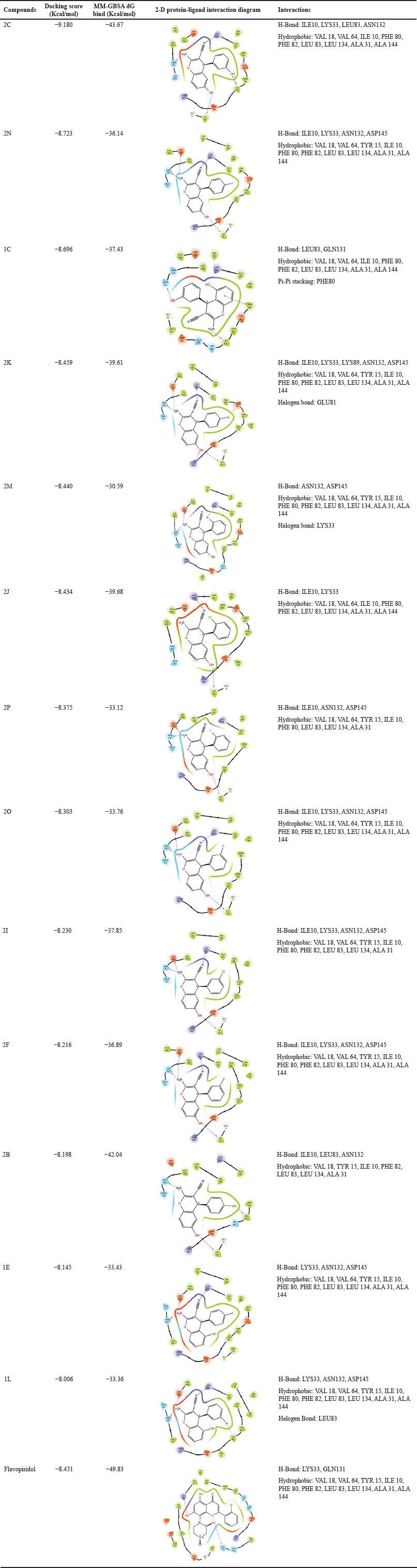 | Table 1. Docking score, prime MM-GBSA bind and protein-ligand 2-D interaction of top 13 compounds and flavopiridol. [Click here to view] |
ADMET analysis
ADMET properties of these 13 drugs were evaluated using the QikProp module and were assessed using various descriptor calculations such as molecular weight (mw), hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), QPLogPo/w (octanol/water partition coefficient), QPLogPB/B (brain-blood barrier partition coefficient), % human oral absorption (HOA), and Lipinski rule of five violation (Table 2). All compounds exhibit the acceptable range for mw, HBD, HBA, QPLogPo/w, and QPLogPB/B. In addition, all compounds have high oral absorption potential, with over 80% exhibiting high permeability, and none of the compounds violate the Rule of 5. Altogether, the result suggests that these compounds show drug-likeness characteristics, but further studies are needed to determine their efficacy and safety.
Chemistry
All the 4-aryl-4H-chromene derivatives were synthesized using one-pot synthesis. For the characterization of these compounds, various spectroscopic techniques were employed. The Fourier transform infrared (FT-IR) spectra revealed characteristic absorption bands for key functional groups. Notably, the –NH2 stretching vibrations appeared around ~3,400–3,500 cm−1, while the −OH stretching vibrations were observed within ~3,100–3,300 cm−1. The Aromatic C–H stretching vibrations appeared in the range of ~3,000–3,100 cm−1, and the –CN stretching vibrations were present around ~2,150–2,200 cm−1. The m/z values were observed with the help of mass spectra also confirmed the agreed range of compounds mw. Furthermore, 1H NMR was carried out for six compounds, i.e., 2C, 2N, 2J, 2P, 2F, and 1L, to elucidate the proton environments. The resonances of aliphatic H–4 protons were detected around ~0.5–3.0 ppm, while the NH2 groups displayed resonances in the range of ~6.4–7.5 ppm. The aromatic protons (H-Ar) exhibited resonances within ~6.5–7.0 ppm. The presence of –OH groups resulted in resonances around ~9.0–10.0 ppm. The spectra of these six compounds are provided in the supplementary material. The physical characteristics and the spectral data provided below are representative and cover all synthesized compounds. Detailed spectra for six compounds can be found in the supplementary material.
