
INTRODUCTION
The most harmful form of dementia, Alzheimer’s disease AD, has been studied for the past 50 years, but its root cause and optimal treatment are still unknown. It has been strongly linked to pathogenic factors including tau, or microtubule-associated protein, and amyloid (Aβ). The combination of both of these risky elements contributes to the emergence of neurofibrillary tangles. [1]. Numerous essential transcription factors (TFs) have a substantial impact on the root causes and emergence of AD [2]. Repressor element-1 silencing transcription factor (REST), generally assigned the name “neuron-restrictive silencer factor, is an essential transcriptional repressor that is typically expressed throughout embryogenesis and plays a critical role in regulating the enormous neural genes that have distinct roles assigned specifically to neurons. It has been discovered that the majority of the genes that regulate neurotransmitter synthetases, synaptic vesicle proteins, transporters, receptors, and some ion channel-modifying genes play substantial functions in the central nervous system (CNS). However, many of the aforementioned facts are still not fully explored due to the lack of minimum research on this particular TF [3–7]. But since REST’s probable role in causing AD is still being critically studied, further research is needed in this area. We do, however, highlight some important research from the body of literature that supports the impact of REST on AD. In AD patients, there was a noticeable decrease in the nuclear REST level in neurons from the memory-focused region, particularly those from the CA1, CA3, and CA4 regions of the hippocampus, as well as prefrontal cortical neurons [8]. In contrast to cognitively healthy control subjects, a recent study underscored the diminished REST levels in plasma neuronal-derived exosomes from stable mild cognitive impairment (MCI) patients, MCI patients who are transitioning to AD, and AD people [9]. REST deficit consequently manifests in sporadic AD and persists in differentiated neurons, indicating that it might be the primary etiology of AD. Nuclear translocation deficit has been established as the defining feature of REST-related diseases, including AD [8] and Parkinson’s disease [10]. However, it was found that augmented REST nuclear translocation from the cytoplasm is a pathological feature in Huntington’s disease [11]. Despite conflicting evidence in this paradigm, the principal impacts of the loss of REST function in the neurons of sporadic AD patients have been recognized as nuclear translocation defect from the cytoplasm to neurons and nuclear lamina disruption [12]. Lue and colleagues showed the neuroprotective role of REST in AD patients by triggering oxidative stress resistance genes and suppressing apoptosis-inducing genes that promote Aβ toxicity [8]. Age-related diseases like AD have been speculated to be significantly influenced by cellular senescence. In the brains of AD patients and AD animal models, the senescence phenomenon attributed to crucial brain components such as neurons, astrocytes, and microglia was visible [13]. Importantly, REST was associated with aging and neurodegenerative conditions. REST was required to halt the senescence characteristic in mouse primary neurons [14]. REST deficit has the extraordinary capacity to alter the autophagy process, cause proteostasis to break down, increase oxidative stress, and accelerate cell mortality, making this TF an essential epigenetic entity during aging [14]. Long-lived daf-2 mutants with loss-of-function mutations of REST ortholog genes spr-3 and spr-4 in the C aenorhabditis elegans, were reported with diminished life expectancies and augmented neuronal excitation [15]. Thus, REST is linked to increased longevity and controls neuronal excitation in the aging brain [15]. Along with suppressing several apoptotic genes such as p38 map kinase MAPK11, Tumor necrosis factor receptor type 1-associated DEATH domain protein, and death domain associated protein (DAXX), REST also targets γ-secretase complex essential components that stimulate Aβ synthesis [8]. The repressive influence of REST on these AD-causing genes states the epigenetic remodeling mechanism crucial to progressive neurodegeneration.
REST has lately been studied in neurodegenerative diseases irrespective of its connection with neurodevelopment [10,12,16,17]. The regulatory mechanism of REST in neurological disorders, especially AD, is not clear and demands more research in this area. In addition, the targets of REST and the essential corepressors of REST have not been entirely explored in AD. Therefore, it is crucial to identify the interacting partners of REST and the resulting biological function that they regulate under the circumstances of AD.
We hypothesize that REST is a key regulatory protein that has a beneficial role in AD, through its interactions with the proteins involved in the progression of AD. However, no studies have been conducted on how REST regulates the differentially expressed genes (DEGs) in AD or how other DEGs in AD regulate REST. Hence, the objective of this study is to investigate the regulatory mechanism of REST via several DEGs in AD including multiple TFs that regulate REST during AD conditions. Similar studies have been carried out in other diseases such as atherosclerosis and cervical cancer, which involve the identification of DEGs and protein–protein interaction networks [18,19]. However, the methodology adopted in this study to delineate the mechanism of a transcriptional regulator is novel and has not been reported elsewhere to the best of our knowledge.
MATERIALS AND METHODS
Gene expression data
The gene expression omnibus (GEO) provided a wealth of knowledge on the expression of genes [20]. For this analysis, we used the microarray gene expression information of 17 human brain samples with AD and 19 healthy control samples (GEO accession number GSE138260). The healthy control samples do not have any history of neurological or psychiatric illness. The DEGs ended up being identified using the GEO2R program, which assessed the raw gene expression data. The p-value of 0.05 and logFC values were used to figure out DEGs that were statistically significant as described previously [19]. DEGs went up when logFC values were positive and diminished when logFC values were negative.
REST regulating genes
The REST regulating genes were identified as described previously [21]. It was feasible to determine the genes governing REST and the genes that it controls from the TF-Gene interactions by using the pertinent data from the TRRUST [22], hTFtarget [23], and transcription co-factors (TcoF-DB) [24] database. TRRUST is a database of transcriptional regulatory networks for humans and mice. This collection contains the TF-target regulatory relationships of 800 human TFs that articulate small-scale experimental studies of transcriptional control. Additionally, it offers details pertaining to the type of regulation (activation or suppression). Large-scale ChIP-Seq data of human TFs (7,190 experiment samples of 659 TFs) in 569 settings (399 types of cell line, 129 classes of tissues or cells, and 141 kinds of therapies) have been compiled by hTFtarget to create complete TF-target regulations. The Database of TcoF-DB and transcription factor interactions makes it easier to investigate the genes encoding for proteins that regulate gene transcription in humans and mice by attaching to regulatory regions of DNA (TFs) and the genes encoding for proteins that have interactions with TFs but are unable to attach directly to regulatory DNA regions (transcription co-factors). For this study, only REST interactions among humans were taken into account from all of these datasets.
