
INTRODUCTION
Access to medicines is a worldwide concern, which is why the manufacture of generics is of great importance in the market (WHO, 2018). The fundamental advantage of these drugs over reference or branded drugs lies in their lower cost and easy access. However, internationally, there is a debate on their quality which casts a shadow on their efficacy and safety (Arcaro et al., 2021; Straka et al., 2017; Torres Serna et al., 2018). The implementation of Good Manufacturing Practices, Good Practice of Laboratory, the studies of bioequivalence, and the inspection by sanitary authorities have promoted the accomplishment of quality standards of generic marketed drugs (WHO, 2020), but inspections have also revealed some quality problems related to antibiotics drugs (WHO, 2017).
Ciprofloxacin (CIP), a class III drug according to the Biopharmaceutical Classification System (Reddy et al., 2017; WHO, 2006), is a broad-spectrum antibiotic belonging to the second generation of the quinolone group, and it is frequently prescribed for the treatment of infectious diseases, mainly of the urinary tract (Chao and Farrah, 2019; Instituto Nacional de Salud de Colombia, 2016). Prolonged-release (PR) formulations of CIP have shown advantages over conventional products in terms of patient compliance and reduced adverse effects (Omari et al., 2011). Nevertheless, in Colombia, there are very few oral CIP PR products authorized for commercialization. Since bioequivalence was not mandatory in Colombia for modified-release (MR) products before 2016, nowadays, there are no bioequivalent CIP PR products available in the country.
The release of an active pharmaceutical ingredient (API) from solid oral formulations is a prerequisite for its absorption, bioavailability, and subsequent clinical response, and it depends on the physicochemical characteristics of the API, on the excipients used, and on the product technology (Qiu et al., 2016). Differences in the formulation among this kind of products can affect their release characteristics, and for MR formulations of antibiotics, it has been shown that changes in the composition affecting the release profile of the drug can also cause variations in the minimum inhibitory concentration and the emergence of bacterial resistance, which is currently a major public health problem (Beg et al., 2012). Considering that the difference between generic and reference drugs is precisely in the composition of the formulation, it would be possible to find variations in the release profiles among MR antibiotics from different manufacturers, early through in vitro dissolution studies, before the in vivo evaluation of bioequivalence (Anderson et al., 1998).
In this study, we aimed to compare the dissolution profiles of two CIP 1,000 mg PR products, namely, the Colombian generic product and reference product, available for use in several Latin American countries, such as Brazil, Costa Rica, Cuba, Ecuador, Panama, Peru, and Venezuela. The results enabled us to conclude about the quality of the tested products, describe their release characteristics, and evaluate for any significant differences between them; this is all important information to consider prior to planning a bioequivalence study.
MATERIALS AND METHODS
Materials, reference substance, and products
Reagents
Sodium acetate, hydrochloric acid 37.30%, and monobasic potassium phosphate were from J.T. Baker (Shanghai, China), HPLC-grade acetonitrile, glacial acetic acid, orthophosphoric acid 85%, potassium chloride, sodium hydroxide, and triethylamine 99% were from Merck (Darmstadt, Germany). Water was purified using a Milli-Q system from Millipore (Bedford, MA).
Reference substance
Secondary standard of CIP hydrochloride traceable to the United States, Europe, and British Pharmacopeial standards, were obtained from Sigma-Aldrich (PHR1044, Lot LRAA8718, Deisenhofen, Germany).
CIP products
Two commercial products of CIP extended-release tablets (1,000 mg) were evaluated and designated as generic medicine (CX) and reference medicine CIPRO XR® (XR). The studies were carried out using formulations from the same batch, which were used within its useful life.
Instruments
Dissolution testing was carried out with a 708-DS dissolution apparatus coupled to 850-DS sampling station, and samples were quantified by UV-Vis spectroscopy using a CARY 60 UV-VIS spectrophotometer, all from Agilent (Santa Clara, CA). CIP assay was carried out using a Chromaster HPLC system (Hitachi, Tokyo, Japan).
Methods
Assay
To obtain the individual monograph of CIP PR tablets, the HPLC assay method from the United States Pharmacopeia (USP) 42-NF 37 (USP, 2019a) was adopted. The mobile phase consisted of a mixture of phosphoric acid adjusted to pH 3.0 with triethylamine and acetonitrile (87:13). Separation was carried out using a Gemini® C18 column, 5 µm, 150 × 4.6 mm (Phenomenex, Torrance, CA) at 30°C. All analyses were carried out under isocratic conditions at a flow rate of 1.5 ml/minute and a detection wavelength of 278 nm.
