
INTRODUCTION
Over 1 billion people worldwide are obese: 650 million adults, 340 million adolescents, and 39 million children. The World Health Organization (WHO) estimates that in 2025, around 167 million adults and children will become unhealthy due to being overweight or obese, so the WHO asks all countries to develop anti-obesity drugs or other drugs [1]. Data in Indonesia shows that around 13.5% of adults aged 18 years and over are overweight. In comparison, 28.7% are obese [body mass index (BMI) ≥ 25], and based on the indicators of the RPJMN (National Medium Term Development Plan) in 2015–2019, 15.4% were obese (BMI ≥ 27). The data results on children aged 5–12 were 18.8% overweight and 10.8% obese [2]. Since 2004, obesity can be described as the “New World Syndrome,” where its prevalence steadily increased globally in all age groups [3]. Obesity is a condition of abnormal or excessive fat accumulation, which can pose a health risk [1].
Many drugs have been used to prevent and treat obesity for many years. The use of anti-obesity drugs, such as phentermine, rimonabant, mazindol, diethylpropion, and sibutramine, has been withdrawn from the market by the Food and Drug Administration (FDA) and European Medicines Agency due to their side effects, such as tachycardia, palpitations, hypertension, fatigue, dizziness, insomnia, excessive excitement, tremors, changes in libido, palpitations, shortness of breath, anxiety, and chest pain. Drugs have a relationship with an increased risk of nonfatal severe cardiovascular events such as stroke and myocardial infarction and even hallucinations to suicidal ideation [4].
Only Orlistat has been approved by the FDA [5] and can be used long-term in treating obesity [4]. Orlistat works by reversibly inhibiting gastric and pancreatic lipase (PNLIP) enzymes. Lipase inactivation prevents hydrolysis of triglycerides so that free fatty acids (FAs) are not absorbed [6]. Several clinical trials have confirmed that Orlistat, a PNLIP inhibitor, can reduce obesity caused by a high-fat diet [7].
In the last 5 years, there has been a global increase in the use of herbal/traditional medicines (TMs) as complementary and alternative medicine developed by developing countries. TMs derived from plants can complement each other and act as an alternative treatment important in maintaining health [8]. Several studies have shown that herbal plants, as TMs, can act as lipase inhibitors in controlling obesity [9]. Indonesia, as a country with high diversity, uses the Hibiscus schizopetalus (Kembang sepatu gantung) as an ornamental plant, and the people of North Maluku use it to induce labor and improve the condition of pregnant women [10]; the people of Colombia use an infusion of H. schizopetalus flowers to treat colds and coughs [11]. In Malaysia, H. schizopetalus is also a medicinal plant because it contains chemical compounds, including anthocyanins and triterpene esters, such as antioxidants, antipyretic, anti-inflammatory, analgesic, hypoglycemic, and hypolipidemic [12]. Hibiscus schizopetalus was chosen in this study because it is suspected to have active compounds that act as lipase inhibitors. However, it is not known which active compounds can act as lipase inhibitors. Hibiscus schizopetalus is a plant from the Plantae kingdom, Tracheophyta division, Magnoliopsida class, Malvaceae family, and Hibiscus L genus [13].
Technological developments have resulted in drug development, starting with the stages of the bio-informatics method (in silico), which is simply a computer-based method and uses several software programs easily available on the public web [14]. In this in silico study, it is generally preferred at an early stage to predict and provide a temporary estimate of a study related to the activity of a ligand/compound. The in silico method does not require expensive costs and does not require a very long time [15]. Searching for target proteins and interactions of obesity proteins with active compounds in the H. schizopetalus plant is a step in this in silico study. The in silico method, especially regarding time and prediction of interactions with target proteins, provides good added value in this study. Furthermore, the results of these interactions will be compared with the commercial drug orlistat.
MATERIALS AND METHODS
Identification and screening of active compounds of H. schizopetalus
Plant active compounds can be obtained from an online database [http://www.knapsackfamily.com/ and http://ijah.apps.cs.ipb.ac.id/]. However, this active compound of H. schizopetalus has yet to be found in the online database because many researchers have not extensively studied this plant. The active compound obtained is an isolated component from the leaves and flowers of H. schizopetalus [16] using several research journals from PubChem web (https://pubchem.ncbi.nlm.nih.gov/), which have succeeded in isolating the active compound [17]. Protein codes of all active compounds were found using a database application [https://www.genecards.org/] [18].
Computer-based methods are often used as the initial criteria for eliminating compounds with poor pharmacokinetic and physiochemical profiles and toxicity levels [19]. The pharmacokinetic phase has four stages: absorption, distribution, metabolism, and excretion (ADME), including toxicity. Lipinksi’s role of five (Ro5) (1997) is used to predict the oral bioavailability of a drug. This rule is widely used as a filter for drug-like properties. This rule is also based on the physicochemical characteristics [20] of the tested compounds, among others: clog P ≤ 5, molecular weight (MW) ≤ 500 g/mol, number of hydrogen bond acceptors (HBA) (sum of N and O atoms) ≤ 10, number of hydrogen bond donors (HBD) (sum of OH and NH groups) ≤ 5, number of rotatable bonds ≤ 10, and polar surface area < 140 Å2 [21].