Protein-protein interactions
The interactions among the REST regulating proteins were identified as discussed previously [18]. We used the STRING database, which is widely recognized for illustrating data pertaining to interactions between proteins [25]. Using this database, we were able to determine the DEGs responsible for the regulatory framework for REST through their interactions (physical and functional links, both direct and indirect). To ensure that all interactions were empirically validated and, therefore, legitimate in order to be taken into consideration for this investigation, the minimum needed interactions score was set to the highest confidence level of 0.9. The first shell interactions that involved the query proteins only were considered. The results were retrieved in the form of tabular text.
Gene annotation
Gene annotation was carried out as per a previously reported study [19]. The annotation information such as biological processes, cellular components, and tissue expression of the genes involved in the regulatory mechanism of REST was retrieved from the database for annotation, visualization, and integrated discovery database [26]. This database provides a full range of functional annotation tools that allow researchers to gain insight into the biological relevance of a huge number of genes.
Network construction and analysis
The networks of protein interactions and REST regulatory systems were built using Cytoscape v3.9.0. [27]. It is a free and open-source software platform for portraying intricate networks and merging them with all attribute data. The maximal clique centrality strategy employed by CytoHubba was implemented to pinpoint the hub proteins in the protein-protein interactions of the DEGs that operate via REST [28]. A number of topological techniques are used by the Cytoscape plugin, CytoHubba to anticipate and explore significant nodes and subnetworks in a given network [18]. The hub proteins have been demonstrated to have greater interactions (degrees) than other proteins, relative to the protein-protein interaction network. During the process of developing new drugs, they provide essential information for selecting or classifying targets. Additionally, another Cytoscape plugin, MCODE, was employed to establish the network’s strongly interconnected regions or clusters [29,18].
RESULTS
Gene expression data analysis
The microarray study involved 19 samples (violet) from AD patients and 17 samples (green) from healthy controls (Fig. 1a). The median of each of the chosen samples has been normalized, and their values are identical. It guarantees that each sample that was chosen could be used for differential expression analysis. The DEGs are depicted using a volcano plot (Fig. 1b). From the Venn diagram, it is evident that 97,457 genes -are differential expressed with padj value <0.05 among the 850,125 total analyzed genes in AD samples when compared with the control samples (Fig. 1C). The logFC was in the range of −.5 to 2.5. There are 256 genes that are downregulated (logFC < −1) and 1,523 genes upregulated (logFC >1).
Regulatory mechanism of REST in AD
Overall, there are 33 DEGs regulating REST and 364 DEGs that are regulated by the REST. Further, two genes (GATA1 and AR) were found to act in a feedback manner, where these two genes have the potential to modulate as well as be modulated by REST (Fig. 2). Among the top 250 DEGs, BRD2 regulates REST, and Chromogranin A (CHGA) and PPP1R16A were found to be regulated by REST (Fig. 3a).
REST-TFs interactions in AD
Among the top 250 DEGs, only SP1 was involved in the regulation of REST (Fig. 3b). However, when the whole set of DEGs was analyzed, 20 crucial TFs were found to be directly involved in the REST regulation (Fig. 4). Out of these 20 TFs, 17 TFs were found to share common targets with REST that are involved in AD (Table 1).
Interactions of REST regulatory proteins in AD
The protein-protein interactions of the DEGs involved in the regulatory mechanism of REST demonstrate that most of them are interconnected with each other and are involved in either physical or functional regulation. Only a few proteins are sparsely spaced apart and are only briefly interconnected. HDAC1, JUN, and HDAC2 showed maximum interaction with other proteins. HDAC1 interacted with 21 other proteins. Likewise, JUN and HDAC2 interacted with 16 and 11 other proteins.
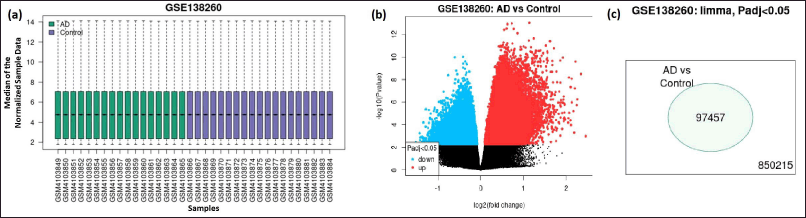 | Figure 1. Gene expression data. (a) Boxplot showing the distribution of the normalizedexpression data of the samples. (Boxplot is used to view the distribution of the values of the selected samples. The samples are colored according to groups. Viewing the distribution can be useful for determining if your selected samples are suitable for differential expression analysis), (b) volcano plot, (c) venn diagram of the overlapping differentially expressed genes (DEGs). [Click here to view] |
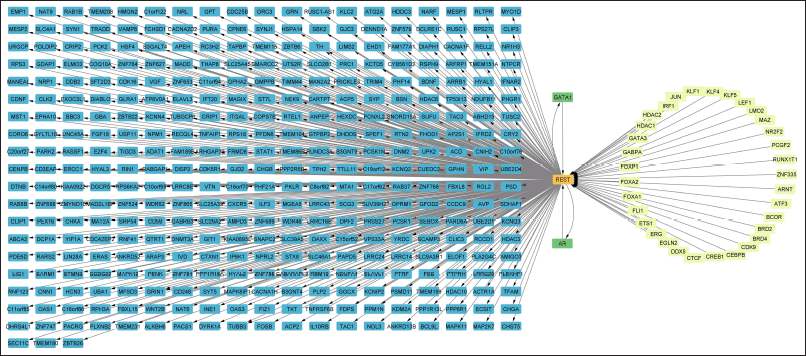 | Figure 2. DEG Regulation related to REST [Click here to view] |
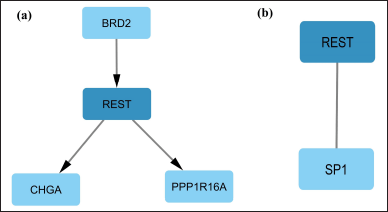 | Figure 3. The role of REST among the top 250 DEGs. a) TF BRD2 regulating REST which in turn regulates genes CHGA and PPP1R16A. b) Interaction of REST and SP1. [Click here to view] |
 | Figure 4. Overall interaction of REST with other TFs among the DEGs in AD [Click here to view] |
Hubs and regulatory modules
CytoHubba identified 10 hub proteins in the REST regulatory network involving the products of DEGs (Fig. 6). In addition, three closely interconnected clusters were obtained from MCODE (Fig. 7). The first module contained five proteins, the second had four proteins, and the third involved three proteins.