To prepare the standard solution, an appropriate amount of the standard powder was weighed and dissolved in the mobile phase. Next, a 1 ml aliquot was placed in a 10 ml volumetric flask and made up to the volume with the mobile phase (0.058 mg/ml).
To prepare the sample solution, 20 tablets of each product were weighed individually, and the average weight was calculated. Next, the tablets were ground into a fine powder and weighed to the equivalent of 125 mg of CIP in triplicate. The powder was then transferred to 250 ml volumetric flasks containing 200 ml of the mobile phase. The solutions were placed on an orbital shaker (J.P. Selecta, Barcelona, Spain) for 15 minutes, sonicated by an ultrasonic bath (Branson-Emerson, St. Louis, MO) for 25 minutes to complete their homogenization, allowed to cool to room temperature, and then made up to the volume with the mobile phase. A portion of each solution was filtered (0.45 μm, nylon), and aliquots of 1 ml were transferred to 10 ml volumetric flasks. The volume was completed with the mobile phase (0.05 mg/ml). Finally, a filtered portion (0.22 µm PTFE) of the dilutions was taken to be injected in triplicate into the chromatograph.
Identification
Identification was carried out by comparing the retention time of the CIP products against that of the standard solution.
Physical tests
Physical tests were carried out on 10 units for each evaluated product. The tested parameters were as follows:
Average weight: the tablets of each product were weighed individually. The average weight and relative standard deviation (RSD) were then calculated.
Dimensions: the length, width, and height of the tablets were measured by using a micrometer. The average values and the RSD were then determined.
Friability: the test was conducted using a friability tester (Nova Ética-Ethik, SP, Brazil), as indicated in Chapter 1216 of the USP. The friability was expressed as a percentage of the weight loss after agitation at 25 r.p.m. for 4 minutes (100 times) (USP, 2019b).
Dosage uniformity
Dosage uniformity of CIP tablets was determined via the weight variation method by calculating the acceptance value (AV) according to the USP Chapter 905 (USP, 2019c).
Validation of the analytical method for dissolution tests
A spectrophotometric method was validated based on the USP monograph for CIP PR (USP, 2019a), including three dissolution media: hydrochloric acid pH 1.2, acetate buffer pH 4.5, and phosphate buffer pH 6.8. To evaluate the linearity, accuracy, and precision of the method, calibration curves of the CIP standard in each medium were prepared, and the parameters were determined as follows.
Linearity was determined using three six-level calibration curves in the range of 0.55–8.57 μg/ml for each medium. For all cases, a goodness-of-fit test was applied considering the values of determination coefficients (r2) ≥0.999, relative error due to regression ≤2.0%, RSD of response factors ≤2.0%, and Cochran’s G test (Gexp ≤ Gtables).
Accuracy was evaluated via the standard addition method, in which samples at three concentration levels (1.5, 3.5, and 7.0 µg/ml) were prepared in triplicate for each dissolution medium and quantified by interpolation from the calibration curves, considering acceptable average recovery percentages between 97.0% and 103.0%. In addition, for each dissolution medium, Student’s t-test was applied, comparing the mean of the recovery percentages with a reference value of 100% recovery (AEFI, 2001).
Precision was determined as repeatability and intermediate precision. Repeatability was evaluated by calculating the RSD values of the concentration obtained for accuracy samples at each level and dissolution media. For intermediate precision, the effect of the interday variability (three different days) and two analysts was evaluated with three replicates for each medium at a nominal CIP concentration of 5.5 µg/ml. RSD values ≤3.0% and ≤6.0% were considered acceptable for repeatability and intermediate precision, respectively.
USP dissolution test
Dissolution was tested according to method 1 of the USP monograph for CIP PR. Six units of each product were subjected to dissolution test under the following conditions: apparatus 2 (paddle) at stirring rate of 50 rpm with 900 ml of acetate buffer (pH 4.5) at 37°C. The sampling times were 30, 60, and 120 minutes after the start of the test. At each time point, 2 ml of sample was withdrawn from each dissolution vessel and replaced with fresh medium. Next, the samples were filtered (0.45 µm, nylon), and aliquots of 50 µl were transferred to 10 ml volumetric flasks that were made up to the volume with the medium. The percentages of dissolved CIP were quantified by UV spectrophotometry at 278 nm using a standard calibration curve and compared against to Q values established in the USP monograph: Q30 min (30%–50%), Q60 min (50%–70%), and Q120 min (not less than 80%) (USP, 2019a).
