
INTRODUCTION
In the last decade, the rapid growth of microbial pathogens and their increasing resistance to antimicrobial drugs have become a major concern of increasing public health (Piccione et al., 2019). The high number of antimicrobial resistances encourages efforts to find new drugs that have more effective antibacterial activity, through either drug synthesis or modification of existing antimicrobial drugs (Bari and Haswani, 2017).
Cationic gemini surfactants are an important type of surfactant consisting of two quaternary ammonium groups linked by a spacer group (Setiawan et al., 2021a). Initiated by Bunton et al. (1971) who synthesized the gemini quaternary ammonium bromide surfactant, this type of surfactant is receiving increasing attention due to its unique properties. The surface properties of gemini surfactants are known to be better than those of monomer analog surfactants. Besides having excellent surface properties, gemini surfactants are also known to act as highly efficient corrosion inhibitors and have good antimicrobial activity (Brycki et al., 2019; Shukla and Tyagi, 2006). The mechanism of inhibition of cationic gemini surfactants is by destroying the cell wall so that it can inhibit the growth of bacteria (Tyagi and Tyagi, 2014).
The process of developing a new drug is a complex, lengthy, and expensive process (Kovalishyn et al., 2018). This process includes the initial concept, synthesis, and testing of its safety and effectiveness in humans until approval to be brought to market. It would take at least 10–15 years and more than £500 million to develop a new drug (Puzyn et al., 2010). To mitigate these limitations, computer-aided drug design (CADD) studies can be used. In recent decades, the CADD approach has emerged as a method that plays an important role in the development of new drug molecules (?iri? Zdravkovi? et al., 2019). One of the CADD approaches is the quantitative structure–activity relationship (QSAR) method (Setiawan et al., 2021b). The QSAR method focuses on known ligands by establishing the relationship between physicochemical properties (descriptors) and biological activity (Roy et al., 2015; Tiwari and Singh, 2017).
In view of the above, the objective of this investigation was to construct a statistically significant QSAR model for gemini quaternary ammonium surfactants (GQAS) that correlates their antibacterial activity against Escherichia coli with their physicochemical properties. The resulting model is able to estimate the antibacterial activity of the newly designed compound.
MATERIALS AND METHODS
Dataset
In this study, a dataset containing 57 molecules of GQAS with antibacterial activity against E. coli was used for the QSAR study (Devinsky et al., 1985, 1987). Antibacterial activity in the form of the minimum inhibitory concentration (MIC) value (minimum concentration of antimicrobial compounds in inhibiting the growth of visible microorganisms) in moles was converted to a negative logarithmic value (pMIC) as an independent variable for QSAR analysis. The pMIC values of the dataset ranged from 1.884 to 4.638. The chemical structure and antibacterial activity of the compounds used are shown in Figure 1 and Table 1.
Molecular modeling and descriptors
The quaternary gemini ammonium surfactant structure was drawn using the Marvin ChemSketch software and saved in .mol format. Furthermore, all molecules were geometrically optimized using quantum chemical methods at the level of the Hartree–Fock (HF) theory and base set 6-311G on the Gaussian software. The resulting geometry optimization structure is used as the basis for calculating various structural parameters (descriptors) such as quantum chemical descriptors, physicochemical descriptors, and 1D–3D molecular descriptors. Based on the optimized three-dimensional structure obtained from molecular modeling at the HF level, 20 descriptors were obtained, including highest occupied molecular orbital energy, lowest occupied molecular orbital, dipole moment, and atomic net charge. The Mordred software was used to calculate 1,825 1D–3D molecular descriptors, which were divided into several groups of descriptors (Moriwaki et al., 2018). Physicochemical descriptors including logP and logS were obtained from SwissADME (http://www.swissadme.ch/) (Daina et al., 2017). In total, 1,842 descriptors were degenerated.
Before the molecular descriptors were used for the development of the QSAR model, descriptors were filtered by eliminating descriptors with constant values and those with correlation values above 0.9. Furthermore, screening was also carried out on descriptors that correlated poorly with antibacterial activity and descriptors that had a value of zero. In the end, 310 remaining descriptors were considered for QSAR modeling using the genetic algorithm-multiple linear regressions (GA-MLR) method.
