
INTRODUCTION
The gastric H+/K+-ATPase pump or the proton pump is the most preferred target in the treatment of Gastric Esophageal Reflux Disease. Proton pump inhibitors (PPIs) achieve gastric acid inhibition through covalent binding with cysteine residues on the enzyme. All PPIs undergo in vivo non-enzymatic activation that involves two protonation steps. The first protonation results in accumulation of the molecules in the parietal cell. This step is followed by a second protonation at the active secretory canaliculus of the parietal cell resulting in the formation of a disulfide bond with cysteine residue on the pump and subsequent acid inhibition (Sachs et al., 2006). The PPI pharmacophore can be described as 2-pyridylmethylsulfinylbenzimidazole with two sites where protonation occurs as shown in Figure 1 (Roche, 2006). The protonation of the pyridine nitrogen is crucial for localization of the molecules in the parietal cell. The benzimidazole nitrogen, present in the pharmacophoric region of all PPIs, plays a crucial role in the in vivo activation of the drugs. The mechanism of the activation of PPIs is shown in Figure 2. The structures of different PPI molecules are very similar to each other except for the substituents on the pyridine and benzimidazole rings.
.png) | Figure 1. The 2-pyridylmethyl sulfinyl benzimidazole PPI pharmacophore. [Click here to view] |
Molecules with similar structures are expected to have similarities in physical and biological properties. The structural similarities of molecules are represented numerically by the descriptors and then assessed and compared with their physical and biological properties using Quantitative Structure Activity/Property Relationship models. These molecular descriptors can be theoretical or experimental and are related to electronic, steric, or topological properties of the molecules. Physicochemical or biological properties of molecules are a result of the net effect of the atomic arrangement in space and the environment in which the molecule is present.
 | Figure 2. The PPI activation and reaction pathway. [Click here to view] |
 | Figure 3. Structures of selected PPIs with the atom numbering system. [Click here to view] |
Nuclear magnetic resonance spectroscopy is a technique that helps in exploration of the atomic arrangement of the molecule. NMR spectroscopy reflects the local environment around a particular atom. In this sense, it mimics the molecular field around the particular nucleus. For this reason, NMR chemical shift may be used as a molecular descriptor. There is a large amount of experimental NMR data available for different molecules; however, there are very few studies that have utilized this data as molecular descriptors. 13C chemical shifts were utilized for predicting the antiradical activity of flavonoids (LuÄić et al., 2014), and lipophilicity of alcohols (Khadikar et al., 2005a). 1H chemical shifts were used for modeling the carbonic anhydrase inhibition activity of benzene sulfonamides (Khadikar et al., 2005b).
However, in the case of nitrogenated molecules like drugs, the use of only 13C or 1H chemical shifts as descriptors may be inadequate. Most interactions of drug molecules that determine their pharmacodynamic or pharmacokinetic properties are mediated through heteroatoms like nitrogen. In PPIs, the pyridine and benzimidazole nitrogens play a very important role in drug activity. In view of their role in mediating interactions, chemical shifts of nitrogens may be more suitable as molecular descriptors for drug molecules. However, 15N-NMR studies are not available for the majority of drug molecules. In the present study, 15N NMR spectroscopic studies of PPIs were carried out. The relationships between 15N and 13C NMR chemical shifts and pharmaceutical properties of PPIs were studied. This study helps in assessing the utility of multinuclear NMR chemical shifts as molecular descriptors in the development of Quantitative Structure Activity/Property Relationship models. The structures of PPI molecules selected for the study are shown in Figure 3.
MATERIALS AND METHODS
Drug samples
All the drug samples were obtained as gift samples in pure form from Quality Control Laboratories of Dr. Reddy’s Laboratories, Hyderabad, Mylan Pharmaceuticals, Bengaluru or Laurus Labs, Visakhapatnam.