Dissolution profiles
The test was carried out on 12 units for each product following the USP method (USP, 2019a) with three dissolution media (hydrochloric acid pH 1.2, acetate buffer pH 4.5, and phosphate buffer pH 6.8). For greater confidence in the data obtained, the 12 units were analyzed in two independent experiments on different days and with six units each. Samples were withdrawn from each dissolution vessel at times of 0.5, 1, 2, 4, 6, 8, 12, 16, 20, and 24 hours, and the dissolved CIP was quantified via UV spectrophotometry at 278 nm for media of pH 1.2 and 4.5 and at 272 nm for that of pH 6.8, using the calibration curves constructed for each medium. In the quantification stage, the percentages of dissolved CIP were calculated, considering only acceptable RSD values ≤20% for samples at the first time point (0.5 hour) and ≤10% for the subsequent times (FDA, 1997; Minsalud Colombia, 2016). In order to describe the drug release in each product, concentration versus time plots were constructed with data represented as mean ± standard deviation (SD) of the 12 units. The dissolution profiles obtained were compared using model-independent and model-dependent methods.
Data analysis
Model-independent method: in the model-independent method, difference factor f1 (Eq. 1), similarity factor f2 (Eq. 2), dissolution efficiency (DE), and the mean dissolution time (MDT) were calculated as follows:
In both equations, Rt and Tt are the percentages of drug dissolved during a period of t time for reference and generic formulations, respectively, and n is the number of experimental points on the dissolution profiles. Factor f1 values of 0–15 reflect the nonexistence of accumulated differences between two profiles at all experimental points. Factor f2 values of 50–100 indicate similarity between the formulations. The Food and Drug Administration, European Medicines Agency, and World Health Organization recommend the use of factor f2 for comparison of dissolution profile (EMA, 2010; FDA, 1997; WHO, 2006).
DE of each product was calculated as the area under the dissolution curve up to time t (AUC0t,t=24 hours and was expressed as a percentage to that of the area of the rectangle described by 100% dissolution at the same time point (Q100.t, Eq. 3) (Aguilar Ros et al., 2014):
MDT was calculated to characterize the dissolution rate of the drug in each dissolution profile using Equation (4), where Ti is a midpoint between two sampling time, ΔQi is the amount of drug dissolved in every interval of time, and Q∞ represents the maximum of drug dissolved (Aguilar Ros et al., 2014).
Model-dependent method: the experimental data obtained for each dissolution profile in the three media were transformed by applying the equations of different kinetic models as follows: order zero, order one, cube root, square root, and Weibull function. The best-fit model was selected considering the values of r2 (close to unity), and the smallest Akaike information criterion value (AIC) was also used as a discriminatory parameter (Eq. 5):
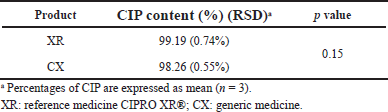 | Table 1. Results of CIP content in the assayed products. [Click here to view] |
AIC = n × In (WSSQ) + 2p (5)
where n is the number of dissolution data points, p is the number of parameters of the model, and WSSQ is the weighted sum of the square of residues (Costa and Sousa Lobo, 2001).
Once the best-fit model was selected, the parameters describing the model were estimated for each product and dissolution media.
Statistical analysis
The software Statgraphics Centurion XVI v.16.1.02 was used for statistical analyses. The means of the results of the tests for the two products were compared using Student’s t-test for two independent samples, with a confidence level of 95% and significance level alpha of 0.05. Normal distribution of the data was verified by the Shapiro–Wilk test and homogeneity of variance by the F test.
RESULTS AND DISCUSSION
Assay, identification, and physical tests
Results of CIP content in the studied products, as determined via HPLC, are presented in Table 1. In all products, the CIP content (%) met the specifications of the monograph (90.0%–110.0%) (USP, 2019a); the generic product was similar in potency to the reference medicine (p > 0.05). It was found a total correspondence between the retention times of CIP in the analyzed products where were compared with that in the reference substance
The results of the physical tests are presented in Table 2. The distribution of the weight of the products tablets ranged between 1,499.40 and 1,638.30 mg with RSD values of ≤0.32% that indicated low variability of this parameter in both products. In addition, the percentages of the drug in the composition of the tablets were not less than 60% of the total weight of the dosage units for the evaluated brands. The dimension test results showed that the values of length, width, and height had an RSD ≤ 1.00%, which indicates that the tablet dimension per product was homogeneous.
According to the results of the friability test, the percentage of weight loss in both products was ?1.0% meeting the pharmacopeial specification and indicating that the tablets exhibit wear resistance during the manufacturing and modification stages. The dose uniformity test showed that the products complied with the maximum allowed AV, L1 = 15.0. This possibly indicates a homogeneous distribution of CIP in the tablets as well as adequate and uniform incorporation of the active substance and excipients during the manufacturing process (USP, 2019b; Qiu et al., 2016).