Compound 2C: (2-amino-6-hydroxy-4-(3-hydroxyphenyl)-4H-chromene-3-carbonitrile)
C16H12N2O3; Brown solid, m.p. 233°C−237°C, Rf 0.35; IR (KBr, cm−1): 3,444 (–NH2), 3,340, 3,113 (–OH), 3,107, 3,086 (Ar –CH), 2,194 (–CN), 1,649 (=CH–CN); LCMS (APCI) (70 eV) m/z: 279 [M]−, 280 [M]; 1H NMR (400 MHz, DMSO-d6): 4.49 (s, 1H, H–4), 6.85–6.52 (s, 2H, NH2), 7.09–6.39 (m, 7H, H– Ar), 9.33–9.3 (s, 2H, OH) (Fig. S1).
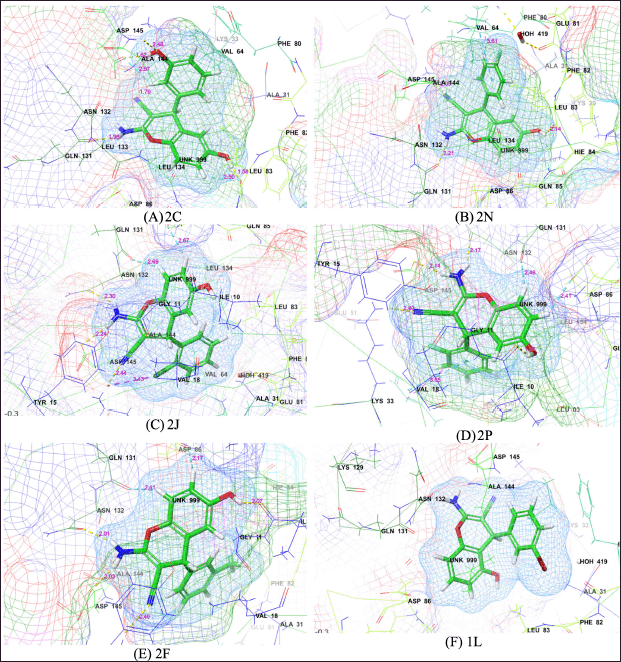 | Figure 3. 3D interaction plot of top IFD poses for different compounds; (A) 2C, (B) 2N, (C) 2J, (D) 2P, (E) 2F, and (F) 1L. [Click here to view] |
Compound 2N: (2-amino-6-hydroxy-4-(4-fluorophenyl) -4H-chromene-3-carbonitrile)
C16H11FN2O2; Brown solid, m.p. 216°C−222°C, Rf 0.47; IR (KBr, cm−1): 3,491 (–NH2), 3,412, 3,215 (–OH), 3,062, 2,966 (Ar –CH), 2,194 (–CN), 1,651 (=CH–CN); LCMS (APCI) (70 eV) m/z: 281 [M]−; 1H NMR (400 MHz, DMSO-d6): 4.69 (s, 1H, H–4), 6.97–6.95 (s, 2H, NH2), 7.39–6.41 (m, 7H, H– Ar), (OH peaks are not visible due to high exchangeability of proton with DMSO) (Fig. S2).
Compound 1C: (2-amino-5-hydroxy-4-(3-hydroxyphenyl)-4H-chromene-3-carbonitrile)
C16H12N2O3; Reddish brown solid, m.p. 208°C−211°C, Rf 0.43; IR (KBr, cm−1): 3,442 (–NH2), 3,338, 3,211 (–OH), 3,107, 3,086 (Ar –CH), 2,194 (–CN stretch), 1,645 (=CH–CN); LCMS (APCI) (70 eV) m/z: 281 [M]+.
Compound 2K: (2-amino-6-hydroxy-4-(4-bromophenyl)-4H-chromene-3-carbonitrile)
C16H11BrN2O2; Yellow solid, m.p. 219°C−221°C, Rf 0.45; IR (KBr, cm−1): 3,437 (–NH2), 3,338 (–OH), 3,078 (Ar –CH), 2,193 (–CN), 1,643 (=CH–CN); LCMS (APCI) (70 eV) m/z: 341 [M]2−, 343 [M], 342 [M]−, 344 [M]+.