Parameters of the physicochemical and pharmacokinetic properties use the application (https://biosig.lab.uq.edu.au/pkcsm/) [22], and the toxicity test can be seen in the application (https://tox-new.charite.de/protox_II/) [23]. Effective medicines depend on the interaction between pharmacokinetics, toxicity, and potency. A substance’s pharmacokinetic profile determines its ADME characteristics [22].
The toxicity is predicted using the application ProTox-II. To predict various toxicity endpoints, such as acute toxins, hepatotoxicity, cytotoxics, carcinogenic, mutagenicities, immunotoxication, adverse outcomes pathways (Tox21), and toxicities targets, ProTox-II incorporates molecular similarity, pharmacophores, fragment propensities, and machine-learning models. The web server accepts a 2-D chemical structure as input. It outputs 33 models with confidence ratings, an overall toxicity radar chart, and the 3 most similar compounds with known acute toxicity. It also reports the chemical’s potential toxicity profile [23].
Obesity target prediction
The obesity gene is obtained from databases such as PubMed and STRING disease in the Cytoscape 3.9.1 application, which stores information for up to thousands of target genes. By using the keyword “obesity,” you will find obesity genes. The confidence value created is 0.9. In addition, to see the similarity of target genes, you can use DisGeNET (https://www.disgenet.org/search) by typing obesity.
This data collection is in a separate place to facilitate data processing. List the target genes in text format or Excel. If the data is complete, then each network construction is made. The target for obesity is done by analyzing the application (https://platform.opentargets.org/). Choose a target protein with an Overall Association Score value above 0.50.
Relation protein target-active compound
After obtaining the protein target of obesity and the protein target of the active compounds of H. schizopetalus, we performed the search of the relationship slices using the Venny diagram (https://bioinfogp.cnb.csic.es/tools/venny/). The entire list of each compound was then collected, and then through the STRING Database in Cytoscape 3.9.1, a retrieving network was carried out with a confidence value of 0.9 to form a target network of H. schizopetalus extract compounds.
The second network, the obesity gene network, is carried out similarly. A list of target genes was collected, and then a retrieving network was carried out with a confidence value of 0.9. The result will form an obesity network.
Next, carry out the process of making a gene slice that is the same between H. schizopetalus extract compounds and the obesity network by selecting “tools,” then “merge,” then selecting the desired network, and selecting “intersection.” Then, target genes will be found that can be used as therapeutic targets.
Protein network construction
Using CytoCluster, the formed network can be grouped into several nearby clusters to facilitate the hypothesis of possible pathways. The cluster is assessed based on closeness centrality, degree, closeness centrality, and betweenness values. The data is processed using an application to see the protein component network and targets (https://cytoscape.org/). This network of protein components and targets was obtained by interacting with active plant compounds with target codes for each active compound using a database [https://www.genecards.org/]. The network construction will show the interaction between the active compound of H. schizopetalus and the target code of obesity.
Pathway and enrichment analysis
Analysis of interactions between genes/proteins can be carried out with the help of the STRING platform https://string-db.org/, STITCH http://stitch.embl.de/, and Human References Interactome http://www.interactome-atlas.org/.
The results of the analysis of STRING can lead researchers to further analyze the possible pathways as interventions from test substances. Accessible platforms for this analysis include DAVID: Functional Annotation Tools (ncifcrf.gov), metascape.org, and the KEGG Pathways database [https://-www.genome.jp/]. To obtain the target protein that has the desired pharmacological effect [24], a specific protein as the best target can be searched using the metascape application (https://metascape.org/) and KEGG (https://www.kegg.jp/).
Macromolecule preparation
The ligand and target structures must be prepared in advance. The target macromolecule/protein attached to the complex can be downloaded via the https://www.rcsb.org/ platform. The structure is then prepared by removing water molecules, adding hydrogen atoms, and optimizing amino acids. Meanwhile, the ligand is released and stored in program database [PDB] format for grid box optimization in the method validation process. This preparation can be done using the Biovia Discovery Studio application. The molecular structure can be obtained by downloading the 3-D structure from Databank (https://www.rcsb.org) [25] and converting it into a PDB file using UCSF Chimera (https://www.cgl.ucsf.edu/chimera/) [26].
Simulation of molecular docking and visualization of docking results
Molecular docking or the blind docking process can be done using the Pyrx Vina application (https://pyrx.sourceforge.io/) [27]. Then, to visualize the docking results, BIOVIA application is used (https://discover.3ds.com/discovery-studio).
Molecular docking validation
The native ligand that had been separated was then re-attached to the macromolecules using the AutoDock 4.0 application. This is done to get the grid box value associated with the active site of the macromolecule.
The appropriate grid box parameter, with a reference value of root mean square deviation (RMSD) < 2 A, will be the benchmark for the next virtual screening.
Validation for molecular docking will be conducted using the auto dock application (AutoDockTools-1.5.7) [28], where at the time of this validation, native ligands are used to determine the grid box that will be used for docking macromolecules and ligands to form a complex ligand-protein. The interactions and binding of these complex ligand proteins will be further analyzed through dynamic molecular simulations.