Validation of the analytical method for dissolution tests
The spectrophotometric method for quantification of dissolved CIP was validated for the three dissolution media used for dissolution profiles, and the results showed that the method had linearity range of 0.55–8.58 µg/ml, with r2 ? 0.999, relative error due to the regression ?2%, RSD of the response factors <2%, and Gexp < Gtables, which indicate the goodness-of-fit of the regression (Table 3). The method was also proven to be accurate as it achieved recovery percentages between 97.0% and 103.0%, with RSD ? 3.0%. t-test was conducted to confirm the accuracy in all cases, where texp < ttables, indicating no significant differences between the average recovery and the 100% recovery. Finally, the method was proven to be precise, with RSD values of <3.0% for repeatability and RSD < 6.0% for intermediate precision (Table 4).
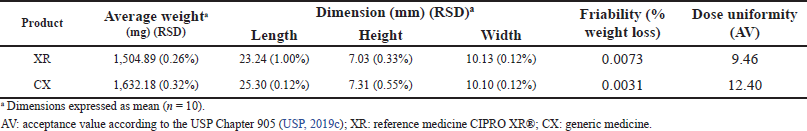 | Table 2. Results of physical tests: average weight, dimension, friability, and dose uniformity (weight variation). [Click here to view] |
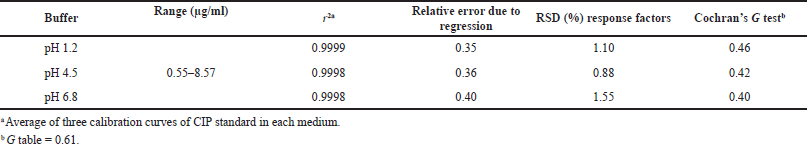 | Table 3. Linearity of the analytical method for dissolution tests of CIP. [Click here to view] |
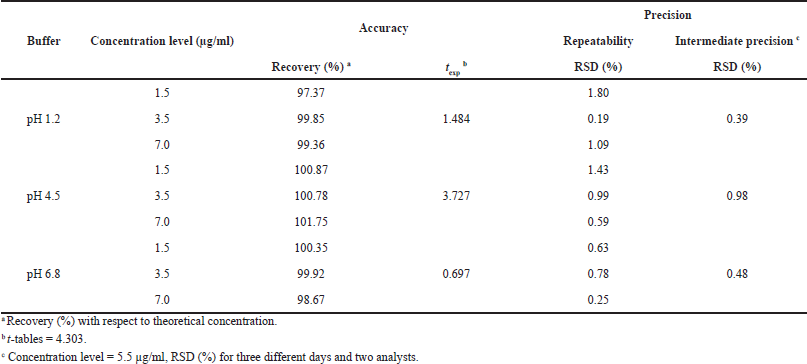 | Table 4. Accuracy and precision of the analytical method for dissolution tests of CIP. [Click here to view] |
 | Table 5. Results of the dissolution test of the CIP PR products according to USP monograph. [Click here to view] |
Dissolution test
In order to evaluate if the analyzed products met the specifications established by the USP monograph for CIP PR, the dissolution test 1 (USP, 2019a) was carried over. The percentage amount of dissolved CIP in the analyzed products met the established range of Qi for the L1 level dissolution criteria (Table 5), showing the good in vitro performance of the products. However, significant differences were observed in the Q30 min values between XR and CX products (p < 0.05).
The generic medicine released about 4% more CIP than the reference within the first 30 minutes; this may be due to differences in the formulation excipients (Omari et al., 2011). Knowing that the dissolution test works as a substitute in vitro for evaluation of the performance of the medicines and it could be an adequate tool for comparisons of generic versus reference products (FDA, 1997; Minsalud Colombia, 2016; Polli, 2008), this result justified the performance of dissolution profiles in the pH range of 1.2 to 6.8 to analyze possible differences between the products.
Dissolution profile
At pH 1.2 and 4.5, both products showed a characteristic profile, corresponding to a slow, controlled, and complete release which is typical of PR systems (Fig. 1). In those conditions, the RSD of the dissolved %CIP at the sampling times met the criteria established with values ≤6.8%; the products released approximately 80% of CIP after 2 hours, with the exception of CX in the pH 1.2 buffer in which it released only 68%, but at the end of the test (24 hours), the released %CIP was greater than 97% for all the products in the media aforementioned. Meanwhile, in the pH 6.8 phosphate buffer medium, the products coincide in showing a poor release profile exhibiting released %CIP values less than 15% (Fig. 1).
