
INTRODUCTION
Zidovudine (azidothymidine) (AZT, Fig. 1a) was the first active pharmaceutical ingredient to be approved by the Food and Drug Administration for the treatment of acquired immunodeficiency syndrome (AIDS) in humans and is still in use as a part of the highly active antiretroviral therapy regimen (Shey et al., 2013). Although its efficacy has long been demonstrated, AZT has various unfavorable aspects that constitute major concerns, such as cellular toxicity (D’Andrea et al., 2008) and suboptimal pharmacokinetic properties (Barbier et al., 2000; Eilers et al., 2008; Narciso et al., 2014). Among the clinical circumstances of AZT toxicity, there are numerous hematological effects, suppression of bone marrow cell functions, liver disorders, and myopathies (Khandazhinskaya and Shirokova, 2013). In addition, AZT exhibits short plasma half-life (t1/2 ≈ 1 hour) (Barbier et al., 2000), low plasma protein-binding capacity (Narciso et al., 2014), and incapacity to reach effective concentrations in viral reservoir tissues (Eilers et al., 2008). For this reason, AZT has to be administered frequently at high doses, thereby increasing the incidence of unwanted side effects that often compromised the adherence of the patient to the anti-HIV treatment (Narciso et al., 2014; Quevedo and Briñón, 2009).
 | Figure 1. Chemical structures of (a) zidovudine (AZT) and (b) AZT-Ac, and atom numbering used in this study. [Click here to view] |
Several strategies have been applied to enhance the oral bioavailability of AZT as well as to prolong its elimination half-life (De Clercq, 2007), with the preparation of prodrugs being a widely applied methodology (D’Andrea et al., 2008; Dalpiaz et al., 2012; Khandazhinskaya and Shirokova, 2013; Moroni et al., 2002; Parang et al., 2000; Quevedo and Briñón, 2009; Quevedo et al., 2008; Raviolo et al., 2009; Ribone et al., 2016; Solyev et al., 2012). Among these prodrugs, 3´-azido-2´,3´-deoxy-5´-O-oxalatoyl-thymidine (AZT-Ac, Fig. 1b) exhibited anti-HIV potency, low cytotoxicity, and improved in vitro permeability. In fact, it permeates the rat intestinal segment at a lower rate than AZT but resists enzymatic hydrolysis with no evidence of saturable transport mechanisms in the jejunum or the proximal ileum (as the case of AZT) and has a extended plasma half-life in rats (Quevedo and Briñón, 2009). AZT-Ac also displayed a high stability under acidic conditions (pH 2) for 48 hours and suffered hydrolysis at pH 7.2, but its calculated t1/2 was 12.8 hours (Ribone et al., 2016).
Considering that AZT-Ac is a prodrug of AZT with improved hydrolytic and pharmacokinetic properties and that thymidine nucleosides such as AZT and stavudine exhibit crystalline polymorphism (Gandhi et al., 2000; Soares et al., 2013), the goal of this study was to investigate the existence of different polymorphs of AZT-Ac to support the development effort. To this aim, we subjected AZT-Ac to a polymorph screen using several solid-form screening techniques. We isolated only two solid forms, which were characterized by means of powder X-ray diffraction (PXRD), solid-state nuclear magnetic resonance (SS 13CNMR), differential scanning calorimetry (DSC), thermogravimetry (TG), and hot-stage microscopy (HSM). The effects of temperature and moisture on the chemical and physical stabilities of both the solid forms of AZT-Ac were investigated at 40°C/75% relative humidity (RH) along with their disc intrinsic dissolution behavior in simulated gastric fluid (SGF) without pepsin at 37°C to compare their solid-state stabilities and dissolution behaviors.
MATERIALS AND METHODS
Materials
AZT was generously provided by Filaxis Laboratories (Buenos Aires, Argentina). AZT-Ac raw material (AZT-Ac-r) was synthesized and purified following a described procedure (Moroni et al., 2002). Precoated silica gel 60 F254 plates (Merck KGaA, Germany), filter paper (2.7 μm, Whatman 542, UK), and nylon membranes (0.45 μm, Pall Corporation, USA) were commercially acquired. All the solvents used were of analytical grade, and Milli-Q→ water (Millipore, Bedford, USA) was also utilized. For thin-layer chromatography (TLC) analyses, ethyl acetate-acetone-methanol 5:3:2 v/v and precoated plates of silica gel 60 F254 (Merck Chemicals) were used as the eluting solvent system and stationary phase, respectively. Spots were visualized with ultraviolet (UV) light and iodide vapors.