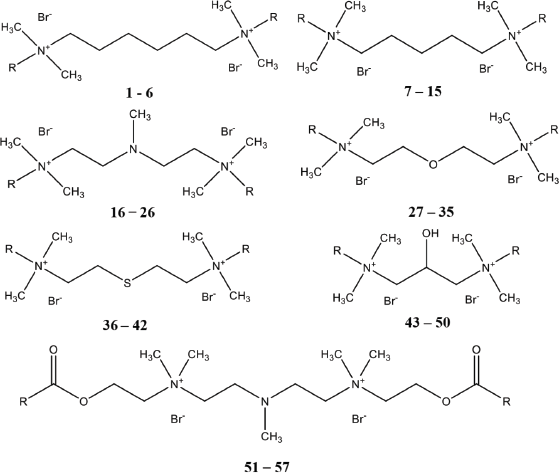 | Figure 1. General structure of GQAS. [Click here to view] |
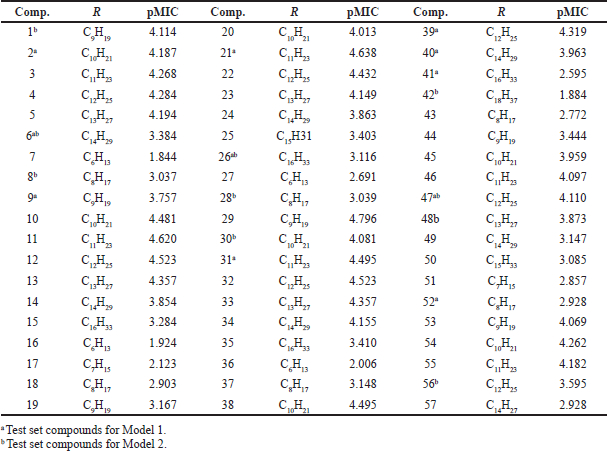 | Table 1. Antibacterial activity of GQAS against E. coli expressed as pMIC. [Click here to view] |
QSAR modeling and validation
In the present work, the QSAR-INSUBRIA (QSARINS) software from the Insubria QSAR Research Unit was used to carry out MLR in combination with the GA technique for variable selection (GA-MLR) (Gramatica et al., 2014, 2013). Two division techniques implemented in the QSARINS software were used to divide the dataset, namely an ordered biological activity-based approach and a structure similarity-based approach (Cassani and Gramatica, 2015). In the division based on the order of biological activity, the molecules were ordered according to the increasing value of antibacterial activity (pMIC), and one out of every three molecules was included in the test set. The division based on structural similarity was obtained from principal component analysis on the available molecular descriptors. Molecules in the dataset were ordered by PC1 score, which explained most of the total structural variance; then, one out of every three molecules was introduced into the test set. Finally, 75% of the compounds as the training set (46 compounds) were used for the development of the QSAR model, and the remaining 25% (11 compounds) were used as the test set for the purpose of validating the QSAR model.
As mentioned above, the QSARINS software was used to generate the GA-MLR model. The quality of the model was internally determined using the fitting criteria [R2, lack-of-fit (LOF), and root mean square error (RMSEtrain)] and robustness (Q2LOO) criteria. The coefficient of determination (R2), Friedman’s LOF, and the calibration error of the mean square root (RMSEtrain) were used as measures of the goodness of fit for the developed model. The cross-validation coefficient (Q2LOO) was used to verify their stability and robustness.
After being internally optimized, stable, and robust, the QSAR model was evaluated externally by a test set using different external validation parameters such as R2test, Q2Fn, and RMSEtest.
Furthermore, Y randomization was conducted to identify and exclude models that might have been obtained by chance, and applicability domain (AD) analysis was carried out through a leverage approach and using William’s plot (Gadaleta et al., 2016; Veerasamy et al., 2011). William’s plot that relates the leverage value (h) versus the standard residual is used to identify compounds that are structural outliers (which have a leverage value greater than the threshold value h) and residual outliers (which have a predicted response value above the specified standard residual limit). The threshold value h (h*) is calculated using the formula:
h* = 3(p + 1)/n,
where p is the number of descriptors in the model and n is the number of training set compounds used to build the QSAR model.