The physicochemical characteristics of CIP could explain the greater release percentages observed in the pH 1.2 and 4.5 media (up to 80%) and the poor release of the drug at pH 6.8 (less than 15%) since CIP exhibits a “U”-shaped pH-solubility profile in which it is highly soluble at pH values below 5 and above 10. This behavior is caused by the fact that CIP is a zwitterion (an electrically neutral chemical compound that has positive and negative formal charges on different atoms), with pKa1 of 6.0 (acid group), pKa2 of 8.8 (basic group), and isoelectric point of 7.2, where it is less soluble (neutral species) (Roca Jalil et al., 2015). The pH of the greatest solubility of CIP agrees with that for the upper gastrointestinal tract, where drug absorption is more efficient (Olivera et al., 2011).
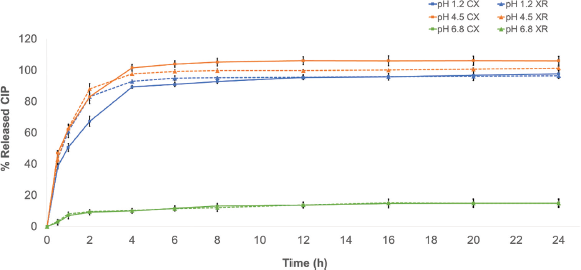 | Figure 1. Dissolution profile of the CIP products in hydrochloric acid buffer medium (pH 1.2), acetate buffer dissolution medium (pH 4.5), and phosphate buffer dissolution medium (pH 6.8). XR: reference medicine CIPRO XR®; CX: generic medicine. Data are expressed as mean ± SD. [Click here to view] |
Model-independent analysis
Table 6 presents the results of profile comparison using the model-independent method, in which only the profiles that met the established RSD criteria for the percentages of dissolved CIP at the sampling times were analyzed.
According to the literature, f2 values ≥50% indicate differences less than 10% in the dissolution profiles of two products, f2 values ≥65% indicate that the differences are reduced to 5%, and f2 values ≥83% indicate that the differences are even smaller than 2% (Gohel et al., 2014; Shah et al., 1998). Moreover, f1 values ≤15 reflect the nonexistence of accumulated differences between the profiles (FDA, 1997). The model-independent analysis showed that the dissolution profile of CX can be considered similar to that of XR in the pH 1.2 and 4.5 media, based on the f1 value less than 5.2 and the f2 value greater than 50.
The values of DE and MDT were calculated in order to preliminarily describe the dissolution rate constant of CIP in the studied formulations. Since DE values can be theoretically correlated to the plasma concentration versus the time curve obtained in vivo, it is a good parameter to compare dissolution profiles (Alkhalidi et al., 2010). At pH 4.5, pretty closer DE% for CX and XR products was observed, which confirmed the similarity between the dissolution profiles.
For his part, MDT characterizes the release rate of the drug from a dosage form, and in MR formulations, it indicates the efficacy of a polymer in delaying drug release (Wadher et al., 2011). According to the manufacturer, CIP XR tablets contain two layers, one of immediate release and another of controlled release consisting of an erosion matrix (Bayer HealthCare, 2021); once it is orally administered, the formulation rapidly releases up to 35% of the total dose and then gradually releases the remaining of the drug by water-swell of the polymer hypromellose, resulting in the erosion of the matrix (Gao et al., 2011; Talan et al., 2004). The products presented similar MDT values in the pH 4.5 medium; this finding can be attributed to the similar nature of the polymer used in the study formulations that delays the release of the drug from the matrix.
In contrast, at pH 1.2, both DE and MDT were statistically different between reference and generic products, but the variation was less than 10% for DE, which is considered acceptable to conclude about the equivalence between dissolution profiles (Anderson et al., 1998). Differences closer to 1 hour between MDT values of CIP PR products were also observed in this pH condition. However, literature reports indicate that even PR formulations with very different values of MDT and mean residence time (MRT) could be bioequivalent (Gomez-Mantilla et al., 2014); thus, this parameter could not affect the equivalence aforementioned.