Annotation of REST regulatory genes
The REST regulatory genes are involved in a variety of bodily functions, and the bulk of them are expressed in the brain and CNS (Table 2).
DISCUSSION
REST is a crucial TF that represses several genes involved in various neurological disorders including AD. Understanding the regulatory mechanism of REST during AD would provide novel insights into the disease mechanism and identify potential disease targets. This could be achieved by analyzing the relation of REST with the DEGs in AD. In addition, it is critical to demonstrate the REST-TF interactions in AD since REST can interact with and influence other TFs that are differently expressed in AD.
Among the top 250 DEGs in AD, CHGA and PPP1R16A are regulated by REST (Fig. 3a). CHGA is a neuroendocrine secretory protein found in neurons and neuroendocrine cells. CHGA levels in the brain tissue of AD patients were substantially greater than those of healthy individuals. [30,31]. It was reported that CHGA could potentially be used as a biomarker for AD [32]. CHGA, expressed in amyloid plaques in AD, activated microglia to a reactive neurotoxic phenotype [33]. This is probably the bridge between AD-related neuronal, glial, and inflammatory pathways. PPP1R16A, which is also known as MYPT3, is involved in actin binding and GCPR signaling [34]. The role of GPCR and actin protein in AD pathology have been discussed elsewhere [35,36]. We have observed that REST is in turn regulated by BRD2. BRD2 is involved in the histone acetylation landscape and its dysregulation aids in the etiology of AD [37]. Although our study showed the interaction between BRD2 and REST, it is still unclear how both TFs regulate CHGA and PPP1R16A.
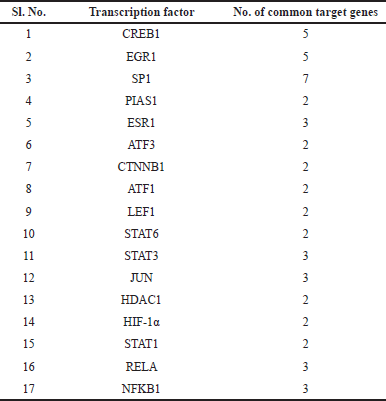 | Table 1. Transcription factors that share common target genes with REST. [Click here to view] |
Yet another important interaction that we have observed among the top 250 DEGs is between REST and TF SP1 (Figs. 3b and 4). SP1 is a REST activator [38] that regulates the genes linked to the development of amyloid plaques and governs the genes relevant to neuronal death, oxidative stress, tau phosphorylation, and inflammation, all of which likely have an impact on the evolution of AD [39,40]. It shares seven common target genes with REST (Table 1).
Apart from the top 250 DEGs, when the overall network was analyzed, we noted that REST interacted with 20 other TFs (Fig. 4) which included SMARCA4 (known as BRG1), RNF2, SIN3A, KDM1A, NCOR1, HDAC family proteins, etc. A chromatin remodeling factor called SMARCA4 is involved in the transcriptional stimulation of genes that is fundamental to neural development. SMARCA4 levels have been detected to be more abundant in individuals with AD than in controls. [41]. RNF2, a transcriptional enhancer, boosts the expression of genes that contribute to brain development by binding to certain DNA regions. The establishment of glutamatergic synapses in the hippocampus is encouraged by RNF2 [42]. SIN3A controls the metabolism of Aβ peptides, and Soluble Aβ peptides accumulated extensively as a consequence of loss-of-function mutations in SIN3A [43]. According to studies on the role of REST in the regulation of these TFs, SMARCA4 is a REST-interacting protein that forms epigenetic repressor complexes during the development of the brain by restricting the activity of specific genes [44]. Similarly, SIN3A acts as a transcriptional corepressor that engages with the REST and serves as crucial to modulating gene expression [45].
 | Figure 5. Physical protein interactions and functional associations of the proteins related to the DEGs regulated by REST. [Click here to view] |
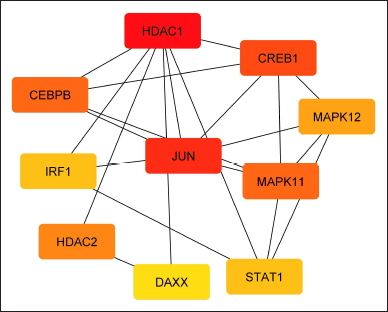 | Figure 6. Interactions of the Hub proteins related to the DEGs regulated by REST. (Generated by CytoHubba). The color indicates the order of ranks, red is rank 1 and yellow is rank 10. [Click here to view] |
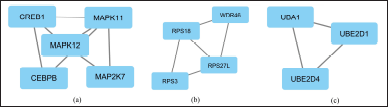 | Figure 7. Clusters (highly interconnected regions) in the interaction network of proteins related to the DEGs regulated by REST. (Generated by MCODE). [Click here to view] |
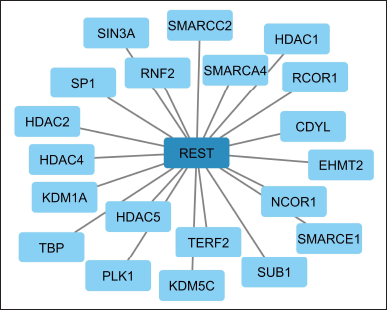
REST actively interacts with the HDAC family of TFs such as HDAC1, HDAC2, HDAC4, and HDAC5 (Fig. 4). HDAC1 is involved in the formation of amyloid plaques and the regulation of tau protein, which are a hallmark of AD [46]. HDAC1 seems to play a part in controlling the expression of specific genes linked to synaptic plasticity. In AD models, HDAC1 was reported to be elevated similarly to HDAC2 [47]. REST interacts with HDAC1 to regulate gene expression. REST binds to HDAC1 and modifies the chromatin structure, which in turn affects gene expression [48–51]. Interestingly, SIN3A recruits its corepressor HDAC2 to the target promoter genes to preserve a suitable repressive state [52]. REST binds to HDAC2 and inhibits its activity, thus preventing the transcription of target genes which is important for maintaining the proper balance of gene expression in the nervous system [48–50]. In our study, HDAC1 and HDAC2 were found to be the crucial hub genes that interact with most of the other proteins involved that come under the REST regulation (Fig. 6). Thus, we speculate that the REST via HDAC1 and HDAC2 regulates the other proteins involved in AD.