Compound 2M: (2-amino-6-hydroxy-4-(2-bromophenyl)-4H-chromene-3-carbonitrile)
C16H11BrN2O2; Yellow solid, m.p. 221°C−225°C, Rf 0.45; IR (KBr, cm−1): 3,437 (–NH2), 3,334, 3,209 (–OH), 3,078 (Ar –CH), 2,193 (–CN), 1,643 (=CH–CN); LCMS (APCI) (70 eV) m/z: 341 [M]2−, 343 [M], 342 [M]−, 344 [M]+.
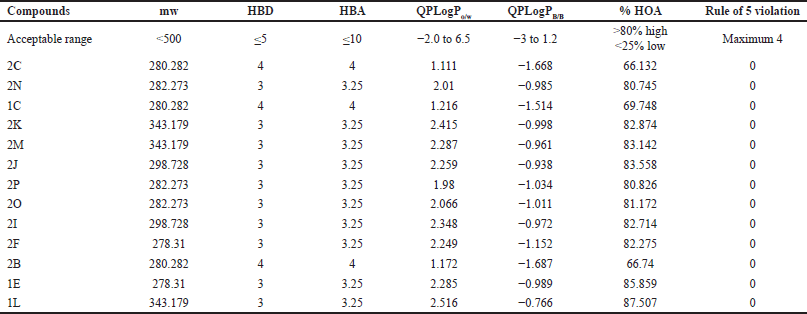 | Table 2. ADMET properties of the top 13 compounds. [Click here to view] |
Compound 2J: (2-amino-6-hydroxy-4-(2-chlorophenyl)-4H-chromene-3-carbonitrile)
C16H11ClN2O2; Orange solid, m.p. 223°C−227°C, Rf 0.45; IR (KBr, cm−1): 3,473 (–NH2), 3,336, 3,253 (–OH), 2,927 (Ar –CH), 2,189 (–CN), 1,635 (=CH–CN); LCMS (APCI) (70 eV) m/z: 297 (100) [M]−; 1H NMR (400 MHz, DMSO-d6): 1.19–1.15 (s, 3H, CH3), 4.49 (s, 1H, H–4), 6.85–6.52 (s, 2H, NH2), 7.09–6.47 (m, 7H, H– Ar), 9.3 (s, 1H, OH) (Fig. S3).
Compound 2P: (2-amino-6-hydroxy-4-(2-fluorophenyl)-4H-chromene-3-carbonitrile)
C16H11FN2O2; Reddish brown solid, m.p. 215°C−220°C, Rf 0.29; IR (KBr, cm−1): 3,510 (–NH2), 3,400, 3,215 (–OH), 3,076 (Ar –CH), 2,185 (–CN), 1,645 (=CH–CN); LCMS (APCI) (70 eV) m/z: 281 [M]−; 1H NMR (400 MHz, DMSO-d6): 4.89 (s, 1H, H–4), 7.16–6.93 (s, 2H, NH2), 7.28–6.40 (m, 7H, H– Ar), (OH peaks are not visible due to high exchangeability of proton with DMSO) (Fig. S4).
Compound 2O: (2-amino-6-hydroxy-4-(3-fluorophenyl)-4H-chromene-3-carbonitrile)
C16H11FN2O2; Reddish brown solid, m.p. 215°C−221°C, Rf 0.31; IR (KBr, cm−1): 3,396 (–NH2), 3,336, 3,215 (–OH), 3,076, 2,933 (Ar –CH), 2,183 (–CN), 1,643 (=CH–CN); LCMS (APCI) (70 eV) m/z: 281 [M]−.
Compound 2I: (2-amino-6-hydroxy-4-(3-chlorophenyl)-4H-chromene-3-carbonitrile)
C16H11ClN2O2; Orange solid, m.p. 227°C−230°C, Rf 0.43; IR (KBr, cm−1): 3,475 (–NH2), 3,336, 3,251 (–OH), 3,076 (Ar –CH), 2,189 (–CN), 1,639 (=CH–CN); LCMS (APCI) (70 eV) m/z: 297 [M]−.
Compound 2F: (2-amino-6-hydroxy-4-(3-methylphenyl)-4H-chromene-3-carbonitrile)
C17H14N2O2; Pale yellow solid, m.p. 223°C−228°C, Rf 0.31; IR (KBr, cm−1): 3,466 (–NH2), 3,383, 3,317 (–OH), 3,192 (Ar –CH), 2,204 (–CN), 1,647 (=CH–CN); LCMS (APCI) (70 eV) m/z: 277 [M]−; 1H NMR (400 MHz, DMSO-d6): 1.19–1.15 (s, 3H, CH3), 4.69 (s, 1H, H–4), 7.06–6.95 (s, 2H, NH2), 7.36–6.41 (m, 7H, H– Ar), 9.75 (s, 1H, OH) (Fig. S5).