RESULTS AND DISCUSSION
Obtaining the QSAR-MLR model
In this study, two separation techniques (biological activity ordered-based and structure-based) were used to divide the dataset (n = 57) into a training set and a test set. In order to check the correctness of the training set and test set molecules selection, a unicolumn analysis was conducted (Table 2). The GA-MLR method was used to select the optimal combination of descriptors and build a linear model. The GA is a selection technique that imitates the natural selection process in its processes, such as inheritance, mutation, selection, and crossover. The GA parameters used a 100 population size, 500 iterations, and a 25% mutation rate. As a result, we obtained the best model with biological activity ordered-based dataset splitting (Model 1) and the best model with structure similarity-based splitting (Model 2). Equations (1) and (2) correspond to the best GA-MLR with different splitting techniques (Model 1 and Model 2, respectively) as follows:
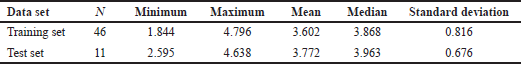 | Table 2. Uni-column analysis for training-set and test-set. [Click here to view] |
pMICEC = 0.211 AATS5m − 12.854 MATS8m + 0.210 PNSA3 + 5.452 IC5 − 18.136 AMID_N − 20.435, (1)
pMICEC = 4.3322 IC5 − 21.5384 MATS2Z − 0.0057 TIC3 − 15.6399. (2)
Based on the statistical parameters in Table 3, both Model 1 and Model 2 have acceptable statistical quality values for many parameters, but Model 1 showed a better model than Model 2, as indicated by higher values for R2, Q2LOO, R2test, and Q2Fn and lower value for Friedman’s LOF parameter and error parameter in both the training and test sets (RMSEtrain and RMSEtest, respectively). Model 1 has an R2 of 0.891, so it has a good degree of fit and significance. Moreover, it has a low LOF parameter of 0.116, which indicates no overfitting in the model. The correlation between the descriptors of Model 1 was acceptable (Table 4). The model has a small error in training calculations and parameter estimation (RMSEtrain = 0.267). The scatter plot of the predicted pMIC value versus the experimental antibacterial activity is shown in Figure 2. It can be seen that the predicted pMIC values were in good agreement with the experimental values.
Based on Equation (1), Model 1 consists of the following descriptors: AATS5m, MATS8m, PNSA3, IC5, and AMID_N. The descriptor AATS5m is Average Broto-Moreau autocorrelation which represents the compounds with larger average molecular weights between atoms of five-bond topological distance, with neither end of the five-bond atoms being a carbon (Prabhakar et al., 2005). The descriptor MATS8m is a 2D descriptor which represents Moran autocorrelation of lag 8 weighted by mass (Melville and Hirst, 2007). The PNSA3 descriptor stands for atom charge weighted negative surface areas. The IC5 is a descriptor which represents the information content index (neighborhood symmetry of 5-order) from the information indices group (Abadi et al., 2016). The last descriptor was AMID_N, which stands for averaged molecular ID on N atoms (Kamiya et al., 2021).
The plot between the standardized residuals versus leverage value was used to describe the AD of the model (Fig. 3). Based on William’s plot in Figure 3, all molecules have a leverage value that is less than the threshold value h (h* = 0.391), which means that they are no outlier compounds.
The results of Y randomization indicate that the resulting model was not inferred by luck because the averages values of R2Yscr and Q2Yscr are ever lower with respect to the R2 and Q2 values of the model (R2Yscr = 0.113 and Q2Yscr = −0.186). Figure 4 shows the values of R2 and Q2 of the model are very far from the averages values of Yscr, which indicates that the model is not obtained because of a random correlation.
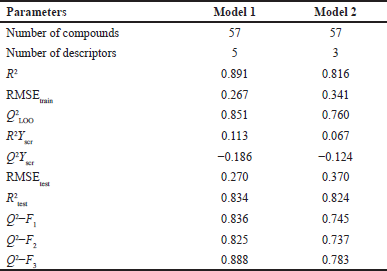 | Table 3. Statistical comparison of Models 1 and Model 2. [Click here to view] |
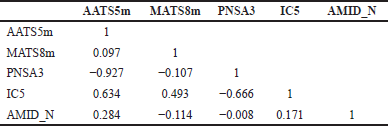 | Table 4. Descriptors correlation matrix of Model 1. [Click here to view] |
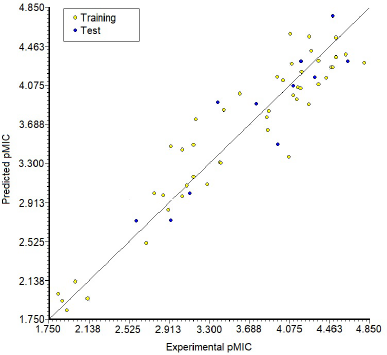 | Figure 2. The scatter plot of the predicted values of pMIC versus the experimental values by Model 1 for the training set and test set. [Click here to view] |
After being validated internally, the model was validated externally by using test set compounds. The external validation of the resulted model showed high values of the coefficient of determination (R2test = 0.834) and low values of the error parameter (RMSEtest = 0.270), which indicates that Model 1 can be used to predict the antibacterial activity of a potential new quaternary gemini ammonium surfactant.