 | Table 2. Biological processes, cellular compartments and the tissues in which the REST regulatory proteins are involved. [Click here to view] |
Two histone deacetylases, HDAC4 and HDAC5, have been tied to the cause of AD. They play an integral part in controlling the regulation of the expression of genes, and the malfunctioning of any of them has been attributed to the appearance of AD. Boosted HDAC4 has been found in AD patients’ brains, according to studies [53], which contributes to the buildup of Aβ. Additionally, transgenic mice with ApoE4, the sole genetic risk factor for late-onset AD, have higher nuclear HDAC4 levels [54]. HDAC4 controls age-associated declines in memory generally. Neuner et al. [55]. HDAC5 gets elevated in AD patients’ brains, hinting that it could be contributing to the emergence of AD [56]. HDAC5 additionally performed an essential part in controlling the process of beta-amyloid synthesis. In addition, it has been established that REST interacts with HDAC5, and regulates the genes necessary for neural differentiation. As a result, REST is vital to the stage of neuronal differentiation [57].
Another REST interacting TF KDM1A, also known as lysine-specific demethylase 1 (LSD1), is a well-characterized co-repressor [58,59] that has been connected to the onset of AD. For instance, in CNS disorders, the repressor complex LSD1-REST-CoREST-HDAC1/2 largely controls the developmental program and modifies neuronal morphology [60]. REST along with LSD1 actively contributes to the repression of REST targets. Similarly, REST along with Chromodomain Y Like (CDYL) represses neuronal genes in non-neuronal tissues CDYL can suppress the transcription of genes [61–63].
REST interacts with NCOR1, which is a transcriptional corepressor and controls the brain’s gene expression related to a variety of processes including synaptic plasticity, learning, and memory as well as neuronal growth. In addition to controlling neurotransmitter release, NCOR1 is also linked to the etiology of AD [64]. REST also interacts with TRF2 or TERF2 whose expression was significantly lower in the peripheral blood serum of AD patients compared to the control. This decrease in expression is likely related to the reduction in telomere length observed in AD patients [65]. TERF2 was suggested to be an important factor in regulating telomere shortening in AD [66].
The protein-protein interaction and network analysis identified certain hub proteins forming clusters (Fig. 5). These hub proteins are DAXX, MAPK11, CREB1, and MAPK12 (Figs. 6 and 7). Understanding the role of these genes would help in elucidating the REST regulation in AD. Because these are the main cell death genes involved in AD pathology [8].
The p38 MAP kinases (MAPK11 and MAPK12) are implicated in tau phosphorylation [8]. CREB1 is crucial for controlling gene expression in the brain and contributes to the onset of AD [67–69]. CREB activation induces REST expression in the context of neuroinflammation [70]. In addition, the TF CREB1 shares five common target genes with REST (Table 1).
REST has also been observed to regulate gene expression via a synergistic mechanism, i.e., REST along with other TFs regulates common genes. For instance, the transcriptional regulator STAT3 is involved in neuroinflammation, transcriptional regulation, and the accumulation of amyloid and tau fragments. STAT1 and its interactions with STAT3 affect tau accumulation and synaptic plasticity [71–75]. STAT3 shares three common target genes with REST (Table 1). Recent research has proposed that STAT6-mediated promotion of M2 polarization of microglial cells may improve the cognitive ability of AD mice [76]. STAT6 shares two common target genes with REST (Table 1).
HIF-1 boosts the formation of Aβ, and diminishes the functioning of microglia, induces neuroinflammation and microglia mortality, all of which contribute to the genesis of AD [77,78]. REST suppresses HIF-1, and this in turn slows the uptake of glucose and lactate generation driven by hypoxia. REST restricts HIF-1-dependent transcription in order to perform its role as a repressor of gene expression [79]. But direct HIF-1α -REST interplay needs to be elucidated in AD. HIF-1α shares two common target genes with REST (Table 1).
The NF-κB pathway plays a negative role in the development of AD with inflammatory characteristics. REST and NF-κB 1 share three target genes in common (Table 1). Aβ and other pro-inflammatory molecules trigger NF-ΚB in AD, which boosts the level of expression of pro-inflammatory cytokines and chemokines. By encouraging neuronal death and hindering synaptic plasticity, this heightened inflammation is believed to speed up the manifestation of AD. NF-κB activation is additionally linked to the occurrence of amyloid plaques, an identifiable feature of AD [80–82].
To summarize, REST mediates the regulation process through other TFs such as NCOR1, TERF2, KDM1A, and HDAC family proteins to regulate hub proteins such as DAXX, MAPK11, CREB1 and MAPK12. In addition, it regulates target gene expression synergistically with other TFs such as HIF-1, STAT1, NF-κB, etc., thus are involved in crucial processes of AD pathology.
CONCLUSION
Understanding the regulation of beneficial TFs like REST in a complex disease like AD would be helpful to comprehend the underlying mechanism of the disease. In the regulatory networks involving DEGs in AD, active TFs influencing REST and the potential molecular mechanism of REST-DEGs interaction were identified. REST was found to physically interact with key TFs like SP1. Ten proteins produced by the DEGs were identified as hubs that might comprise direct (physical) and indirect (functional) associations with most of the proteins involved in the regulatory mechanism of REST. These hubs are critically involved in the pathogenesis of AD. Three clusters that are tightly regulated and involved in the REST regulation were identified. Seventeen TFs in the DEGs were found to share common targets with REST. The proteins in the REST regulatory network were involved in the biological processes associated with neuronal death.