Compound 2B: (2-amino-6-hydroxy-4-(4-hydroxyphenyl)-4H-chromene-3-carbonitrile)
C16H12N2O3; Brown solid, m.p. 235°C−239°C, Rf 0.36; IR (KBr, cm−1): 3.442 (–NH2), 3.338, 3.113 (–OH), 3.209, 3.113 (Ar –CH), 2.194 (–CN), 1.645 (=CH–CN); LCMS (APCI) (70 eV) m/z: 279 [M]−.
Compound 1E: (2-amino-5-hydroxy-4-(4-methylphenyl)-4H-chromene-3-carbonitrile)
C17H14N2O2; Yellow solid, m.p. 223°C−227°C, Rf 0.41; IR (KBr, cm−1): 3,464 (–NH2), 3,317, 3,192 (–OH), 3,192 (Ar –CH), 2,204 (–CN), 1,645 (=CH–CN); LCMS (APCI) (70 eV) m/z: 277 [M]−.
Compound 1L: (2-amino-5-hydroxy-4-(3-bromophenyl)-4H-chromene-3-carbonitrile)
C16H11BrN2O2; Brown solid, m.p. 221°C−225°C, Rf 0.45; IR (KBr, cm−1): 3,435 (–NH2), 3,334, 3,029 (–OH), 3,076 (Ar –CH), 2,193 (–CN), 1,641 (=CH–CN); LCMS (APCI) (70 eV) m/z: 341 [M]2−, 343 [M], 342 [M]−, 344 [M]+; 1H NMR (400 MHz, DMSO-d6): 4.68 (s, 1H, H–4), 7.34–6.97 (s, 2H, NH2), 7.42–6.41 (m, 7H, H– Ar), 9.77 (s, 1H, OH) (Fig. S6).
Biological evaluation
Antioxidant assay
The effect of antioxidants on DPPH radical and ABTS radical was thought to be due to their hydrogen-donating ability. As shown in Table 3, all the compounds showed antioxidant activity. However, in the DPPH assay, compound 2M had the most potent radical scavenging activity with IC50 of 24.44 μM, followed by 2I (95.66 μM) and 1C (128.65 μM), and the rest showed moderate activity.
In the case of the ABTS assay, 2C has the strongest antioxidant activity with IC50 of 39.03 μM, followed by 2K, 2B, 1C, and 2N with IC50 less than 200 μM. The remaining compounds showed moderate activity. The results suggest that the compounds exhibit stronger scavenging ability in the ABTS assay compared to the DPPH assay, indicating that electron-donating substituents in the aryl ring, such as hydroxy groups found in compounds 2C, 2B, and 1C, have a more significant positive impact in the ABTS test. The results of these assays indicate that all synthesized compounds exhibited antioxidant activity, suggesting their potential to counteract oxidative stress associated with cancer progression.
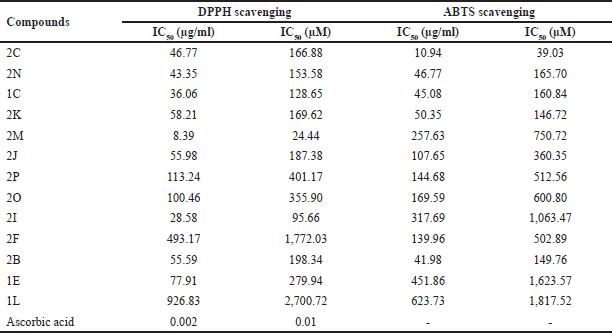 | Table 3. Antioxidant activities of the 13 synthesized derivatives as determined by calculation of IC50 in μM and μg/ml range from DPPH and ABTS radical scavenging activity. [Click here to view] |
Cell proliferation assay
The SRB assay, conducted on the MCF-7 breast cancer cell line, was carried out for the assessment of the compounds’ anticancer potential. Figure 4 illustrates the results obtained for all 13 compounds by this assay. Compound 1L demonstrated the highest potency, exhibiting a significant inhibition of growth with an IC50 value of 0.2 μM. Conversely, compound 2I displayed the lowest potency, indicating weak GI with an IC50 of 202.1 μM. Compounds 2O, 2K, 1C, 2M, and 2J exhibited promising inhibitory activity at concentrations below 30 μM. The remaining compounds displayed moderate to low potency, suggesting the need for further optimization to enhance their effectiveness. The remaining compounds exhibited moderate to low potency with an IC50 less than 80 μM, indicating a need for further optimization to improve their potency.