Preparation of AZT-Ac forms
In search of different polymorphs for AZT-Ac, a variety of solid-form screening techniques were used, which included crystallization (slow and fast) from solution, antisolvent precipitation, vapor diffusion crystallization (Cunha, 2008), polymer-induced heteronucleation (Price et al., 2005), and lyophilization (Kassuha et al., 2015). Samples generated by these techniques were initially analyzed by means of TLC for chemical purity, finding that they were chromatographically pure, as no other spots than that of AZT-Ac were detected. Eventually, only two different solid forms were obtained, which will be reported in this paper. These two forms were named hereafter as AZT-Ac-C and AZT-Ac-A and were reproducibly obtained using the following procedures:
AZT-Ac-C: a saturated solution of AZT-Ac-r was prepared in acetonitrile at 20°C–25°C room temperature (RT) and filtered (Whatman 532 paper) into a beaker. The beaker was covered with filter paper and allowed to evaporate at the (RT, 20°C–25°C) in the dark. Then, the isolated crystals were stored in a desiccator under CaCl2.
AZT-Ac-A: a t-butanol-water (20:80, v/v) solution of AZT-Ac-r (30 mg/ml) was frozen in air liquid and lyophilized at −40°C for 24 hours (FreeZone 6, Labconco→, USA). The resultant solid was subjected to a secondary drying in a desiccator (vacuum, RT, P2O4) for 24 hours and stored at −20°C in a tightly closed vial until analyzed.
PXRD
PXRD patterns were collected at RT on a X´PERT PRO X-ray diffractometer (PANalytical, The Netherlands) fitted with a copper tube (Cu Kα = 1.54178 Å) and a Ni filter, with the X-ray generator being set at a voltage of 40 kV and a current of 30 mA. The samples were analyzed with a step size of 0.05° 2θ and step time of 3 seconds from 3° to 35° 2θ, using a 25-mm diameter Si single crystal holder.
SS 13CNMR
SS 13CNMR spectra were recorded at the RT [Bruker Avance II spectrometer operating at 300.13 MHz (protons) and 75.47 MHz (carbons), equipped with a 4-mm Magic Angle Spinning (MAS) probe] using the cross polarization (CP)/MAS sequence with proton decoupling during acquisition. Adamantane was used as an external reference for the 13C spectra and to set the Hartmann−Hahn matching condition in the cross-polarization experiments. The spinning rate, recycling time, contact time during CP, and the acquisition time were 9 kHz, 30 seconds, 2 ms, and 41 ms, respectively. The numbers of transients were 256, and the SPINAL-64 sequence was used for decoupling during acquisition with the proton decoupling field H1H satisfying ω1H/(2π) = γH1H/(2π) = 78 kHz. The quaternary carbon edition spectra were acquired with the nonquaternary suppression (NQS) sequence, with the 1H and 13C radio-frequency fields being removed during 40 μs after CP and before the acquisition. This delay allowed the carbon magnetization to decay because of 1H−13C dipolar coupling, resulting in spectra where CH and CH2 were substantially removed. Assignment of the 13C resonances was accomplished with the aid of NQS experiments and residual dipolar coupling in the solid-state and by comparison with the 13C spectrum (DMSO-d6) of AZT-Ac-r.
DSC, TG, and HSM
DSC and TG measurements were recorded on Modulated Differential Scanning Calorimetry (MDSC) 2920 and TG 2950 analyzers (TA Instruments Inc., USA), respectively. Samples (1–2 mg) were heated in nonhermetically sealed aluminum pans, using a heating rate of 10°C/min and nitrogen (99.99%) purge of 50 ml/min. The DSC and TG temperature axes were calibrated with indium (99.99%, m.p.156.60°C) and the Curie point of Ni (358.14°C), respectively. Empty aluminum pans were used as references. The data were analyzed using the Universal Analysis 2000 software (TA Instruments Inc.). The physical and morphological changes that occurred in the samples during heating were observed through a microscope fitted with a Kofler hot stage (Leitz, Wetzlar, Germany) at a constant rate of 8°C/min.