Further in-depth research on the regulatory mechanism of REST for the treatment of AD under both in vitro and in vivo conditions can be carried out using this study as a stepping stone. In addition, because REST is a member of the category of epigenetic repressors, it is crucial to establish the relevance of other vital repressors and their corepressors under the epigenetic perspective of AD. Investigations on the inhibition of hub proteins reported in this study, which are involved in the pathogenesis of AD, can be performed to develop novel therapeutic molecules. Similar studies can be carried out to delineate the regulatory role of key TFsin other diseases.
ACKNOWLEDGMENTS
The authors acknowledge the Council of Scientific & Industrial Research, Government of India, New Delhi, for providing the Senior Research Fellowship to Mr. Ajmal Nassar and Manipal Academy of Higher Education, Manipal for providing financial and infrastructural support for the research activities. Ms. Gayathri S acknowledges the Indian Council of Medical Research (ICMR), for providing the Senior Research Fellowship.
AUTHOR CONTRIBUTIONS
Fayaz Shaik Mahammad: Conceptualization, Methodology, Formal analysis, Investigation, Writing, Reviewing & Editing, Project administration. Ajmal Nassar: Methodology, Investigation, Writing, Reviewing & Editing. Gayathri S: Methodology, Investigation, Writing, Reviewing & Editing. Prasada Chowdari Gurram: Data collection and Reviewing. Sairaj Satarker: Data collection and Reviewing. Madhavan Nampoothiri: Methodology, Formal analysis, Writing, Reviewing & Editing. Dinesh Upadhya: Methodology, Formal analysis, Writing, Reviewing & Editing.
FINANCIAL SUPPORT
This work was supported by the Council of Scientific & Industrial Research 08/602(0007)2019-EMR-1 and the Manipal Academy of Higher Education. Ms. Gayathri S has received financial support from the Indian Council of Medical Research (ICMR) (Grant No: BMI/11(65)/2020).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Metaxas A, Kempf SJ. Neurofibrillary tangles in Alzheimer’s disease: elucidation of the molecular mechanism by immunohistochemistry and tau protein phospho-proteomics. Neural Regen Res. 2016;11(10):1579–81. doi: https://doi.org/10.4103/1673-5374.193234
2. Nassar A, Kodi T, Satarker S, Chowdari Gurram P, Upadhya D, Sm F, et al. Astrocytic MicroRNAs and transcription factors in Alzheimer’s disease and therapeutic interventions. Cells. 2022;11(24):4111. doi: https://doi.org/10.3390/cells11244111
3. Andrés ME, Burger C, Peral-Rubio MJ, Battaglioli E, Anderson ME, Grimes J, et al. CoREST: c. Proc Natl Acad Sci U S A. 1999;96(17):9873–78. doi: https://doi.org/10.1073/pnas.96.17.9873
4. Chong JA, Tapia-Ramírez J, Kim S, Toledo-Aral JJ, Zheng Y, Boutros MC, et al. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80(6):949–57. doi: https://doi.org/10.1016/0092-8674(95)90298-8
5. Kuwabara T, Hsieh J, Nakashima K, Taira K, Gage FH. A small modulatory DsRNA specifies the fate of adult neural stem cells. Cell. 2004;116(6):779–93. doi: https://doi.org/10.1016/S0092-8674(04)00248-X
6. Roopra A, Qazi R, Schoenike B, Daley TJ, Morrison JF.. Localized domains of g9a-mediated histone methylation are required for silencing of neuronal genes. Mol Cell. 2004;14(6):727–38. doi: https://doi.org/10.1016/j.molcel.2004.05.026
7. Schoenherr CJ, Anderson DJ. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science. 1995;267(5202):1360–63. doi: https://doi.org/10.1126/science.7871435
8. Lu T, Aron L, Zullo J, Pan Y, Kim H, Chen Y, et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature. 2014;507(7493):448–54. doi: https://doi.org/10.1038/nature13163
9. Winston CN, Goetzl EJ, Akers JC, Carter BS, Rockenstein EM, Galasko D, et al. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimer’s Dement (Amst). 2016;3(1):63–72. doi: https://doi.org/10.1016/j.dadm.2016.04.001
10. Kawamura M, Sato S, Matsumoto G, Fukuda T, Shiba-Fukushima K, Noda S, et al. Loss of nuclear REST/NRSF in aged-dopaminergic neurons in parkinson’s disease patients. Neurosci Lett. 2019;699:59–63. doi: https://doi.org/10.1016/j.neulet.2019.01.042
11. de Souza JM, Abd-Elrahman KS, Ribeiro FM, Ferguson SSG. MGluR5 regulates REST/NRSF signaling through N-Cadherin/β-catenin complex in huntington’s disease. Mol Brain. 2020;13(1):118. doi: https://doi.org/10.1186/s13041-020-00657-7
12. Meyer K, Feldman HM, Lu T, Drake D, Lim ET, Ling KH,. REST and neural gene network dysregulation in IPSC models of Alzheimer’s disease. Cell Rep. 2019;26(5):1112–7.e9. doi: https://doi.org/10.1016/j.celrep.2019.01.023
13. Liu RM. Aging, cellular senescence, and Alzheimer’s disease. Int J Mol Sci. 2022;23(4):1989. doi: https://doi.org/10.3390/ijms23041989
14. Rocchi A, Carminati E, De Fusco A, Kowalska JA, Floss T, Benfenati F. REST/NRSF deficiency impairs autophagy and leads to cellular senescence in neurons. Aging Cell. 2021;20(10). doi: https://doi.org/10.1111/acel.13471
15. Zullo JM, Drake D, Aron L, O’Hern P, Dhamne SC, Davidsohn N, et al. Regulation of lifespan by neural excitation and REST. Nature. 2019;574(7778):359–64. doi: https://doi.org/10.1038/s41586-019-1647-8
16. Shimojo M. Huntingtin regulates RE1-silencing transcription factor/neuron-restrictive silencer factor (REST/NRSF) nuclear trafficking indirectly through a complex with REST/NRSF-interacting LIM domain protein (RILP) and dynactin P150 Glued. J Biolog Chem.2008; 283(50):34880–86. doi: https://doi.org/10.1074/jbc.M804183200
17. Su XJ, Shen BD, Wang K, Song QX, Yang X, Wu DS, et al. Roles of the neuron-restrictive silencer factor in the pathophysiological process of the central nervous system. Front Cell Dev Biol. 2022;10:834620. doi: https://doi.org/10.3389/fcell.2022.834620
18. Oany AR, Mia M, Pervin T, Alyami SA, Moni MA. Integrative systems biology approaches to identify potential biomarkers and pathways of cervical cancer. J Pers Med. 2021;11(5):363. doi: https://doi.org/10.3390/jpm11050363
19. Udhaya Kumar S, Thirumal Kumar D, Bithia R, Sankar S, Magesh R, Sidenna M, et al. Analysis of differentially expressed genes and molecular pathways in familial hypercholesterolemia involved in atherosclerosis: a systematic and bioinformatics approach. Front Genet. 2020;11:734. doi: https://doi.org/10.3389/fgene.2020.00734