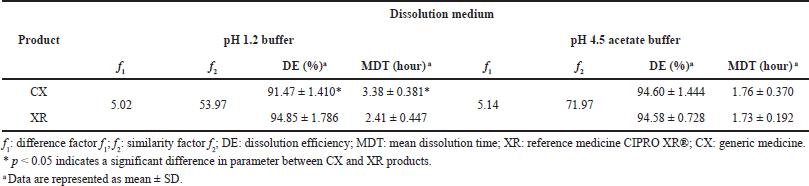 | Table 6. Model-independent method of the CIP PR products. [Click here to view] |
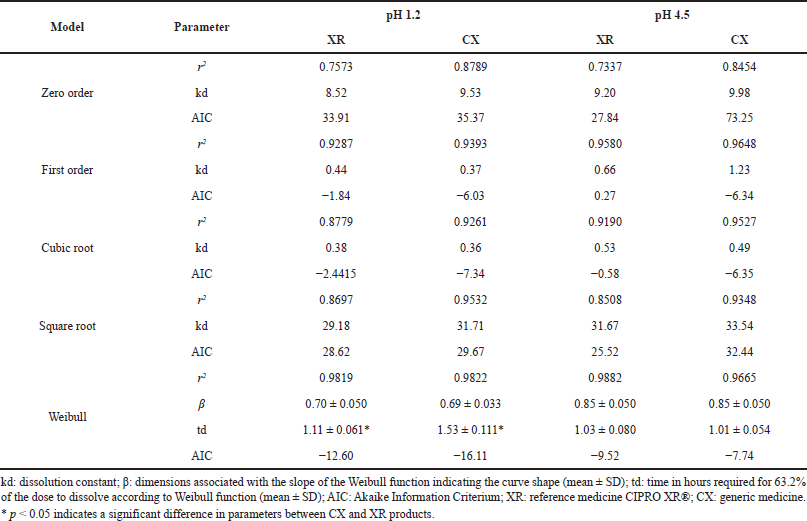 | Table 7. Model-dependent analysis of dissolution of CIP PR products. [Click here to view] |
Model-dependent analysis
The model-dependent method was applied to compare the parameters describing the dissolution profiles of the evaluated CIP PR products; since the drug was poorly released at pH 6.8, modeling was only made for the pH 1.2 and 4.5 media. According to r2 and AIC values, the profiles were best-fit to a Weibull function with the parameters presented in Table 7.
Weibull model has been widely used to analyze drug release from solid dosage forms since it could be applied to describe almost all types of dissolution curves when the drug release mechanism is the class Fickian diffusion (proportional to the gradient of the drug concentration), especially for a curve that exhibits fast and complete dissolution of the drug (Dokoumetzidis et al., 2006; Papadopoulou et al., 2006). In this function, β characterizes the shape of the dissolution curve as either exponential (β = 1), sigmoid/S-shaped (β > 1), or parabolic (β < 1), and td represents the time required for release 63.2% of the drug (Costa and Sousa Lobo, 2001). The shape of dissolution profiles of CIP from XR and CX products was parabolic for both pH media, while td values were equivalent at pH 4.5 but exhibited significant differences between products at the more acidic pH being less for the reference medicine, which also presented the lowest MDT and the greater DE from the model-independent analysis at pH 1.2 (Table 6).
In vivo comparison studies have proven the convenience of the use of CIP PR once a day against two doses of CIP-immediate release, since PR formulation provided more favorable plasma concentrations, less variability between patients, and reduction of frequency of nausea and diarrhea (Meagher et al., 2004; Omari et al., 2011; Rashid et al., 2011; Talan et al., 2004), attributes that could improve patient adherence to treatment and thus reduce the risk of recurrence infections or bacterial resistance.
Comparative studies between branded and generic drugs can contribute to improving the confidence of healthcare personnel and the community in all the medical technologies available on the market. It is important to have MR antibiotics accessible to the public given the aforementioned advantages.
CONCLUSION
In this study, the results of the quality control tests, dissolution profiles, and the release-kinetic parameters of 1,000 mg CIP PR tablets commercially available in Colombia were compared. The products CX and XR evaluated met all the quality specifications established in USP 42-NF 37, and the dissolution profile analysis showed that the drug release of the products was similar at pH 1.2 and 4.5 media.
Considering that CIP XR and CX products contain identical amounts of the API in the same chemical forms (hydrochloride and base), along with the f2 and f1 values, the modeling and the quality control assays result, both products can be considered pharmaceutically equivalent, and it is necessary to test their bioequivalence in an in vivo study in order to comply with the requirements established for MR formulations in most of the countries.
ACKNOWLEDGMENT
This work was supported by a grant from Impacto Caribe from Vicerrectoría de Investigación, Extensión y Proyección Social, Universidad del Atlántico (Colombia).
CONFLICT OF INTEREST
The authors state that they have no conflict of interest with the products mentioned in the manuscript.