Solid-state stability
To investigate the effect of storage temperature and moisture on the physical and chemical stabilities of the samples, 200 mg of each solid form (n = 2) was stored at 40°C/75% RH (open glass vials inside a drying pistol (ISV, Argentina) filled with a saturated aqueous solution of NaCl) and analyzed through TLC and PXRD at various time intervals to evaluate chemical purity and phase changes.
Disc intrinsic dissolution measurements
Intrinsic dissolution experiments were performed using a rotating disc apparatus that meets United States Pharmacopeia (USP) specifications (USP, 2012) on a Hanson SR6 dissolution tester (Hanson Research, Chatsworth, CA). The powdered samples (100.0 mg) of AZT-Ac-C and AZT-Ac-A were directly compacted into the stainless steel cylinder of the rotating disc apparatus (resulting in discs at one side of the cylinder with surface area of 0.5 cm2) at 118 MPa (dwell time of 1 minute), a compromise compression that produced nondisintegrating discs and did not induce polymorphic transitions and devitrification, as indicated by PXRD analyses. Disc intrinsic dissolution was carried out at 50 rpm in 250 ml of degassed SGF without pepsin at 37°C ± 0.5°C. This medium and its volume were established by considering the chemical stability of AZT-Ac in acidic media (Ribone et al., 2016), its solubility (∼8 mg/ml, pH 1.2 at 37°C), and the minimum volume required to completely immerse the die. The 250 ml volume maintained sink conditions during the experiment (since the concentrations of AZT-Ac-C and AZT-Ac-A after 25 minutes were 0.028 and 0.035 mg/ml, respectively) and completely immersed the die. Therefore, the concentrations applied in the experiments were below 0.8 mg/ml (10% of the saturation concentration). Aliquots of 5 ml were withdrawn (with replacement) at time intervals of 5, 10, 15, 20, and 25 minutes, filtered (0.45 μm), and analyzed by UV spectroscopy at 267 nm. In all cases, the first ml was discarded. The dissolution tests were performed in triplicate (AZT-Ac-A) and duplicate (AZT-AC-C). For the quantification of the concentration of AZT-Ac in the dissolution medium, a standard curve was prepared using five concentration levels. The cumulative amount of dissolved test specimen per initial area was plotted against time. The intrinsic dissolution rate (IDR) (or mass flux (J) in mg/cm2/min) was calculated from the slope of the regression line (USP, 2012).
RESULTS AND DISCUSSION
Identification of AZT-Ac polymorphs
Figure 2 displays the powder patterns for the raw material (Fig. 2a), AZT-Ac-C (Fig. 2b), and AZT-Ac-A (Fig. 2c). As shown in Figure 2(a) and (b), the powder patterns of the raw material and AZT-Ac-C were coincident in the position of the diffraction peaks, and no additional reflections were visualized, indicating that they represented the same phase. In contrast, the powder pattern of AZT-Ac-A (Fig. 2c) had a broad halo and an absence of diffraction peaks, revealing that it was X-ray amorphous.
It is worth mentioning that the PXRD patterns of samples obtained using other crystallization techniques such as fast evaporation from different solvents, vapor diffusion crystallization with petroleum ether (35°C‒60°C) as antisolvent, and polymer-induced heteronucleation were coincident (Figures not shown) to those of Figure 2(a) and (b), indicating a low likelihood for several crystalline forms of AZT-Ac. Unfortunately, we could not obtain crystals suitable for resolving the crystalline structure of AZT-Ac-C; thus, to gain structural information on this form, its SS 13CNMR spectrum was obtained (Figure 3).