Compounds 2N, 1C, 2K, and 2M, with halogen substituents on the 4-aryl ring, show higher activity than compound 2I with a chloro-substituent. The position of halogen also has an influence on activity, with compound 2O exhibiting higher activity than 2N. Compound 1L, with a bromine substituent at the 3rd position of the aryl and a hydroxyl group at the 5th position, shows the strongest antiproliferative activity. The presence of a hydroxyl group at the 5-position in compounds 1C, 2C, and 2B also contributes to their favorable activity. However, the methyl group in compound 1E appears to reduce its potency (Supplementary file: Figs. S7–S11). This observation suggests that the varying potencies observed among the compounds underscore the significance of structural modifications in influencing their effectiveness in the inhibition of cell growth.
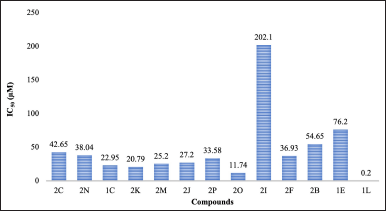 | Figure 4. IC50 values are expressed in (μM) of the synthesised compounds against the MCF-7 cell line. [Click here to view] |
CONCLUSION
In conclusion, a total of thirty-eight, 4-aryl-4H-chromene derivatives were designed and evaluated as inhibitors for CDK-2, a target for breast cancer therapy. Molecular docking studies identified 13 compounds as potential inhibitors, which exhibited strong binding with the target protein through hydrogen bonds and hydrophobic interactions and had favorable ADMET properties that are associated with good drug candidates. These interactions suggest a plausible mechanism through which the compounds could interfere with CDK-2 activity, ultimately contributing to cell GI in breast cancer.
The 13 compounds were then synthesized and characterized using FT-IR, Liquid chromatography-mass spectrometry (LC-MS), and Nuclear Magnetic Resonance (NMR) spectroscopy. Later, in biological evaluation of these synthesized compounds notably, compound 1L, distinguished by its 3-bromo substitution on the 4-aryl ring, demonstrated remarkable inhibitory activity against MCF-7 cell growth (IC50 = 0.2 μM), indicating a potential role in disrupting cancer cell proliferation. The synergy between halogen-substituted compounds (2O, 2K, 1C, 2M, and 2J) and their promising activity suggests the significance of electronegative substitutions in conferring potent anti-cancer effects with IC50 values ranging from 11.74 to 27.2 μM. In addition, compounds 2M and 2C showed notable antioxidant potential with IC50 values of 24.44 and 39.03 μM, respectively, in DPPH and ABTS assays, which is an attribute that may contribute to their cytotoxicity against cancer cells, given the established interplay between oxidative stress and cancer progression. While the precise pathways remain speculative at this stage, it is plausible that these compounds engage in intricate interactions with critical cellular components, potentially impacting cell cycle regulation and redox balance. These mechanisms could synergistically contribute to their observed anticancer efficacy. The elucidation of these mechanisms could unlock insights into novel strategies for breast cancer treatment. Overall, the results suggest that 4-aryl-4H-chromene derivatives with electronegative substitutions can be regarded as potential lead candidates for breast cancer treatment, particularly compounds 1L, 2O, 2K, 1C, 2M, and 2J, which exhibited promising biological activities in the evaluated assays. These compounds may provide a basis for further optimization and development of new, more effective therapies for breast cancer.