Molecular dynamics (MDs) simulation
MDs simulation was carried out using the YASARA Dynamics software package (http://www.yasara.org/). The hit’s finest pose from the virtual showing was chosen, and YASARA Structure’s scene mode was then set up using the default mode [29]. Complex ligand-protein from the AutoDock application will be uploaded in YASARA software. It will use NaCl 0.9%, and the temperature will be set at 298K for simulation. Water density is 0.997, and to represent solvation on water molecules using a cell shape cubic box. The whole system was neutralized to pH 7.4 and used at normal speed. The length of the MD run was 34.60 ns, and the forcefield used AMBER14. Hydrogen bonds, RMSD of protein (backbone and side chains), and root mean square fluctuation (RMSF) of amino acid residues were used to evaluate the ligand-protein interaction [30,31].
RESULT AND DISCUSSIONS
Active compounds of H. schizopetalus
The physicochemical and pharmacokinetic properties of active compounds were predicted, as shown in Tables 1 and 2 [17]. Each of these compounds is then downloaded in SMILES canonical form, 2-D and 3-D structures in spatial data file format using the facilities available in PubChem (https://pubchem.ncbi.nlm.nih.gov). When choosing the most suitable structure is important to match the MW and molecular formula according to the LC/GC-MS information for the extract. Each compound was then selected for its bioavailability value through the Swiss ADME platform (http://www.swissadme.ch/index.php) and SCFBio (http://www.scfbio-iitd.res.in/software/drugdesign/lipinski.jsp. The result is 14 compounds found in H. schizopetalus with a bioavailability value of 0.17–0.55, as shown in Table 2.
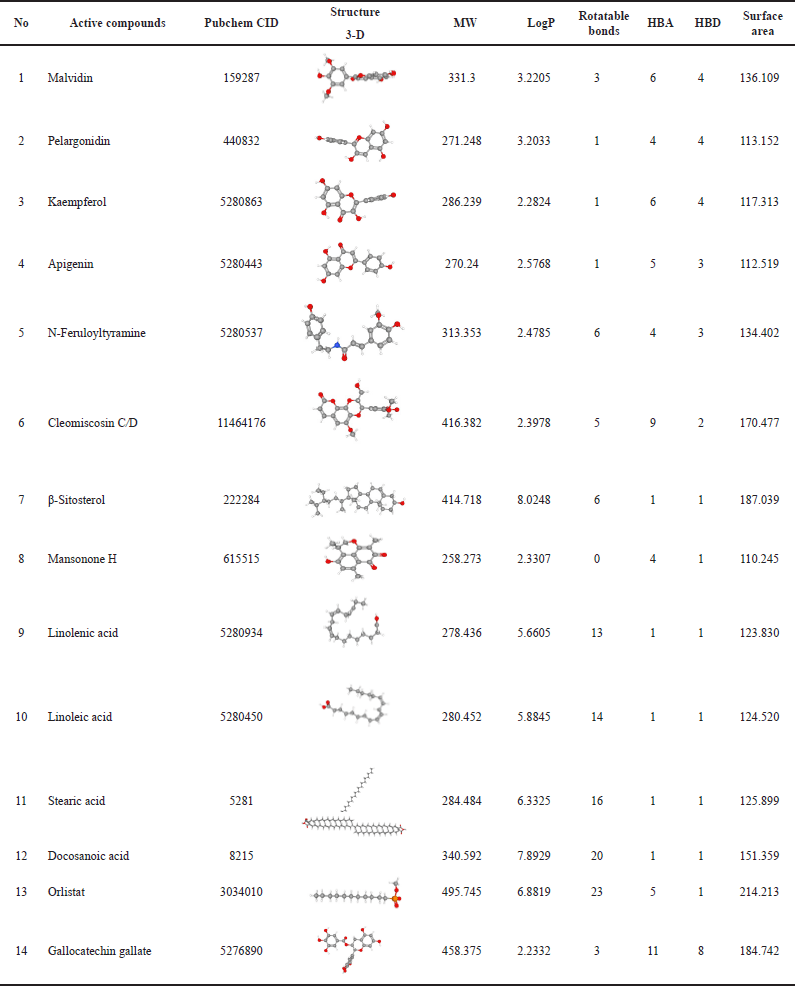 | Table 1. Physicochemistry properties. [Click here to view] |
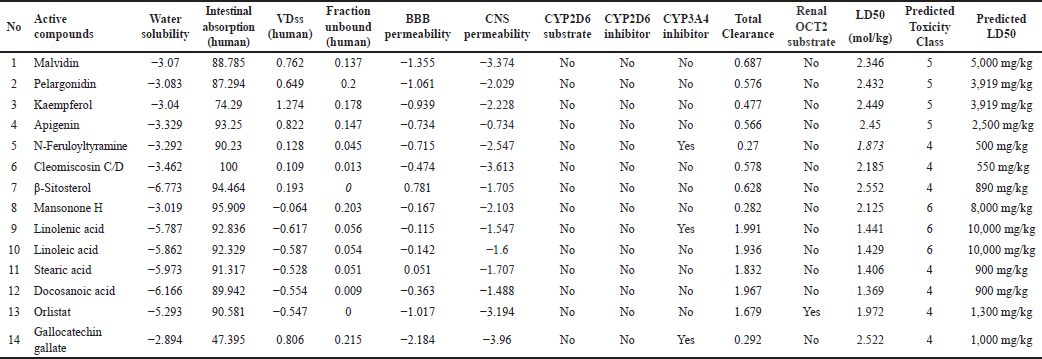 | Table 2. Pharmacokinetics properties. [Click here to view] |
Relation protein target obesity-active compound
There are 165 genes related to the pathogenesis of obesity and 196 genes that are the target of H. schizopetalus compounds. Some of the web-based servers used are SwissPrediction (http://www.swisstargetprediction.ch/), PharmMapper (http://www.lilab-ecust.cn/pharmmapper/), GeneCards V4.12 (https://www.genecards.org/), OMIM (https://omim.org/) Pharmgkb (https://www.pharmgkb.org/), TTD (http://db.idrblab.net/ttd/), (http://www.way2drug.com/passonline/), and Pharmacology Browser 2 (PPB2) https://ppb2.gdb.tools/. Retrieval of target genes is carried out on more than one web-based platform because not all compound structures can be identified.