AUTHORS’ CONTRIBUTIONS
Concept and design, GPDM, TSRA, and IBPB; data acquisition, AVDG and RMRI; data analysis/ interpretation AVDG, RMRI, IBPB, and GPDM; drafting manuscript, AVDG; critical revision of the manuscript, GPDM, TSRA, and IBPB; statistical analysis, AVDG; final approval, GPDM and IBPB; funding acquisition, GPDM, TSRA, and IBPB. All authors have read and agreed to the published version of the manuscript.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included within this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Aguilar-Ros A, Camaño-Somoza M, Martin-Martin, FR, Montejo-Rubio MC. Biofarmacia y farmacocinética: ejercicios y problemas resueltos. 2nd edition, Elsevier, Barcelona, España, 2014.
Alkhalidi BA, Alkhatib HS, Khdair AA. Comparative dissolution of diltiazem immediate and extended release products using conventional USP and innovative dissolution paddles. Open Drug Deliv J, 2010; 4(SPEC. ISSUE 1):48–54. CrossRef
Anderson NH, Bauer M, Boussac N, Khan-Malek R, Munden P, Sardaro M. An evaluation of fit factors and dissolution efficiency for the comparison of in vitro dissolution profiles. J Pharm Biomed Anal, 1998; 17(4–5):811–22. CrossRef
Arcaro R, da Veiga CRP, da Silva WV, da Veiga CP. Attitude and purchase intention to generic drugs. Int J Environ Res Public Health, 2021; 18:4579. CrossRef
Asociación Española de Farmacéuticos de la Industria (AEFI). Validación de métodos analíticos. Asociación Española de Farmacéuticos de la Industria, Barcelona, España, 2001.
Bayer HealthCare. CIPRO XR. 2021. [ONLINE] Available via https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/021473s043lbl.pdf (Accesed 20 August 2021).
Beg S, Nayak AK, Kohli K, Swain S, Hasnain MS. Antimicrobial activity assessment of time-dependent release bilayer tablets of amoxicillin trihydrate. Braz J Pharm Sci, 2012; 48(2):265–72. CrossRef
Chao YS, Farrah K. Fluoroquinolones for the treatment of respiratory tract infections: a review of clinical effectiveness, cost-effectiveness, and guidelines. Canadian Agency for Drugs and Technologies in Health, Ottawa, Canada, 2019.
Costa P, Sousa Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci, 2001; 13:123–33. CrossRef
Dokoumetzidis A, Papadopoulou V, Macheras P. Analysis of dissolution data using modified versions of Noyes–Whitney equation and the Weibull function. Pharm Res, 2006; 23(2):256–61.
European Medicine Agency (EMA). Guideline on the investigation of bioequivalence discussion. 2010. [ONLINE] Available via https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf (Accesed 16 October 2019).
Food and Drug Administration (FDA). Guidance for industry, dissolution testing of immediate release solid oral dosage forms. 1997. [ONLINE] Available via https://doi.org/10.14227/DT040497P15 (Accesed 16 October 2019). CrossRef
Gao P, Nie X, Zou M, Shi Y, Cheng G. Recent advances in materials for extended-release antibiotic delivery system. J Antibiot (Tokyo), 2011; 64(9):625–34. CrossRef
Gohel MC, Ramkishan A, Patel TM, Pandya R, Suthar V, Koradia H, Madat DV, Bariya S, Mehta T. Nomogram for computing the value of similarity factor. Indian J Pharm Sci, 2014; 76(3):245–51.
Gomez-Mantilla JD, Schaefer UF, Casabo VG, Lehr T, Lehr CM. Statistical comparison of dissolution profiles to predict the bioequivalence of extended release formulations. AAPS J, 2014; 16(4):791–801. CrossRef
Instituto Nacional de Salud de Colombia (INS). Informe Final de evento consumo de antibióticos en el ámbito hospitalario, Colombia, 2016. 2016. [ONLINE] Available via https://www.ins.gov.co/buscador-eventos/Informesdeevento/Consumo de antibiotico intrahospitalario 2016.pdf (Accesed 14 October 2019).
Meagher AK, Forrest A, Dalhoff A, Stass H, Schentag JJ. Novel pharmacokinetic-pharmacodynamic model for prediction of outcomes with an extended-release formulation of ciprofloxacin. Antimicrob Agents Chemother, 2004; 48(6):2061–8. CrossRef
Ministerio de Salud y Protección Social, República de Colombia (Minsalud Colombia). Resolución 1124 de 2016. 2016. [ONLINE] Available via https://www.invima.gov.co/documents/20143/453029/Resolución+1124+de+2016.pdf/97ecb574-0101-09a1-9eb1-d7594a5bd8e2?t=1540931891213 (Accesed 14 October 2019).