ACKNOWLEDGMENT
The authors are thankful to the Department of Pharmacology, Manipal College of Pharmaceutical Sciences, for their help in cell line studies. The authors are thankful to Manipal—Schrödinger Centre for Molecular Simulations, Manipal Academy of Higher Education, and Manipal College of Pharmaceutical Sciences, Manipal for providing the necessary resources and facilities for this study.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
ETHICAL APPROVALS
The study does not involve experiments on humans or animals.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality Worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49. CrossRef
2. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–66. CrossRef
3. Bhullar KS, Lagarón NO, McGowan EM, Parmar I, Jha A, Hubbard BP, et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer. 2018;17(1):1–20. CrossRef
4. Tadesse S, Anshabo AT, Portman N, Lim E, Tilley W, Caldon CE, et al. Targeting CDK2 in cancer: challenges and opportunities for therapy. Drug Discov Today. 2020;25(2):406–13. CrossRef
5. Zhang M, Zhang L, Hei R, Li X, Cai H, Wu X, et al. CDK inhibitors in cancer therapy, an overview of recent development. Am J Cancer Res. 2021;11(5):1913–35.
6. Zhong L, Li Y, Xiong L, Wang W, Wu M, Yuan T, et al. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal Transduct Target Ther. 2021;6(1):34–41. CrossRef
7. Zheng L, Yang Y, Bao J, He L, Qi Y, Zhang JZH. Discovery of novel inhibitors of CDK2 using docking and physics-based binding free energy calculation. Chem Biol Drug Des. 2022;99(5):662–73. CrossRef
8. Arun AK, Mohan K, Riyaz S. Structure guided inhibitor designing of CDK2 and discovery of potential leads against cancer. J Mol Model. 2013;19(9):3581–9. CrossRef
9. Ali Chohan T, Qian H, Pan Y, Chen JZ. Cyclin-dependent kinase-2 as a target for cancer therapy: progress in the development of CDK2 inhibitors as anti-cancer agents. Curr Med Chem. 2015;22:237–63. CrossRef
10. Pandey K, Park N, Park KS, Hur J, Cho YB, Kang M, et al. Combined cdk2 and cdk4/6 inhibition overcomes palbociclib resistance in breast cancer by enhancing senescence. Cancers (Basel). 2020;12(12):1–17. CrossRef
11. Basati G, Saffari-Chaleshtori J, Abbaszadeh S, Asadi-Samani M, Ashrafi-Dehkordi K. Molecular dynamics mechanisms of the inhibitory effects of abemaciclib, hymenialdisine, and indirubin on CDK-6. Curr Drug Res Rev. 2019;11(2):135–41. CrossRef
12. Roncato R, Angelini J, Pani A, Cecchin E, Sartore-Bianchi A, Siena S, et al. Cdk4/6 inhibitors in breast cancer treatment: potential interactions with drug, gene, and pathophysiological conditions. Int J Mol Sci. 2020;21(17):1–36. CrossRef
13. Senderowicz AM. Flavopiridol: the first cyclin-dependent kinase inhibitor in human clinical trials. Invest New Drugs. 1999;17(3):313–20. CrossRef
14. Xu H, Yu S, Liu Q, Yuan X, Mani S, Pestell RG, et al. Recent advances of highly selective CDK4/6 inhibitors in breast cancer. J Hematol Oncol. 2017;10(1):1–12. CrossRef
15. Whittaker SR, Mallinger A, Workman P, Clarke PA. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther. 2017;173:83–105. CrossRef
16. Schettini F, De Santo I, Rea CG, De Placido P, Formisano L, Giuliano M, et al. CDK 4/6 inhibitors as single agent in advanced solid tumors. Front Oncol. 2018;8:1–12. CrossRef
17. Sarhan MO, Abd El-Karim SS, Anwar MM, Gouda RH, Zaghary WA, Khedr MA. Discovery of new coumarin-based lead with potential anticancer, cdk4 inhibition and selective radiotheranostic effect: synthesis, 2d & 3d qsar, molecular dynamics, in vitro cytotoxicity, radioiodination, and biodistribution studies. Molecules. 2021;26(8):22–31. CrossRef
18. Abdelall EKA, Elshemy HA, Labib MB, Mohamed FE. Characterization of novel heterocyclic compounds based on 4-aryl-4H-chromene scaffold as anticancer agents: design, synthesis, antiprofilerative activity against resistant cancer cells, dual β-tubulin/c-Src inhibition, cell cycle arrest and apoptosis induction. Bioorg Chem. 2022;(120:10):55–91. CrossRef