The results of the description of the number of genes involved using https://bioinformatics.psb.ugent.be/webtools/Venn/ can be seen in Figure 1.
It was seen that 11 genes were the same between obesity targets and H. schizopetalus compounds. If calculated, as many as 7.14% of obesity genes are targets of H. schizopetalus compounds. This means that H. schizopetalus can interfere with many of the genes involved in obesity.
From here, it can then be filtered for genes that tend to have a close relationship with one another based on the values of closeness centrality, degree, closeness centrality, and betweenness values. The grouping above uses CytoCluster, the choice of ClusterONE algorithm, and then other parameters based on default. Then, several clusters with different confidence values will be formed, so Cluster 5 is chosen, which has a p-value of 0.001, as shown in Figure 2a and b.
Protein network construction
Based on the Venn diagram in Figure 1 and the construction of the target protein and active compounds in Figure 2a and b, it is obtained that 11 target proteins can be further analyzed. The target proteins are CREB binding protein (CREBBP), E1A binding protein p300 (EP300), peroxisome proliferator-activated receptor gamma (PPARG), cannabinoid receptor 1 (CNR1), PNLIP, prostaglandin-endoperoxide synthase 2 (PTGS2), vitamin D receptor (VDR), carbonic anhydrase 2 (CA2), prostaglandin-endoperoxide synthase 1 (PTGS1), estrogen receptor 1 (ESR1), and estrogen receptor 2 (ESR2). From the 11 target proteins, we then did visualization using Cytoscape (https://cytoscape.org/) as shown in Figure 3 below.
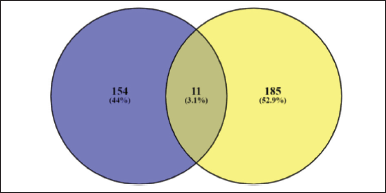 | Figure 1. Venny diagram of protein target obesity (blue) and H. schizopetalus (yellow). [Click here to view] |
 | Figure 2. (a) Network construction obesity and active compound. (b) Combined network construction and active compound. [Click here to view] |
From the 11 target proteins above, the protein code is searched using the protein database [https://www.rcsb.org/], and the data obtained are shown in Table 3.
The topology above can be interpreted as an identification of the role of genes in the network. The degree can be interpreted as whether the gene is directly connected to other genes in the network; closeness centrality is how close the gene is to other genes indirectly.
Pathway and enrichment analysis
KEGG pathway
The gene is then analyzed in terms of its enrichment and gene ontology to see the most likely pathways and to see that interaction analysis between genes/proteins can be carried out with the help of the STRING platform https://string-db.org/, DAVID: Functional Annotation Tools (ncifcrf.gov) and metascape.org, which is linked to other gene ontology servers such as the GAD Disease Genetic Association Disease Database [https://geneticassociationdb.nih.gov/] and the KEGG Pathways database [https://-www.genome.jp/] and Human References Interactome http://www.interactome-atlas.org/. The group can identify overrepresented genes in obesity networks using KEGG pathways [23]. The result from KEGG is known to have four signaling pathways: glycerolipid metabolism, metabolic pathway, pancreatic secretion, fat digestion and absorption, and vitamin digestion and absorption. Then, each signal pathway is analyzed, and the most likely pathway associated with obesity is the fat digestion and absorption pathway, as shown in Figure 4 below.
According to Figure 4 above, it can be seen that lipase plays an important role in the upstream section. Lipase is the main lipase enzyme secreted from the pancreas, which hydrolyzes dietary fats in the digestive system, converting triacylglycerol (TG) substrates found in digested oils into monoglycerides and free fats. After the human body digests the fat-containing food, the lipids containing triglycerides are first hydrolyzed by lipase into monoglycerides, glyceryl esters, and free FAs, and the content of 1,2-glycolide and FAs in the product is higher. Then, the fat enters the body and is further hydrolyzed into monoacylglycerol (MG) and FA by gastric lipase (10%~30% decomposition) and PNLIP (50%~70% decomposition) in the digestive tract and small intestine such as in Figure 4. Then, cholesterol and lipoproteins (LPAs) are formed in the body. MG, FA, cholesterol, LPA, and bile acids are absorbed by the small intestine, and then TG is re-synthesized as energy storage for adipose tissue. Then this adipose tissue will secrete excess fat, which, if not removed, will result in fat accumulation and can cause obesity [7].