Design for new GQAS with antibacterial activity
Relying on the GA-MLR models, based on cationic gemini surfactants’ structure from Shukla and Tyagi (2006), several new GQAS have been designed to enhance antibacterial activity of gemini quaternary ammonium surfactants (Table 5 and Figure 5). We designed 30 new GQAS based on 2 factors. First, GQAS in the dataset with medium chain lengths, C10−C14, show the optimal antimicrobial activity, so we designed new gemini ammonium surfactants with chain lengths C10, C12, and C14. Second, many research studies have revealed that the antibacterial activity of gemini ammonium surfactants depends on the nature of the spacers (Andrzejewska et al., 2017; Negm et al., 2014; Pérez et al., 2002), so we designed new gemini ammonium surfactants with several kinds of spacer group. The newly predicted structures 1d and 2d showed higher activity (pMIC = 6.630 and 7.425, respectively) than compound 29 (the most active compound of the series pMIC = 4.796). New compounds with spacer group s1–s4 present high predicted activities, which means that the predicted compounds can almost be more effective than the compounds of the database.
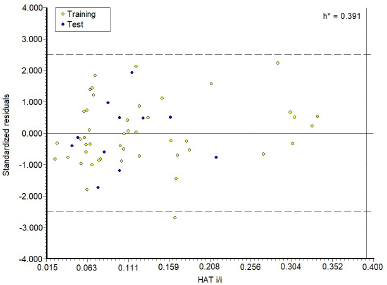 | Figure 3. William’s plot for the AD of the model. [Click here to view] |
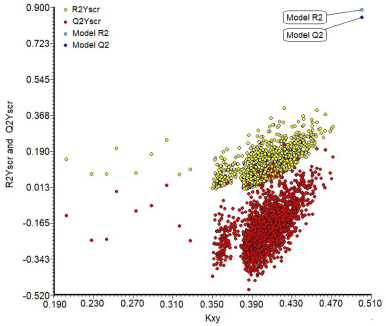 | Figure 4. Y-scrambling graph in the internal validation. [Click here to view] |
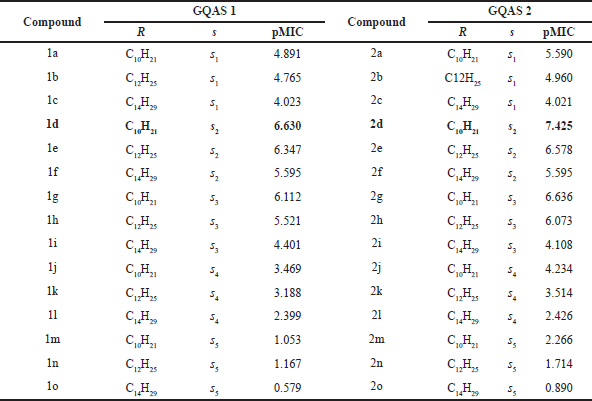 | Table 5. Chemical structure of newly GQAS and their predicted pMIC based on Model 1. [Click here to view] |
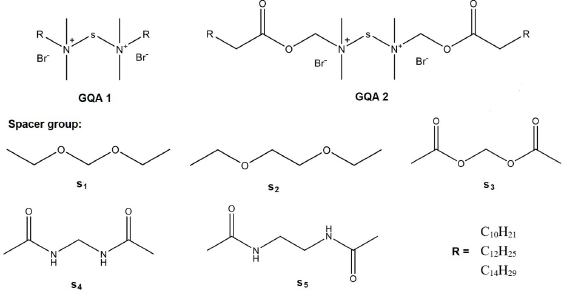 | Figure 5. The general structure of designed compounds (GQAS 1 and GQAS 2) with various spacer groups (s1 -s5) and R = C10H21, C12H25, and C14H29 based on cationic gemini surfactants’ structure from Shukla and Tyagi (2006) . [Click here to view] |
CONCLUSION
The QSAR study of antibacterial activity data against E. coli for 57 GQAS was reported for the first time. Two different splitting techniques were used to divide the dataset; consequently, two GA-MLR models were generated. The best model has five descriptors with good predictive performance with acceptable statistic quality (R2 = 0.891, Q2LOO = 0.851; the prediction R2 = 0.834, RMSEtest = 0.269). A newly designed compound of 30 GQAS was predicted by the developed GA-MLR model in this study. Sixteen newly designed GQAS with promising antibacterial activity have been proposed.