20. Mathé E, Davis S, editors. Statistical Genomics. New York, NY: Springer New York; 2016.Vol. 1418.
21. Nampoothiri SS, Fayaz SM, Rajanikant GK. A novel five-node feed-forward loop unravels MiRNA-gene-TF regulatory relationships in ischemic stroke. Mol Neurobiol. 2018;55(11):8251–62. doi: https://doi.org/10.1007/s12035-018-0963-6
22. Han H, Shim H, Shin D, Shim JE, Ko Y, Shin J, et al. TRRUST: a reference database of human transcriptional regulatory interactions. Sci Rep. 2015;5(1):11432. doi: https://doi.org/10.1038/srep11432
23. Zhang H, Wang X, Xu P, Ji X, Chi T, Liu P, et al. Tolfenamic acid inhibits GSK-3β and PP2A mediated tau hyperphosphorylation in Alzheimer’s disease models. J Physiol Sci. 2020;70(1):29. doi: https://doi.org/10.1186/s12576-020-00757-y
24. Schaefer U, Schmeier S, Bajic VB. TcoF-DB: dragon database for human transcription co-factors and transcription factor interacting proteins. Nucleic Acids Rese. 2011;39(Database):D106–10. doi: https://doi.org/10.1093/nar/gkq945
25. Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605–12. doi: https://doi.org/10.1093/nar/gkaa1074
26. Sherman BT, Hao M, Qiu J, Jiao X, Baseler MW, Lane HC, et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 Update). Nucleic Acids Res. 2022;50(W1):W216–21. doi: https://doi.org/10.1093/nar/gkac194
27. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi: 10.1101/gr.1239303.
28. Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. CytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(S4):S11. doi: https://doi.org/10.1186/1752-0509-8-S4-S11
29. Chen J, Chiang Y. Applying the minimal common oncology data elements (MCODE) to the Asia-Pacific Region. JCO Clin Cancer Inform. 2021;(5):252–53. doi: https://doi.org/10.1200/CCI.20.00181
30. Weiler R, Lassmann H, Fischer P, Jellinger K, Winkler H. A high ratio of chromogranin a to synaptin/synaptophysin is a common feature of brains in Alzheimer and pick disease. FEBS Lett. 1990;263(2):337–39. doi: https://doi.org/10.1016/0014-5793(90)81408-G
31. Duits FH, Brinkmalm G, Teunissen CE, Brinkmalm A, Scheltens P, Van der Flier WM, et al. Synaptic proteins in CSF as potential novel biomarkers for prognosis in prodromal Alzheimer’s disease. Alzheimer Res Ther. 2018;10(1):5. doi: https://doi.org/10.1186/s13195-017-0335-x
32. Lechner T, Adlassnig C, Humpel C, Kaufmann WA, Maier H, Reinstadler-Kramer K, et al. Chromogranin peptides in Alzheimer’s disease. Exp Gerontol. 2004;39(1):101–3. doi: https://doi.org/10.1016/j.exger.2003.09.018
33. Taylor DL, Diemel LT, Cuzner ML, Pocock JM. Activation of group II metabotropic glutamate receptors underlies microglial reactivity and neurotoxicity following stimulation with chromogranin A, a peptide up-regulated in Alzheimer’s disease. J Neurochem. 2002;82(5):1179–91. doi: https://doi.org/10.1046/j.1471-4159.2002.01062.x
34. Amlie-Wolf A, Tang M, Way J, Dombroski B, Jiang M, Vrettos N, et al. Inferring the molecular mechanisms of noncoding Alzheimer’s disease-associated genetic variants. J Alzheimer’s Dis. 2019;72(1):301–18. doi: https://doi.org/10.3233/JAD-190568
35. Pelucchi S, Vandermeulen L, Pizzamiglio L, Aksan B, Yan J, Konietzny A, et al. Cyclase-associated protein 2 dimerization regulates cofilin in synaptic plasticity and Alzheimer’s disease. Brain Commun. 2020;2(2). doi: https://doi.org/10.1093/braincomms/fcaa086
36. Thathiah A, De Strooper B. The role of G protein-coupled receptors in the pathology of Alzheimer’s disease. Nat Rev Neurosci. 2011;12(2):73–87. doi: https://doi.org/10.1038/nrn2977
37. Benito E, Ramachandran B, Schroeder H, Schmidt G, Urbanke H, Burkhardt S, et al. The BET/BRD inhibitor JQ1 improves brain plasticity in WT and APP mice. Transl Psychiatry. 2017;7(9):e1239. doi: https://doi.org/10.1038/tp.2017.202
38. Ravache M, Weber C, Mérienne K, Trottier Y. Transcriptional activation of REST by Sp1 in huntington’s disease models.PLoS One. 2010;5(12):e14311. doi: https://doi.org/10.1371/journal.pone.0014311
39. Qin J, Zhang X, Wang Z, Li J, Zhang Z, Gao L, et al. Presenilin 2 deficiency facilitates Aβ-induced neuroinflammation and injury by upregulating P2X7 expression. Sci China Life Sci 2017;60(2):189–201. doi: https://doi.org/10.1007/s11427-016-0347-4
40. Zhang Q, Liu W, Zhang HM, Xie GY, Miao YR, Xia M, et al. HTFtarget: a comprehensive database for regulations of human transcription factors and their targets. Genomics Proteomics Bioinformatics. 2020;18(2):120–28. doi: https://doi.org/10.1016/j.gpb.2019.09.006
41. Kong W, Mou X, Deng J, Di B, Zhong R, Wang S, et al. Differences of immune disorders between Alzheimer’s disease and breast cancer based on transcriptional regulation. PLoS One. 2017;12(7):e0180337. doi: https://doi.org/10.1371/journal.pone.0180337