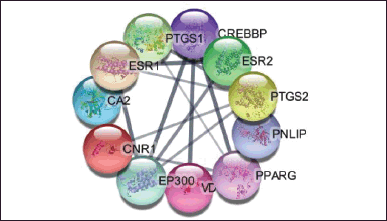 | Figure 3. Protein target of obesity-active compound. [Click here to view] |
Figure 5 shows that the enrichment analysis related to the metabolic process is carried out by the PNLIP protein through the olefinic compound metabolic pathway so that this PNLIP protein will be further analyzed.
Simulation molecular docking protein targets and active compounds
The PNLIP (PDB ID: 1LPB) macromolecule [42] was obtained from the information contained in the active compounds in H. schizopetalus extract, where analysis has been carried out related to the target of obesity. Native ligands methoxyundecylphosphinic acid C12H27O3P derived from 3-D macromolecular structures (PDB ID: 1LPB).
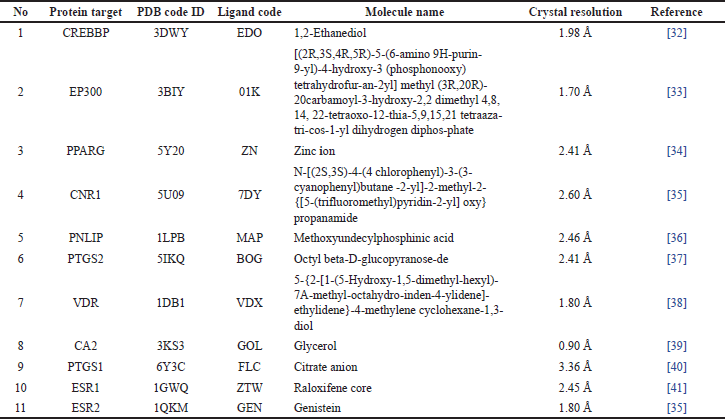 | Table 3. Complex proteins and ligands from the PDB database. [Click here to view] |
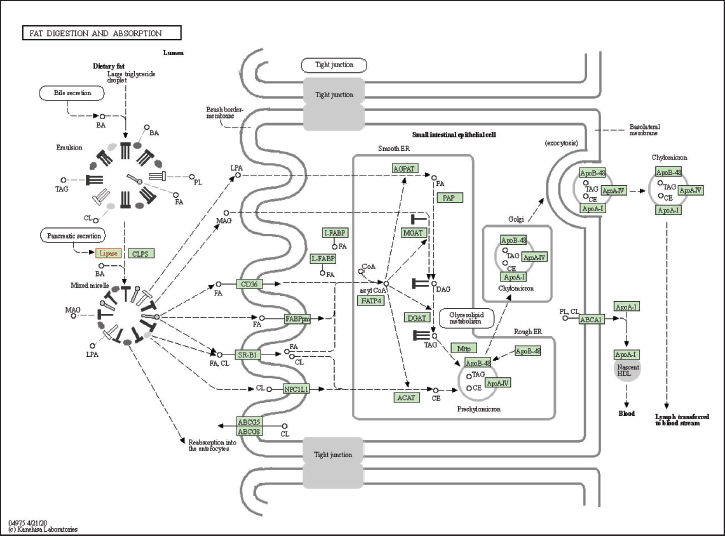 | Figure 4. KEGG pathway of fat digestion and absorption. [Click here to view] |
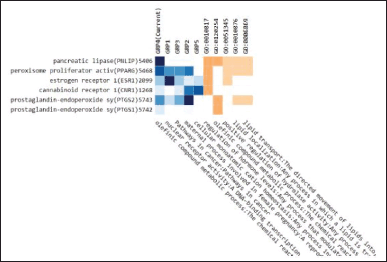 | Figure 5. Enrichment analysis from 11 proteins. [Click here to view] |
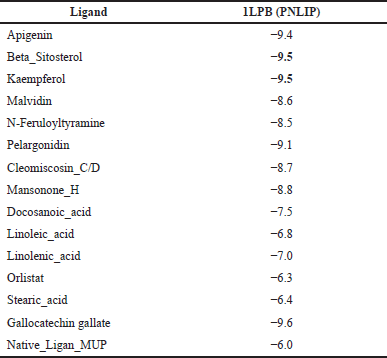 | Table 4. Binding affinity of the protein target and active compound of H. schizopetalus. [Click here to view] |
The protein target (PNLIP) was then docked with 14 active compounds with known pharmacokinetic and physiochemical properties and 1 native ligand to determine which target protein has the most powerful role in obesity. The PNLIP (PDB ID: 1LPB) macromolecule [42] was obtained from the information contained in the active compounds in H. schizopetalus extract. An analysis has been carried out related to the target of obesity. The results of the docking can be seen in Table 4.