 | Figure 2. Overlay PXRD data for three representative samples of AZT-Ac: (a) AZT-Ac raw material. (b) AZT-Ac-C (crystallized from acetonitrile). (c) AZT-Ac-A (obtained by lyophilization from t-butanol-water, 20:80 v/v). [Click here to view] |
As shown in Figure 3(a), the spectrum of AZT-Ac-C exhibited well-resolved signals for the 11 carbons of the molecule (Fig. 1b), with their line widths being all in the range of 30–50 Hz, indicating an ordered and crystalline form (Monti et al., 2014). The resonances at 83.3, 139.9, 149.9, and 166.9 ppm showed a broadened and split structure, but these effects were due to coupling to 14N quadrupolar nuclei not due to the presence of more than one molecule in the asymmetric unit of AZT-Ac, which, thus, comprised one molecule of AZT-Ac. In particular, the resonances at 83.5, 139.9, and 166.9 ppm consisted of two lines with relative intensities of 2:1, which is typical of 13C nuclei coupled to a single 14N (Olivieri et al., 1988). Thus, these were assigned to C1´, C6, and C4, respectively (Fig. 1b, Table S1). In contrast, the resonance at 149.9 ppm consisted of four lines, which is the characteristic of a 13C nucleus coupled to a pair of 14N (Olivieri et al., 1988); hence, this resonance was assigned to C2 (Table S1). According to the NQS spectrum (Fig. 3b), the quaternary carbons of AZT-Ac-C resonated at 110.7 (C5), 149.9 (C2), 157.5 (C1´´), 159.1 (C2´´), and 166.9 ppm (C4) (Table S1), and it is worth mentioning that the methine carbons (C2´ and C5´´) were also observed in the NQS spectrum (Fig. 3b) because their resonance intensities were severe affected but not totally suppressed during the delay time. C1´ (83.5 ppm) and C4´ (82.9 ppm) (Table S1) were assigned without ambiguity, as the C1´ resonance line shape revealed a residual dipolar interaction structure due to bonding of C1´ to N1, whereas the one of C4´ did not. In the case of C3´ (59.1 ppm), no residual dipolar coupling was observed, probably due to the mobility of the azide group or the puckering motion of the furanose ring (Kolodziejski and Klinowski, 1999), which can contribute to average out the coupling.
 | Figure 3. (a) 13C CP/MAS NMR and (b) NQS spectra of AZT-Ac-C. [Click here to view] |
DSC, TG, and HSM
The behavior on heating of AZT-Ac-C and AZT-Ac-A was investigated by DSC and TG (Figure 4). The DSC curve of AZT-Ac-C (Fig. 4a) exhibited, in the temperature range of 25°C–160°C, an endotherm at 92.1°C (extrapolated onset temperature), followed by several small broad endothermic effects. According to the TG data (Fig. 4a), the endotherm at 92.1°C was associated with weight loss, suggesting that the sample melted with decomposition. The interpretation of the DSC events just described was assisted by HSM and TLC. Microscopic observations revealed that the transparent prismatic particles (Figure S1, Supplementary material) of AZT-Ac-C (not embedded in silicon oil to provide experimental conditions similar to those in the DSC and TG experiments) melted at about 93°C, and on further heating, the molten phase diminished in size and became brownish, a typical feature of a decomposition process (Bruno et al., 2010; Cuffini et al., 2007; Sperandeo and de Bertorello, 2001; Sperandeo et al., 2005). TLC analysis of the obtained residue confirmed the decomposition of the sample as several spots were detected, with AZT-Ac being absent.
 | Figure 4. DSC (nonhermetically sealed pan) and TG (open pan) curves (10°C/min, flowing N2 at 50 ml/min) of: (a) AZT-Ac form I and (b) AZT-Ac- A. [Click here to view] |
The DSC curve of AZT-Ac-A (Fig. 4b) showed, in the temperature range of 25°C–110°C, only a very broad endotherm at 48.5°C (Tpeak), resembling a glass transition overlapped with a desolvation process (Kassuha et al., 2015) as the sample was X-ray amorphous and the respective TG curve (Fig. 4b) exhibited a gradual weight loss. Taking into account that AZT-Ac-A was obtained by lyophilization from t-butanol-water and both solvents can remain in lyophilized solids, as it was observed for other lyophilized samples (Kassuha et al., 2015; Teagarden and Baker, 2002), the occurrence of a desolvation process was considered. Above 110°C, the DSC curve displayed another very small broad endotherm superimposed with a large exothermic effect that continued at temperatures above 170°C. Both the effects had associated a TG weight loss (Fig. 4b) that continued above 170°C, typical of a decomposition process that continued at higher temperatures (Bruno et al., 2010; Cuffini et al., 2007; Sperandeo and de Bertorello, 2001). Thus, AZT-Ac-A was visually examined by HSM to assist in the interpretation of the DSC events. Heating the white opaque particles (Figure S1, Supplementary material) of AZT-Ac-A (not immersed in silicon oil) from 25°C revealed that they became almost colorless and compacted at about 60°C, which was consistent with a glass transition concomitant with a desorption process. The sample started to move (at about 80°C) and became liquefied (at about 103°C), and on further heating, the molten phase diminished in size and brownish. TLC analysis of the obtained residue confirmed the decomposition of the sample as the AZT-Ac spot was not detected. Further studies using other analytical techniques should be necessary to determine the glass transition temperature of AZT-Ac-A as well as to identify the residual solvents.