42. Kumari P, Srinivasan B, Banerjee S. Modulation of hippocampal synapse maturation by activity-regulated E3 ligase via non-canonical pathway. Neuroscience. 2017;364:226–41. doi: https://doi.org/10.1016/j.neuroscience.2017.08.057
43. Cao W, Song HJ, Gangi T, Kelkar A, Antani I, Garza D, et al. Identification of novel genes that modify phenotypes induced by Alzheimer’s β-Amyloid overexpression in drosophila. Genetics. 2008;178(3):1457–71. doi: https://doi.org/10.1534/genetics.107.078394
44. Jayaprakash S, Drakulic S, Zhao Z, Sander B, Golas MM. The ATPase BRG1/SMARCA4 is a protein interaction platform that recruits BAF subunits and the transcriptional repressor REST/NRSF in neural progenitor cells. Mol Cell Biochem. 2019;461(1–2):171–82. doi: https://doi.org/10.1007/s11010-019-03600-0
45. Halder D, Lee CH, Hyun JY, Chang GE, Cheong E, Shin I. Suppression of Sin3A activity promotes differentiation of pluripotent cells into functional neurons. Sci Rep. 2017;7(1):44818. doi: https://doi.org/10.1038/srep44818
46. Tao CC, Hsu WL, Ma YL, Cheng SJ, Lee EH.. Epigenetic regulation of HDAC1 SUMOylation as an endogenous neuroprotection against Aβ toxicity in a mouse model of Alzheimer’s disease. Cell Death Differ. 2017;24(4):597–614. doi: https://doi.org/10.1038/cdd.2016.161
47. Han Y, Chen L, Guo Y, Wang C, Zhang C, Kong L, et al. Class I HDAC inhibitor improves synaptic proteins and repairs cytoskeleton through regulating synapse-related genes in vitro and in vivo. Front Aging Neurosci. 2021;12:619866. doi: https://doi.org/10.3389/fnagi.2020.619866
48. Formisano L, Guida N, Valsecchi V, Cantile M, Cuomo O, Vinciguerra A, et al. Sp3/REST/HDAC1/HDAC2 complex represses and sp1/hif-1/p300 complex activates ncx1 gene transcription, in brain ischemia and in ischemic brain preconditioning, by epigenetic mechanism. J Neurosci. 2015;35(19):7332–48. doi: https://doi.org/10.1523/JNEUROSCI.2174-14.2015
49. Inui K, Zhao Z, Yuan J, Jayaprakash S, Le LTM, Drakulic S, et al. Stepwise assembly of functional C-Terminal REST/NRSF transcriptional repressor complexes as a drug target. Protein Sci. 2017;26(5):997–1011. doi: https://doi.org/10.1002/pro.3142
50. Roopra A, Sharling L, Wood IC, Briggs T, Bachfischer U, Paquette AJ, et al. Transcriptional repression by neuron-restrictive silencer factor is mediated via the Sin3-histone deacetylase complex. Mol Cell Biol. 2000;20(6):2147–57. doi: https://doi.org/10.1128/MCB.20.6.2147-2157.2000
51. Zhang J, Wang S, Yuan L, Yang Y, Zhang B, Liu Q, et al. Neuron-restrictive silencer factor (NRSF) represses cocaine- and amphetamine-regulated transcript (CART) transcription and antagonizes CAMP-response element-binding protein signaling through a dual NRSE mechanism. J Biol Chem. 2012;287(51):42574–87. doi: https://doi.org/10.1074/jbc.M112.376590
52. Srivas S, Thakur MK. Transcriptional Co-repressor SIN3A silencing rescues decline in memory consolidation during scopolamine-induced amnesia. J Neurochem. 2018;145(3):204–16. doi: https://doi.org/10.1111/jnc.14320
53. Shen X, Chen J, Li J, Kofler J, Herrup K. Neurons in vulnerable regions of the Alzheimer’s disease brain display reduced ATM signaling. Eneuro. 2016;3(1):ENEURO.0124-15.2016. doi: https://doi.org/10.1523/ENEURO.0124-15.2016
54. Sen A, Nelson TJ, Alkon DL. ApoE4 and A oligomers reduce BDNF expression via HDAC nuclear translocation. J Neurosci 2015;35(19):7538–51. doi: https://doi.org/10.1523/JNEUROSCI.0260-15.2015
55. Neuner SM, Wilmott LA, Hoffmann BR, Mozhui K, Kaczorowski CC. Hippocampal proteomics defines pathways associated with memory decline and resilience in normal aging and Alzheimer’s disease mouse models. Behav Brain Res. 2017;322:288–98. doi: https://doi.org/10.1016/j.bbr.2016.06.002
56. Anderson KW, Chen J, Wang M, Mast N, Pikuleva IA, Turko IV. Quantification of histone deacetylase isoforms in human frontal cortex, human retina, and mouse brain. PLoS One. 2015;10(5):e0126592. doi: https://doi.org/10.1371/journal.pone.0126592
57. Lee N, Park SJ, Haddad G, Kim DK, Park SM, Park SK, et al. Interactomic analysis of REST/NRSF and implications of its functional links with the transcription suppressor TRIM28 during neuronal differentiation. Scie Rep. 2016;6(1):39049. doi: https://doi.org/10.1038/srep39049
58. Forneris F, Binda C, Vanoni MA, Battaglioli E, Mattevi A. Human histone demethylase LSD1 reads the histone code. J Biol Chem. 2005;280(50):41360–65. doi: https://doi.org/10.1074/jbc.M509549200
59. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–53. doi: https://doi.org/10.1016/j.cell.2004.12.012
60. Song Y, Dagil L, Fairall L, Robertson N, Wu M, Ragan TJ,et al. Mechanism of crosstalk between the LSD1 demethylase and HDAC1 deacetylase in the CoREST complex. Cell Rep. 2020;30(8):2699–711.e8. doi: https://doi.org/10.1016/j.celrep.2020.01.091
61. Caron C, Pivot-Pajot C, van Grunsven LA, Col E, Lestrat C, Rousseaux S, et al. Cdyl: a new transcriptional Co-repressor. EMBO Rep. 2003;4(9):877–2. doi: https://doi.org/10.1038/sj.embor.embor917