Preparation of ligands from active compounds of H. schizopetalus
The molecular structure of the active compounds from H. schizopetalus and Orlistats as commercial drugs were obtained by downloading the 3-D structure from the database [https://pubchem.ncbi.nlm.nih.gov/] and has been converted to PDB format. The structures of best hit compounds can be seen in Table 1. The docking scores and binding activity of the hits compound are shown in Table 5
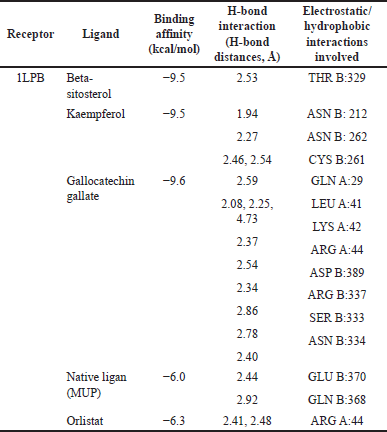 | Table 5. Docking scores, H-bond interactions, electrostatic/hydrophobic interactions, and the binding affinity of the best hit compound BIOVIA Discovery Studio. [Click here to view] |
Visualization of docking results
Interaction of best hit compounds with ligand on receptor using BIOVIA Discovery Studio
Based on the results of this bioinformatics study, one of the most potent compounds is gallocatechin gallate, as shown in Figure 6a–e. (−)-gallocatechin gallate is a gallate ester formed by the formal condensation of gallic acid’s carboxy group with the (3R)-hydroxy group of (−)-gallocatechin. Green tea contains a natural substance. It functions as an inhibitor of EC 3.4.22.69 (severe acute respiratory syndrome coronavirus major proteinase), a human xenobiotic metabolite, an antineoplastic drug, and a plant metabolite. It contains gallate ester, polyphenol, and catechin. It is structurally similar to (−)-gallocatechin and gallic acid. It is the (+)-gallocatechin gallate enantiomer.
Molecular docking validation using AutoDock tools
Grid box validation requires the native ligand MUP structure and 1LPB macromolecule in PDB format. This validation is necessary for the formation of a ligand-protein complex. Validation is done with three types of grid box sizes, as shown in Table 6. The grid box was set to the center position of the ligand.
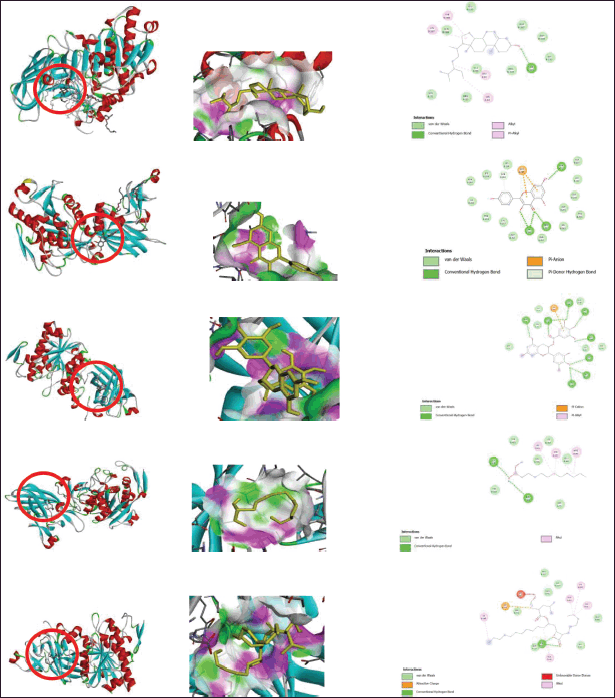 | Figure 6. (a) Interaction receptor 1LPB with beta-sitosterol 2-D (left) and 3-D (right). (b) Interaction receptor 1LPB with kaempferol 2-D (left) and 3-D (right). (c) Interaction receptor 1LPB with gallocatechin gallate 2-D (left) and 3-D (right). (d) Interaction receptor 1LPB with native ligand MUP 2-D (left) and 3-D (right). (e) Interaction receptor 1LPB with Orlistat 2-D (left) and 3-D (right). [Click here to view] |
Redocking using AutoDock tools
The RMSD parameter of heavy atoms between the molecular docking conformation and experimental findings (crystallography), which should be no more than 2–3 Å, can be used to evaluate the validity of a molecular docking process. The grid box dimensions were 60 each. The target protein was prepared, and the grid box was set to the center position of the ligands, as shown in Table 7.
MDs simulation
Based on the docking results of the formation of the complex ligand-protein, it was known that the active compound gallocatechin gallate has the highest binding energy, so this active compound will be analyzed for MD analysis. The results of the simulation of MDs at 34.60 ns are shown in Table 8.
MDs simulation results of PNLIP proteins against the active compound gallocatechin gallate showed binding stability at times above 25 ns. RMSF shows the binding site’s stability between the compounds with the target protein [43]. RMSF in Table 8 shows a value close to zero, which means no residue was detected on the molecular bond above.
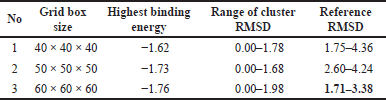 | Table 6. Docking validation result. [Click here to view] |
 | Table 7. Molecular docking using AutoDock tools. [Click here to view] |
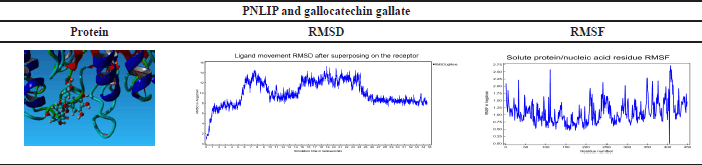 | Table 8. RMSD and RMSF result of PNLIP and gallocatechin gallate MDs. [Click here to view] |
CONCLUSION
The study showed the interaction of protein in obesity with active compounds in the H. schizopetalus plant. The search for interactions of obesity proteins and active compounds was obtained using computational techniques from online databases. The results showed that 11 proteins interact with each other. Then, the target protein search was supported by enrichment analysis, and it was known that PNLIP protein has a role in obesity. The group used PDB ID: 1LPB as a macromolecule to dock the 14 active compounds.