ACKNOWLEDGMENTS
This project was financially supported by Universitas Gadjah Mada (UGM) through a Rekognisi Tugas Akhir (RTA) program in 2020.
LIST OF ABBREVIATIONS
AATS5m: Average Broto—Moreau autocorrelation—lag 5/weighted by mass; AD: Applicability domain; AMID_N: Averaged molecular ID on N atoms; CADD: Computer-aided drug design; GA: Genetic algorithm; HF: Hartree–Fock; IC5: Information content index (neighborhood symmetry of 5-order); LOF: Lack-of-fit; LOO: Leave-one-out; MATS8m: Moran autocorrelation of lag 8 weighted by mass; MIC: Minimum inhibitory concentration; MLR: Multiple linear regression; PNSA3: Atomic charge weighted partial negative surface area; QSAR: Quantitative structure–activity relationship; QSARINS: QSAR-INSUBRIA; RMSE: Root mean square error.
CONFLICTS OF INTEREST
The authors declared that they have no conflicts of interest.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included within this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Abadi RSK, Alizadehdakhel A, Paskiabei ST. A DFT and QSAR study of several sulfonamide derivatives in gas and solvent. J Korean Chem Soc, 2016; 60:225–34; doi:10.5012/JKCS.2016.60.4.225. CrossRef
Andrzejewska W, Wilkowska M, Chrab?szczewska M, Kozak M. The study of complexation between dicationic surfactants and the DNA duplex using structural and spectroscopic methods. RSC Adv, 2017; 7:26006–18; doi:10.1039/c6ra24978g. CrossRef
Bari SB, Haswani NG. Design, synthesis and molecular docking study of thienopyrimidin-4(3H)-thiones as antifungal agents. J Saudi Chem Soc, 2017; 21:S264–74; doi:10.1016/j.jscs.2014.02.011. CrossRef
Brycki B, Szulc A, Koenig H, Kowalczyk I, Pospieszny T, Górka S. Effect of the alkyl chain length on micelle formation for bis(N-alkyl-N,N-dimethylethylammonium)ether dibromides. Comptes Rendus Chim, 2019; 22:386–92; doi:10.1016/j.crci.2019.04.002. CrossRef
Bunton CA, Robinson L, Stam MF, Schaak J. Catalysis of nucleophilic substitutions by micelles of dicationic detergents. J Org Chem, 1971; 36:2346–50; doi:10.1021/jo00815a033. CrossRef
Cassani S, Gramatica P. Identification of potential PBT behavior of personal care products by structural approaches. Sustain Chem Pharm, 2015; 1:19–27; doi:10.1016/j.scp.2015.10.002. CrossRef
?iri? Zdravkovi? S, Pavlovi? M, Apostlovi? S, Kora?evi? G, Šalinger Martinovi? S, Stanojevi? D, Sokolovi? D, Veselinovi? AM. Development and design of novel cardiovascular therapeutics based on Rho kinase inhibition—in silico approach. Comput Biol Chem, 2019; 79:55–62; doi:10.1016/j.compbiolchem.2019.01.007. CrossRef
Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep, 2017; 7:1–13; doi:10.1038/srep42717. CrossRef
Devinsky F, Lacko I, Bittererova F, Mlynarcik D. Quaternary ammonium salts XVIII. Preparation and relationship between structure, IR spectral characteristics, and antimicrobial activity of some new bis-quaternary isosters of 1, 5-pentanediammonium dibromides. Chem Pap, 1987; 41:803–14.