15N-NMR Experiments
15N-NMR spectroscopic studies of lansoprazole, pantoprazole, and ilaprazole were performed at natural abundance. The sensitivity of 15N nucleus in NMR is very low due to its low gyromagnetic ratio. Furthermore, the 15N isotope has a low natural abundance. Hence, the detection of 15N nucleus at natural abundance is very difficult. No signals were detected using direct detection or 1D experiments like Insensitive Nuclei Enhanced by Polarization Transfer (INEPT). However, we were able to detect signals using 1H-15N Heteronuclear Multiple Bond Correlation (HMBC) experiments. The molecules were, thus, studied by 1H-15N HMBC-NMR experiments, carried out in dimethylsulphoxide (DMSO-d6) using Bruker Avance 400 MHz instrument operating at 40 MHz for 15N nucleus at a temperature of 298.2 K. The chemical shifts are reported with reference to liq. ammonia at 25°C. The doubly bonded nitrogen in the benzimidazole ring was detected by this method in all the three molecules. By employing 1H-15N Heteronuclear Single Quantum Coherence (HSQC)-NMR, we were able to detect the peak of the second benzimidazole nitrogen in lansoprazole. The 15N channel projection of the 1H-15N HMBC spectra of the three molecules, ilaprazole, lansoprazole, and pantoprazole and the 15N channel projection of the 1H-15N HSQC spectrum of lansoprazole are shown in Figures 4–7. 15N chemical shifts of all the molecules, viz., omeprazole, pantoprazole, lansoprazole, ilaprazole, and rabeprazole were also predicted using ACD Labs N-NMR predictor.
 | Figure 4. 15N channel projection of the 1H-15N HMBC spectrum of Ilaprazole. [Click here to view] |
13C-NMR data
The 13C NMR chemical shifts of all the five molecules, omeprazole, ilaprazole, pantoprazole, lansoprazole, and rabeprazole were collected from the literature (Naidu, 2016).
 | Figure 5. 15N channel projection of the 1H-15N HMBC spectrum of Lansoprazole. [Click here to view] |
 | Figure 6. 15N channel projection of the 1H-15N HSQC spectrum of Lansoprazole. [Click here to view] |
 | Figure 7. 15N channel projection of the 1H-15N HMBC spectrum of Pantoprazole. [Click here to view] |
Correlation coefficients
Correlation coefficient, r, gives a measure of the strength of the linear relationship between two variables. The correlation coefficients between NMR chemical shifts and different properties were determined using the CORREL function in Microsoft Excel. Correlation coefficient varies between −1 and +1. A value between 1 and 0.9 indicates a very strong negative or positive correlation depending on whether the sign is negative or positive. A value from 0.7 to 0.9 indicates a strong correlation, while a value between 0.5 and 0.7 indicates a moderate correlation. Values below 0.5 indicate poor correlation, while those below 0.3 indicate negligible correlation.
RESULTS AND DISCUSSION
There are no reported 15N-NMR studies for any of the PPI molecules, except for omeprazole (Claramunt et al., 2006). This is the first report of 15N-NMR studies for lansoprazole, pantoprazole, and ilaprazole. The assignment of experimental 15N chemical shifts and the predicted values, along with those reported for omeprazole in the literature (Claramunt et al., 2006), are shown in Table 1.
 | Table 1. Predicted and experimental 15N chemical shifts. [Click here to view] |
 | Table 2. Sum of 13C chemical shifts. [Click here to view] |
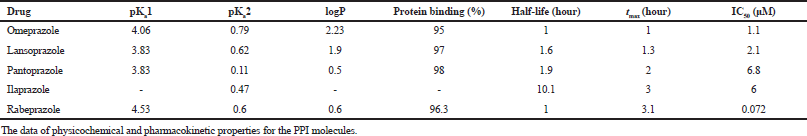 | Table 3. Pharmacokinetic properties of selected molecules. [Click here to view] |
The sum of 13C NMR chemical shifts of the two moieties in PPIs, the pyridine moiety and the benzimidazole moiety, along with that for the whole molecule are presented in Table 2.
Comparison between predicted and experimental 15N chemical shifts
The predicted 15N chemical shifts show strong correlation (r = 0.87) with experimental chemical shifts. However, as shown in Table 1, the predicted values are not differentiating between the molecules. In all the five molecules, both the benzimidazole nitrogens were predicted to resonate at 224.3 ppm. However, the same was found to vary between 146.3 and 236.83 ppm in four of the molecules. Similarly, the pyridine nitrogen was shown to resonate at 313.6 ppm in three out of five molecules, whereas the actual values range from 294.6 to 306.2 ppm.
Relationships between 15N and 13C chemical shifts and physicochemical and pharmacokinetic properties
Verma and Hansch (2011) reviewed the use of 13C chemical shifts as molecular descriptors. They had described several approaches for utilizing chemical shifts as descriptors. Some of the methods described are using sum of chemical shifts, using chemical shift of a particular atom common to all the molecules, using the difference between chemical shift of a particular atom in the parent molecule and in an analog, etc. In the current study, some of these methods were employed to investigate spectral data droperty relationships in PPIs, using 15N and 13C chemical shifts. Wherever sum of 15N chemical shifts were used, the predicted value of the chemical shift for the N1 nitrogen and experimental values for N3 and N6 nitogens were used in all the molecules. The data for various physicochemical and pharmacokinetic properties were collected from the literature (Beil et al., 1992; de Bortoli et al., 2013; Kwon et al., 2001; Morii et al., 1990; Nagaya et al, 1991; Roche, 2006; Smolka et al., 2004) and summarized in Table 3.