Orlistat is a commercial drug already circulating on the market and used as a comparison drug of active compounds. Using network pharmacology shows that gallocatechin gallate has a higher binding energy than Orlistat. This research needs to continue to the next stage of the study in vivo to prove the truthfulness of this in-silico method.
AUTHORS CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
The Directorate of Research and Development, Universitas Indonesia, funded this research under Hibah PUTI 2022 Batch 3 (Grant No. NKB-1371/UN2.RST/HKP.05.00/2022).
CONFLICTS OF INTERESTS
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published
REFERENCES
1. WHO. World obesity day 2022—accelerating action to stop obesity. Geneva, Switzerland: WHO; 2022. pp 13–4.
2. Ayuningtyas D, Kusuma D, Amir V, Tjandrarini DH, Andarwati P. Disparities in obesity rates among adults: analysis of 514 districts in Indonesia. Nutrients. 2022;14(16):3332. CrossRef
3. Nammi S, Koka S, Chinnala KM, Boini KM. Obesity: an overview on its current perspectives and treatment options. Nutr J. 2004;3:1–8. CrossRef
4. Seyedan A, Alshawsh MA, Alshagga MA, Koosha S, Mohamed Z. Medicinal plants and their inhibitory activities against pancreatic lipase: a review. Evid Based Complement Altern Med. 2015;2015:973143. CrossRef
5. Idrees Z, Cancarevic I, Huang L. FDA-approved pharmacotherapy for weight loss over the last decade. Cureus. 2022;14(9):e29262. CrossRef
6. Bansal AB, Al Khalili Y. Orlistat. Treasure Island, FL: StatPearls Publishing; 2022. pp 1–10.
7. Liu TT, Liu XT, Chen QX, Shi Y. Lipase inhibitors for obesity: a review. Biomed Pharmacother. 2020 Aug;128:110314. CrossRef
8. WHO. National policy on traditional medicine and regulation of herbal medicines. Report of a WHO global survey. Geneva, Switzerland: WHO; 2005. p 168.
9. Sun W, Shahrajabian MH, Cheng Q. Natural dietary and medicinal plants with anti-obesity therapeutics activities for treatment and prevention of obesity during lock down and in post-COVID-19 era. Appl Sci. 2021;11(17):7889. CrossRef
10. Wijaya NR, Dewi TF. Keanekaragaman Spesies Tumbuhan Obat untuk Perawatan Sebelum dan Sesudah Persalinan pada Beberapa Suku di Maluku Utara. Bul Plasma Nutfah. 2020;26(2):145. CrossRef
11. Zahid H, Rizwani GH, Shareef H, Khursheed R, Huma A, Hasan SMF. Antihyperglycemic and hypolipidemic effects of Hibiscus schizopetalus (Mast) Hook in alloxan-induced diabetic rats. Pak J Pharm Sci. 2014;27(1):83–9.
12. Wong SK, Chan EWC, Chan HT. A review on the phytochemistry and pharmacology of two lesser-known Hibiscus species: H. taiwanensis and H. schizopetalus. Int J Pharmacogn Phytochem Res. 2016;8(8):1341–6.
13. Ahmed A, Eisa S, Mohamed AEH, Al-Shamey I. Taxonomic revision and numerical analysis of Hibiscus L. in Egypt. Arab Univ J Agric Sci. 2020. CrossRef
14. Suharna S, Suharna S. Studi in silico Senyawa Turunan Flavonoid Terhadap Penghambatan Enzim Tirosinase [Undergraduate thesis]. Makassar, Indonesia: Universitas Islam Negeri Alauddin Makassar; 2012.
15. Mirza DM. Studi in silico dan in vitro aktivitas antineuroinflamasi ekstrak etanol 96% daun Marsilea crenata C Presl. [Undergraduate thesis]. Malang, Indonesia: Universitas Islam Negeri Maulana Malik Ibrahim; 2019. pp 1–134.