Devinsky F, Lacko I, Mlynarcik D, Racansky V, Krasnec L. Relationship Between critical micelle concentrations and minimum inhibitory concentrations for some non-aromatic quaternary ammonium salts and amine oxides. Tenside Deterg, 1985; 22:10–5; doi:10.1515/tsd-1985-220105. CrossRef
Gadaleta D, Mangiatordi GF, Catto M, Carotti A, Nicolotti O. Applicability domain for QSAR models. Int J Quant Struct Relat, 2016; 1:45–63; doi:10.4018/ijqspr.2016010102. CrossRef
Gramatica P, Cassani S, Chirico N. QSARINS-chem: insubria datasets and new QSAR/QSPR models for environmental pollutants in QSARINS. J Comput Chem, 2014; 35:1036–44; doi:10.1002/jcc.23576. CrossRef
Gramatica P, Chirico N, Papa E, Cassani S, Kovarich S. QSARINS: a new software for the development, analysis, and validation of QSAR MLR models. J Comput Chem, 2013; 34:2121–32; doi:10.1002/JCC.23361. CrossRef
Kamiya Y, Omura A, Hayasaka R, Saito R, Sano I, Handa K, Ohori J, Kitajima M, Shono F, Funatsu K, Yamazaki H. Prediction of permeability across intestinal cell monolayers for 219 disparate chemicals using in vitro experimental coefficients in a pH gradient system and in silico analyses by trivariate linear regressions and machine learning. Biochem Pharmacol, 2021; 192:114749; doi:10.1016/J.BCP.2021.114749. CrossRef
Kovalishyn V, Grouleff J, Semenyuta I, Sinenko VO, Slivchuk SR, Hodyna D, Brovarets V, Blagodatny V, Poda G, Tetko IV, Metelytsia L. Rational design of isonicotinic acid hydrazide derivatives with antitubercular activity: machine learning, molecular docking, synthesis and biological testing. Chem Biol Drug Des, 2018; 92:1272–8; doi:10.1111/cbdd.13188. CrossRef
Melville JL, Hirst JD. TMACC: interpretable correlation descriptors for quantitative structure-activity relationships. J Chem Inf Model, 2007; 47:626–34; doi:10.1021/ci6004178. CrossRef
Moriwaki H, Tian YS, Kawashita N, Takagi T. Mordred: a molecular descriptor calculator. J Cheminform, 2018; 10:1–14; doi:10.1186/s13321-018-0258-y. CrossRef
Negm NA, El Farargy AF, Youssif MA, Mohamed S, Said MM. Antibacterial evaluation of cationic surfactants. Househ Pers Care Today, 2014; 9:48–53.
Pérez L, Garcia MT, Ribosa I, Vinardell MP, Manresa A, Infante MR. Biological properties of arginine-based gemini cationic surfactants. Environ Toxicol Chem, 2002; 21:1279–85; doi:10.1002/etc.5620210624. CrossRef
Piccione D, Mirabelli S, Minto N, Bouklas T. Difficult but not impossible: in search of an anti-candida vaccine. Curr Trop Med Rep, 2019; 6:42–9; doi:10.1007/s40475-019-00173-2. CrossRef
Prabhakar Y, Rawal R, Gupta M, Solomon V, Katti S. Topological descriptors in modeling the HIV inhibitory activity of 2-aryl-3- pyridyl-thiazolidin-4-ones. Comb Chem High Throughput Screen, 2005; 8:431–7; doi:10.2174/1386207054546531. CrossRef
Puzyn T, Leszczynski J, Cronin MTD. Recent advances in QSAR studies. Methods and applications. Springer, Dordrecht, The Netherlands; New York, NY, pp 3 -11, 2010. CrossRef
Roy K, Kar S, Das RN. A primer on QSAR/QSPR modeling: fundamental concepts. SpringerBriefs in Molecular Science, Springer, Cham, Switzerland, 2015. CrossRef
Setiawan E, Wijaya K, Mudasir M. Generic QSPR study for predicting critical micelle concentration of gemini cationic surfactants using the online chemical modeling environment (OCHEM). In: AIP Conference Proceedings, 2021a, vol. 2349, pp 020027; doi:10.1063/5.0051623. CrossRef
Setiawan E, Wijaya K, Mudasir M. QSAR modeling for predicting the antifungal activities of gemini imidazolium surfactants against Candida albicans using GA-MLR methods. J Appl Pharm Sci, 2021b; 11:022–7; doi:10.7324/JAPS.2021.110404.
Shukla D, Tyagi VK. Cationic gemini surfactants: a review. J Oleo Sci, 2006; 55:381–90; doi:10.5650/jos.55.381. CrossRef
Tiwari P, Singh VK. Computer assisted drug designing?: quantitative structure activity relationship studies on mono- and bis- thiazolium salts having potent antimalarial activity. Int J Sci Res Publ, 2017; 7:213–35.
Tyagi S, Tyagi VK. Novel cationic gemini surfactants and methods for determination of their antimicrobial activity—review. Tenside Surfactants Deterg, 2014; 51:379–86; doi:10.3139/113.110319. CrossRef
Veerasamy R, Rajak H, Jain A, Sivadasan S, Varghese CP, Agrawal RK. Validation of QSAR models—strategies and importance. Int J Drug Des Discovery, 2011; 2:511–9.