Relationship between chemical shifts and pKa
PPIs can exhibit two pKa values, one for the protonation of the pyridine nitrogen and the other for the protonation of the benzimidazole nitrogen. The pKa of the pyridine nitrogen in the PPI molecules is referred to as pKa1. The chemical shift of the pyridine nitrogen (N6) shows a small variation, within 12 ppm, varying from 294.6 ppm in ilaprazole to 306.2 ppm in omeprazole. The sum of 13C chemical shifts of the pyridine moiety show moderate positive correlation (r = 0.61) with pKa1.
The value of pKa2 demonstrates the ability of the doubly bonded nitrogen of the benzimidazole moiety (N3) to undergo protonation, which is the first step in the activation of the PPI molecules. Electron donating substituents on the benzimidazole ring increase the nucleophilic character of the nitrogen at N3. This results in enhancement of the rate of activation of the PPI molecules. The diflouromethoxy substitution on the benzimidazole ring in pantoprazole has a strong electron withdrawing effect and reduces the ability of N3 to undergo protonation. In omeprazole, the methoxy substitution results in enhanced protonation of N3. The pKa2 values of the molecules reflect the changes in the nucleophilic character of the N3 nitrogen caused by different substituents on the benzimidazole ring. The trends observed in the chemical shift of the N3 nitrogen also reflect similar changes. The 15N chemical shift shows a large variation, ranging from 146.3 ppm in omeprazole to 236.8 ppm in pantoprazole (about 90 ppm variation). As the nitrogen is shielded, the pKa2 value increases. The correlation between N3 chemical shift and pKa2 is very strong (r = −0.96). However, there is negligible correlation between sum of 13C chemical shifts and pKa2 values. The variations in the 15N chemical shift of N3 nitrogen, thus, adequately reflect the changes in the value of pKa2.
Relationship between chemical shifts and half-life of elimination
Half-life exhibits a very strong positive correlation (r = 0.94) with the sum of 15N chemical shifts of the molecules as well as with the sum 13C chemical shifts of the molecules (r = 0.97). The sum of 13C chemical shifts of the benzimidazole moiety also shows a very strong positive correlation (r = 0.96) with half-life. The half-life of the molecule can, thus, be considered more dependent on the nature of substituents on the benzimidazole moiety.
Relationship between chemical shifts and tmax
The time taken to reach maximum concentration in vivo, or tmax, shows strong negative correlation with 15N chemical shift of the pyridine nitrogen (r = −0.87) and very strong positive correlation with the sum of 15N chemical shifts of the molecule (r = 0.98). tmax depends on the ability of the molecules to be localized in the parietal cell and activation into the active cyclic sulfenamide form. Both these steps are dependent on the ability of the nitrogens to get protonated. The same is reflected in the strong correlation with the sum of 15N chemical shifts. tmax, however, shows poor correlation with 13C chemical shifts of the molecules.
Relationship between chemical shifts and protein binding
The experimental value for protein binding (%) was not available for ilaprazole. Hence, a comparison was made between the other three with 15N chemical shifts and including rabeprazole when using 13C chemical shifts. Protein binding exhibits very strong negative correlation with 15N chemical shift of the pyridine nitrogen (r = −0.98), strong positive correlation with 15N chemical shift of the benzimidazole nitrogen (r = 0.78), and moderately positive correlation (r = 0.63) with sum 13C chemical shifts of the molecules.
Relationship between chemical shifts and logP
Experimental logP values are not available for ilaprazole. Hence, a comparison was made with the 15N chemical shifts of the other three molecules and rabeprazole was included for comparison with 13C chemical shifts. logP shows very strong negative correlation (r = −0.99) with 15N chemical shift of the benzimidazole nitrogen, sum of 15N chemical shifts of the molecules (r = −0.97), and sum of 13C chemical shifts of the molecules (r = −0.99).
CONCLUSION
This work examines the relationships between multinuclear NMR chemical shifts and properties of molecules in order to evaluate the utility of chemical shifts as molecular descriptors in Quantitative Structure/Spectral Data Property Relationship models. It is also the first report of 15N NMR studies of pantoprazole, lansoprazole, and ilaprazole. This work shows that NMR chemical shifts of PPIs exhibit correlation with pharmacokinetic and physicochemical properties of the molecules. The relationship between NMR chemical shifts and properties of molecules demonstrates the utility of NMR chemical shifts as molecular descriptors in Quantitative Structure/Spectral Data Property Relationship models. More importantly, use of 15N chemical shifts in such models can increase their efficiency and reliability since the nitrogens are frequently involved in the interactions of the molecules with the surrounding environment.