16. El-Shiekh RA, Abdelmohsen UR, Ashour HM, Ashour RM. Novel antiviral and antibacterial activities of Hibiscus schizopetalus. Antibiotics. 2020;9(11):1–16. CrossRef
17. El-Shiekh RA, Abdelmohsen UR, Ashour HM, Ashour RM. Novel antiviral and antibacterial activities of Hibiscus schizopetalus. Antibiotics. 2020;9(11):1–16. CrossRef
18. Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, et al. GeneCards version 3: the human gene integrator. Database. 2010;2010:baq020. CrossRef
19. Ntie-Kang F, Lifongo LL, Mbah JA, Owono Owono LC, Megnassan E, Mbaze LM, et al. In silico drug metabolism and pharmacokinetic profiles of natural products from medicinal plants in the Congo basin. In Silico Pharmacol. 2013 Aug 8;1:12. CrossRef
20. Vilar S, Chakrabarti M, Costanzi S. Prediction of passive blood-brain partitioning: straightforward and effective classification models based on in silico derived physicochemical descriptors. J Mol Graph Model. 2010;28(8):899–903. CrossRef
21. de Brito MA. Pharmacokinetic study with computational tools in the medicinal chemistry course. Brazil J Pharm Sci. 2011;47(4):797–805. CrossRef
22. Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58(9):4066–72. CrossRef
23. Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46(W1):W257–63. CrossRef
24. Tipney H, Hunter L. An introduction to effective use of enrichment analysis software. Hum Genomics. 2010;4(3):202–6. CrossRef
25. Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, et al. The protein data bank. Acta Crystallogr D Biol Crystallogr. 2002 Jun;58(Pt 6 No 1):899–907. CrossRef
26. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004 Oct;25(13):1605–12. CrossRef
27. Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. In: Hempel J, Williams C, Hong C, editors. Chemical biology. Methods in molecular biology. New York, NY: Humana Press; 2015. pp 243–50.
28. Kintamani E, Batubara I, Kusmana C, Tiryana T, Mirmanto E. Andaliman (Zanthoxylum acanthopodium) sebagai anti SARS-COV-2 berdasarkan penambatan molekul [The potential of Andaliman (Zanthoxylum acanthopodium) essential leaves oil as anti SARS-Cov-2 based on molecular docking]. Life. 2022;3:167–78.
29. Prasasty VD, Istyastono EP. Structure-based design and molecular dynamics simulations of pentapeptide aeytr as a potential acetylcholinesterase inhibitor. Indones J Chem. 2020;20(4):953–59. CrossRef
30. Kwofie SK, Broni E, Asiedu SO, Kwarko GB, Dankwa B, Enninful KS, et al. Cheminformatics-based identification of potential novel anti-SARS-CoV-2 natural compounds of African origin. Molecules. 2021 Jan 14;26(2):406. CrossRef
31. Zuhri UM, Purwaningsih EH, Fadilah F, Yuliana ND. Network pharmacology integrated molecular dynamics reveals the bioactive compounds and potential targets of Tinospora crispa Linn. as insulin sensitizer. PLoS One. 2022;17(6):1–15. CrossRef
32. Spiliotopoulos D, Caflisch A. Molecular dynamics simulations of bromodomains reveal binding-site flexibility and multiple binding modes of the natural ligand acetyl-lysine. Israel J Chem. 2014;54(8–9):1084–92. CrossRef
33. Atmajani W, Buyung Kurniawan A, Hapsari R, Santoso B. In silico study of agonists of PPAR gamma-receptor protein (5Y2O) as anthiperglicemia using dock 6. Urecol. 2019;9:193–200.
34. Atmajani W, Kurniawan AB, Hapsari R, Santoso B. Kajian in silico agonis PPAR gamma-receptor protein (5y2o) sebagai antihiperglikemia menggunakan dock6. The 9th University Research Colloqium 2019 Universitas Muhammadiyah Purworejo, 2019, vol. 9, no. 5, pp 193–200.
35. Fauzi FA, Goh MS, Johari SATT, Hashim F, Hassim MFN. Dream over life: psychedelic terphenyl derivative induce hallucination via cannabinoid receptor 1. IOP Conf Ser Mater Sci Eng. 2018;440(1):012045. CrossRef
36. Chen KY, Sen Chang S, Chen CYC. In silico identification of potent pancreatic triacylglycerol lipase inhibitors from traditional Chinese medicine. PLoS One. 2012;7(9):1–11. CrossRef
37. Pratama AB, Herowati R, Ansory HM. Studi docking molekuler senyawa dalam minyak atsiri pala (Myristica fragrans H.) dan senyawa turunan miristisin terhadap target terapi kanker kulit. Maj Farm. 2021;17(2):233. CrossRef
38. Kawar N, Maclaughlan S, Horan TC, Uzun A, Lange TS, Kim KK, et al. PT19c, another nonhypercalcemic vitamin D2 derivative, demonstrates antitumor efficacy in epithelial ovarian and endometrial cancer models. Genes Cancer. 2013 Nov;4(11–2):524–34. CrossRef
39. Lomelino CL, McKenna R. Carbonic anhydrase II in complex with carboxylic acid-based inhibitors. Acta Crystallogr Sect F Struct Biol Commun. 2019;75:166–70. CrossRef
40. Miciaccia M, Belviso BD, Iaselli M, Cingolani G, Ferorelli S, Cappellari M, et al. Three-dimensional structure of human cyclooxygenase (hCOX)-1. Sci Rep. 2021;11(1):1–19. CrossRef
41. Hung TC, Lee WY, Chen KB, Chan YC, Chen CYC. Investigation of estrogen receptor (ESR1) for breast cancer from traditional Chinese medicine. Biomed Res Int. 2014;2014:321486. CrossRef
42. Gandhi SP, Lokhande KB, Swamy VK, Nanda RK, Chitlange SS. Computational data of phytoconstituents from Hibiscus rosa-sinensis on various anti-obesity targets. Data Brief. 2019;24:103994. CrossRef
43. Decherchi S, Grisoni F, Tiwary P, Cavalli A. Editorial: molecular dynamics and machine learning in drug discovery. Front Mol Biosci. 2021;8:673773.