Olivera ME, Manzo RH, Junginger HE, Midha KK, Shah VP, Stavchansky S, Dressman JB, Barends DM. Biowaiver monographs for immediate release solid oral dosage forms: Ciprofloxacin hydrochloride. J Pharm Sci, 2011; 100(1):22–33. CrossRef
Omari DM, Johary D, Salem II, Najib N, Sallam AA. Bioequivalence of two oral extended release formulations of ciprofloxacin tablets in healthymale volunteers under fed and fasting conditions. J Bioequivalence Bioavailab, 2011; 3(2):038–42. CrossRef
Papadopoulou V, Kosmidis K, Vlachou M, Macheras P. On the use of the Weibull function for the discernment of drug release mechanisms. Int J Pharm, 2006; 309(1–2):44–50. CrossRef
Polli JE. In vitro studies are sometimes better than conventional human pharmacokinetic in vivo studies in assessing bioequivalence of immediate-release solid oral dosage forms. AAPS J, 2008; 10(2):289–99. CrossRef
Qiu Y, Chen Y, Zhang G, Yu L, Mantri RV. Developing solid oral dosage forms: pharmaceutical theory and practice. 2nd edition, Boston, MA, Academic Press, 2016.
Rashid M, Weintraub A, Nord CE. Comparative effects of the immediate and the extended release formulations of ciprofloxacin on normal human intestinal microflora. J Chemother, 2011; 23(3):145–9. CrossRef
Reddy NHS, Patnala S, Kanfer I. Investigation of biowaivers for immediate release formulations containing BCS III drugs, acyclovir, atenolol, and ciprofloxacin hydrochloride, using dissolution testing. AAPS PharmSciTech, 2017; 18(2):424–31. CrossRef
Roca Jalil ME, Baschini M, Sapag K. Influence of pH and antibiotic solubility on the removal of ciprofloxacin from aqueous media using montmorillonite. Appl Clay Sci, 2015; 114:69–76. CrossRef
Shah VP, Tsong Y, Sathe P, Liu JP. In vitro dissolution profile comparison—statistics and analysis of the similarity factor, f2. Pharm Res, 1998; 15(6):889–96. CrossRef
Straka RJ, Keohane DJ, Liu LZ. Potential clinical and economic impact of switching branded medications to generics. Am J Ther, 2017; 24(3):e278–89. CrossRef
Talan DA, Naber KG, Palou J, Elkharrat D. Extended-release ciprofloxacin (Cipro XR) for treatment of urinary tract infections. Int J Antimicrob Agents, 2004; 23(SUPPL. 1):54–66. CrossRef
United States Pharmacopeial Convention (USP). Ciprofloxacino, tabletas de liberación prolongada. In: The United States Pharmacopeia, 42nd edition, USP, Rockville, MD, pp 1012–15, 2019a. [Spanish version].
United States Pharmacopeial Convention (USP). (1216) Friabilidad de las tabletas. In: The United States Pharmacopeia, 42nd edition, USP, Rockville, MD, pp 8355–56, 2019b. [Spanish version].
United States Pharmacopeial Convention (USP). (905) Uniformidad de unidades de dosificación. In: The United States Pharmacopeia, 42nd edition, USP, Rockville, MD, pp 7239–42, 2019c. [Spanish version].
Torres Serna C, Ángel Medina JC, Klinger Torres HV, Márquez Florez V, Micolta Bejarano JM, Sánchez Suescún JJ. Generic drugs, perception of the doctors. cali, colombia. Rev Científica Cienc Méd, 2018; 21(1):40–9. CrossRef
Wadher KJ, Kakde RB, Umekar MJ. Formulation and evaluation of a sustained-release tablets of metformin hydrochloride using hydrophilic synthetic and hydrophobic natural polymers. Indian J Pharm Sci, 2011; 73(2):208–15. CrossRef
World Health Organization (WHO). WHO Expert Committee on specifications for pharmaceutical preparations. 2006. [ONLINE] Available via https://apps.who.int/iris/handle/10665/43443 (Accesed 14 October 2019).
World Health Organization (WHO). WHO Global surveillance and monitoring system for substandard and falsified medical products. 2017. [ONLINE] Available via https://ahpsr.who.int/publications/i/item/978-92-4-151342-5 (Accesed 13 October 2019).
World Health Organization (WHO). Addressing the global shortage of, and access to, medicines and vaccines: Report by the Director-General. 2018. [ONLINE] Available via http://apps.who.int/gb/ebwha/pdf_files/EB142/B142_13-en.pdf (Accesed 13 October 2019).
World Health Organization (WHO). Expert committee on specifications for pharmaceutical preparations - technical report series. World Health Organization, Geneva, Switzerland, 2020.