The study also offers insights into the additional information that NMR chemical shifts can provide. For example, as observed in the case of pyridine nitrogen of PPI molecules, the variations in chemical shift of particular atoms or lack thereof can be used as a constraint while developing newer molecules by making changes in the parent structure. Also, by studying the NMR chemical shift variations in different moieties of molecules separately, we can make inferences on which moiety influences which property of the molecule, thus enabling us to tweak that fragment to design molecules with desirable properties.
ACKNOWLEDGMENTS
The authors would like to thank Prof. K. V. Ramanathan, NMR Research Center, IISc, Bangalore for providing the facility for conducting 15N-NMR experiments.
FINANCIAL SUPPORT
WOS-A grant, Department of Science and Technology, Government of India.
CONFLICT OF INTEREST
None.
REFERENCES
Beil W, Staar U, Sewing KF. Pantoprazole: a novel H+/K(+)-ATPase inhibitor with an improved pH stability. Eur J Pharmacol, 1992; 218(2–3):265–71. CrossRef
Claramunt RM, López C, Elguero J. The structure of omeprazole in the solid state: a 13C and 15N NMR/CPMAS study. ARKIVOC, 2006; (v):5–11.
de Bortoli N, Martinucci I, Giacchino M, Blandizzi C, Marchi S, Savarino V, Savarino E. The pharmacokinetics of ilaprazole for gastro-esophageal reflux treatment. Expert Opin Drug Metab Toxicol, 2013; 9(10):1361–9. CrossRef
Khadikar PV, Sharma V, Varma BG. Novel estimation of lipophilicity using 13C NMR chemical shift as molecular descriptor. Bioorgan Med Chem Lett, 2005a; 15:421–5. CrossRef
Khadikar PV, Sharma V, Karmakar S, Supuran CT. Novel use of chemical shift in NMR as molecular descriptor: a first report on modeling carbonic anhydrase inhibitory activity and related parameters. Bioorgan Med Chem Lett, 2005b; 15:931–6. CrossRef
Kwon D, Chae JB, Park CW, Kim YS, Lee SM, Kim EJ, Huh IH, Kim DY, Cho KD. Effects of IY-81149, a newly developed proton pump inhibitor, on gastric acid secretion in vitro and in vivo. Arzneimittelforschung, 2001; 51(3):204–13. CrossRef
LuÄić B, Stepanić V, Plavšić D, Amić A, Amić D. Correlation between 13C NMR chemical shifts and antiradical activity of flavonoids. Monatsh Chem, 2014; 145(3):457–63. CrossRef
Morii M, Takata H, Fujisaki H, Takegucht N. The potency of substituted benzimidazoles such as E3810, omeprazole, Ro 18-5364 to inhibit gastric H+, K+-ATPase is correlated with the rate of acid-activation of the inhibitor. Biochem Pharmacol, 1990; 39(4):661–7. CrossRef
Nagaya H, Inatomi N, Nohara A, Satoh H. Effects of the enantiomers of lansoprazole (AG-1749) on H, K-ATPase activity in canine gastric microsomes and acid formation in isolated canine parietal cells. Biochem Pharmacol, 1991; 42:1875–8. CrossRef
Naidu GG. Identification of active sites on selected anticancer, proton pump inhibitor and antihypertensive drugs using UV-visible and NMR spectroscopy. PhD thesis, Andhra University, Visakhapatnam, 2016.
Roche VF. The chemically elegant proton pump inhibitors. Am J Pharm Educ, 2006; 70(5):Article 101. CrossRef
Sachs G, Shin JM, Howden CW. Review Article: The clinical pharmacology of proton pump inhibitors. Aliment. Pharmacol Ther, 2006; 23(Suppl 2):2–8. CrossRef
Smolka AJ, Goldenring JR, Gupta S, Hammond CE. Inhibition of Gastric H, K-ATPase activity and gastric epithelial cell IL-8 secretion by the pyrrolizine derivative ML 3000. BMC Gastroenterol, 2004; 4(4):1–11. CrossRef
Verma RP, Hansch C. Use of 13C NMR chemical shift as QSAR/QSPR descriptor. Chem Rev, 2011; 111:2865–99. CrossRef