19. Ashtekar SS, Bhatia NM. Synthesis of benzopyrans and evaluation of cytotoxicity against ER-MCF-7 cell lines. J Mol Struct. 2022;1268:72–81. CrossRef
20. Elshemy HAH, Zaki MA, Mahmoud AM, Khan SI, Chittiboyina AG, Kamal AM. Development of potential anticancer agents and apoptotic inducers based on 4-aryl-4H chromene scaffold: design, synthesis, biological evaluation and insight on their proliferation inhibition mechanism. Bioorg Chem. 2022;(11):11–8. CrossRef
21. Piyush K, Kuldeep S, Azizur RM, Misbahul HS, Pranay W. A review of benzopyran derivatives in pharmacotherapy of breast cancer. Asian J Pharm Clin Res. 2018;11(7):43–6. CrossRef
22. Raj V, Lee J. 2H/4H-Chromenes—a versatile biologically attractive scaffold. Front Chem. 2020;8:1–23. CrossRef
23. Zhan W, Lin S, Chen J, Dong X, Chu J, Du W. Design, synthesis, biological evaluation, and molecular docking of novel benzopyran and phenylpyrazole derivatives as Akt inhibitors. Chem Biol Drug Des. 2015;85(6):770–9. CrossRef
24. Xiu C, Hua Z, Xiao BS, Tang WJ, Zhou HP, Liu XH. Novel benzopyran derivatives and their therapeutic applications: a patent review (2009–2016). Expert Opin Ther Pat. 2017;27(9):1031–45. CrossRef
25. Gourdeau H, Leblond L, Hamelin B, Desputeau C, Dong K, Kianicka I, et al. Antivascular and antitumor evaluation of 2-amino-4-(3-bromo-4,5-dimethoxy-phenyl)-3-cyano-4H-chromenes, a novel series of anticancer agents. Mol Cancer Ther. 2004;3(11):1375–83. CrossRef
26. Kemnitzer W, Drewe J, Jiang S, Zhang H, Wang Y, Zhao J, et al. Discovery of 4-aryl-4H-chromenes as a new series of apoptosis inducers using a cell- and caspase-based high-throughput screening assay. 1. Structure-activity relationships of the 4-aryl group. J Med Chem. 2004;47(25):6299–310. CrossRef
27. Xi J, Zhu X, Feng Y, Huang N, Luo G, Mao Y, et al. Development of a novel class of tubulin inhibitors with promising anticancer activities. Mol Cancer Res. 2013;11(8):856–64. CrossRef
28. Pattar SV, Adhoni SA, Kamanavalli CM, Kumbar SS. In silico molecular docking studies and MM/GBSA analysis of coumarin-carbonodithioate hybrid derivatives divulge the anticancer potential against breast cancer. Beni-Suef Univ J Basic Appl Sci. 2020;9(1):1–12. CrossRef
29. Mali SN, Sawant S, Chaudhari HK, Mandewale MC. In silico appraisal, synthesis, antibacterial screening and DNA cleavage for 1,2,5-thiadiazole derivative. Curr Comput Aided Drug Des. 2019;15(5):445–55. CrossRef
30. Allegra M, Tutone M, Tesoriere L, Attanzio A, Culletta G, Almerico AM. Evaluation of the IKKβ binding of indicaxanthin by induced-fit docking, binding pose metadynamics, and molecular dynamics. Front Pharmacol. 2021;12:1–13. CrossRef
31. Srinivasa MG, Paithankar JG, Saheb Birangal SR, Pai A, Pai V, Deshpande SN, et al. Novel hybrids of thiazolidinedione-1,3,4-oxadiazole derivatives: synthesis, molecular docking, MD simulations, ADMET study, in vitro, and in vivo anti-diabetic assessment. RSC Adv. 2023;13(3):1567–79. CrossRef
32. Vosooghi M, Rajabalian S, Sorkhi M, Badinloo M, Nakhjiri M, Negahbani AS, et al. Synthesis and cytotoxic activity of some 2-amino-4-aryl-3-cyano-7- (dimethylamino)-4H-chromenes. Res Pharm Sci. 2010;5(1):13–8.
33. Matuszewska A, Jaszek M, Stefaniuk D, Ciszewski T, Matuszewski ?. Anticancer, antioxidant, and antibacterial activities of low molecular weight bioactive subfractions isolated from cultures of wood degrading fungus Cerrena unicolor. PLoS One. 2018;13(6):1–14. CrossRef
34. Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112–6. CrossRef
SUPPLEMENTARY MATERIAL
Supplementary data can be downloaded from the journal’s website [https://japsonline.com/admin/php/uploadss/4222_pdf.pdf]