62. Mulligan P, Westbrook TF, Ottinger M, Pavlova N, Chang B, Macia E, et al. CDYL Bridges REST and histone methyltransferases for gene repression and suppression of cellular transformation. Mol Cell. 2008;32(5):718–26. doi: https://doi.org/10.1016/j.molcel.2008.10.025
63. Zhang Y, Yang X, Gui B, Xie G, Zhang D, Shang Y, et al. Corepressor protein CDYL functions as a molecular bridge between polycomb repressor complex 2 and repressive chromatin mark trimethylated histone lysine 27. J Biolog Chem. 2011;286(49):42414–25. doi: https://doi.org/10.1074/jbc.M111.271064
64. Zhou W, He Y, Rehman AU, Kong Y, Hong S, Ding G, et al. Loss of function of NCOR1 and NCOR2 impairs memory through a Novel GABaergic hypothalamus–CA3 projection. Nature Neurosci. 2019;22(2):205–17. doi: https://doi.org/10.1038/s41593-018-0311-1
65. Wu Q, Han D, Zhang J, Li X. Expression of telomere repeat binding factor 1 and TRF2 in Alzheimer’s disease and correlation with clinical parameters. Neurol Res. 2019;41(6):504–9. doi: https://doi.org/10.1080/01616412.2019.1580456
66. Wang P, Wang P, Luan H, Wu Y, Chen Y. Midazolam alleviates cellular senescence in SH-SY5Y neuronal cells in Alzheimer’s disease. Brain and Behavior. 2023;13(1)e2822. doi: https://doi.org/10.1002/brb3.2822
67. Amidfar M, de Oliveira J, Kucharska E, Budni J, Kim YK. The role of CREB and BDNF in neurobiology and treatment of Alzheimer’s disease. Life Sci. 2020;257:118020. doi: https://doi.org/10.1016/j.lfs.2020.118020
68. Saura CA, Valero J. The role of CREB signaling in Alzheimer’s disease and other cognitive disorders. Revneuro. 2011;22(2):153–69. doi: https://doi.org/10.1515/rns.2011.018
69. Yin Y, Gao D, Wang Y, Wang ZH, Wang X, Ye J, et al. Tau accumulation induces synaptic impairment and memory deficit by calcineurin-mediated inactivation of nuclear CaMKIV/CREB signaling. Proc Natl Acad Sci. 2016;113(26). doi: https://doi.org/10.1073/pnas.1604519113
70. Buffolo F, Petrosino V, Albini M, Moschetta M, Carlini F, Floss T, et al. Neuroinflammation induces synaptic scaling through il-1β-mediated activation of the transcriptional repressor REST/NRSF. Cell Death Dis. 2021;12(2):180. doi: https://doi.org/10.1038/s41419-021-03465-6
71. Hong XY, Wan HL, Li T, Zhang BG, Li XG, Wang X, et al. STAT3 Ameliorates cognitive deficits by positively regulating the expression of NMDARs in a mouse model of FTDP-17. Signal Transduct Target Ther. 2020;5(1):295. doi: https://doi.org/10.1038/s41392-020-00290-9
72. Li XG, Hong XY, Wang YL, Zhang SJ, Zhang JF, Li XC, et al. Tau accumulation triggers
73. Millot P, San C, Bennana E, Hugon J, Paquet C, Hosten B, et al. STAT3 inhibition reverses neuroinflammation and Aβ Metabolism induced by systemic inflammation. Alzheimer’s Dementia. 2020;16(S2). doi: https://doi.org/10.1002/alz.041019
74. Reichenbach N, Delekate A, Plescher M, Schmitt F, Krauss S, Blank N, et al. Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer’s disease model. EMBO Mol Med.2019;11(2). doi: https://doi.org/10.15252/emmm.201809665
75. Zhang Z, Li XG, Wang ZH, Song M, Yu SP, Kang SS, et al. δ-Secretase-cleaved tau stimulates Aβ production via upregulating STAT1-BACE1 signaling in Alzheimer’s disease. Mol Psychiatry. 2021;26(2):586–603. doi: https://doi.org/10.1038/s41380-018-0286-z
76. Shen H, He Z, Pei H, Zhai L, Guan Q, Wang G. Aurantiamide promotes M2 polarization of microglial cells to improve the cognitive ability of mice with Alzheimer’s disease. Phytother Res. 2023;37(1):101–10. doi: https://doi.org/10.1002/ptr.7597
77. Steinbrecher L, Wendeln AC, Brösamle D, Liu P, Wild K, Haesler LM. The role of microglial HIF-1α in Aβ pathology. Alzheimer’s Dement. 2020;16(S2). doi: https://doi.org/10.1002/alz.042904
78. Wang YY, Huang ZT, Yuan MH, Jing F, Cai RL, Zou Q. Role of hypoxia inducible factor-1α in Alzheimer’s disease. J Alzheimer’s Dis. 2021;80(3):949–61. doi: https://doi.org/10.3233/JAD-201448
79. Cavadas MA, Mesnieres M, Crifo B, Manresa MC, Selfridge AC, Scholz CC, et al. REST mediates resolution of HIF-dependent gene expression in prolonged hypoxia. Sci Rep. 2015;5(1):17851. doi: https://doi.org/10.1038/srep17851
80. Kitamura Y, Shimohama S, Ota T, Matsuoka Y, Nomura Y, Taniguchi T. Alteration of transcription factors NF-ΚB and STAT1 in Alzheimer’s disease brains. Neurosci Lett. 1997;237(1):17–20. doi: https://doi.org/10.1016/S0304-3940(97)00797-0
81. Sun E, Motolani A, Campos L, Lu T. The pivotal role of NF-KB in the pathogenesis and therapeutics of Alzheimer’s disease. Int J Mol Sci. 2022;23(16):8972. doi: https://doi.org/10.3390/ijms23168972
82. Thawkar BS, Kaur G. Inhibitors of NF-ΚB and P2X7/NLRP3/caspase 1 pathway in microglia: novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J Neuroimmunol. 2019;326:62–74. doi: https://doi.org/10.1016/j.jneuroim.2018.11.010