Solid-state stability
The solid-state stabilities of AZT-Ac-C and AZT-Ac-A were evaluated at 40°C/75% RH. Exposure of AZT-Ac-C to the assayed conditions for 14 days did not result in polymorphic conversions as indicated by PXRD. In fact, the powder patterns of AZT-Ac-C (Fig. 5a), collected after 7 and 14 days of storage at 40°C/75% RH, did not exhibit new reflections attributable to hydrate formation or polymorphic conversions. However, TLC analysis of an aliquot taken at day 14 revealed the presence of free AZT, indicating that the ester bond of AZT-Ac-C had partially hydrolyzed affording its parent compound; hence, its stability was not investigated further. It should be noted that the characteristic peaks of AZT could not be visualized in Figure 5(a), suggesting that its quantity was below to the detection limit of PXRD (1%–5%) (Pecharsky and Zavalij, 2003). In contrast, exposure of AZT-Ac-A to temperature and moisture provoked its devitrification to AZT-Ac-C as indicated by PXRD. Indeed, the X-ray diffraction patterns registered at day 7 and 14 (Fig. 5b) exhibited various low-intensity peaks of AZT-Ac-C superimposed to the amorphous halo, indicating that AZT-Ac-A partly devitrified to its crystalline counterpart. In addition, TLC analyses revealed the presence of free AZT at day 14, indicating that the temperature and humidity promoted its hydrolysis to its parent compound. Therefore, AZT-Ac-C and AZT-Ac-A cannot be stored in the typical conditions of a hot and humid climate without any special storage and handling instructions.
Intrinsic dissolution behavior
An intrinsic dissolution is a pharmacopeial method (EP, 2013; USP, 2012) for the evaluation of drug powders, and it has wide application in characterizing new chemical entities (NCEs) during the formulation development since parameters which control the rate of dissolution, such as surface area exposed to the medium, temperature, stirring rate, and pH, are kept constant (Kaplan, 1972; Löbmann et al., 2014; Sehic et al., 2010; USP, 2012). In addition, Kaplan (1972) had suggested that IDRs (J), determined in the range of 1–7 pH at 50 rpm and 37°C, may be useful to predict absorption problems as compounds with J > 1 mg/min/cm2 are usually not prone to dissolution rate-limited absorption, with J < 0.1 mg/min/cm2 usually exhibit dissolution rate-limited absorption, and with J between 0.1 and 1.0 mg/min/cm2 are borderline compounds; thus, additional information would be required to ascertain the effect of dissolution on the absorption rate (Du et al., 2013).
 | Figure 5. PXRD patterns of (a) AZT-Ac-C and (b) AZT-Ac-A after storage at 40°C/75% RH. To allow a visual comparison, the powder pattern of AZT-Ac-C was included at the top of (b). [Click here to view] |
The amounts released per initial compact area (mg/cm2) of AZT-Ac-C and AZT-Ac-A are shown in Figure 6. The profile of AZT-Ac-C (Fig. 6a) was a typical linear intrinsic dissolution plot (R2 > 0.999), indicating good linearity between time and amount released per unit area. The IDR, calculated from the slope of the regression line, was found to be 0.494 mg/min/cm2. In contrast, the intrinsic dissolution profile of AZT-Ac-A was not linear (Fig. 6b), showing an upward curvature that is typical of experimental problems such as physical degradation of the compact by cracking or erosion (USP, 2012). Regression analysis of all the data points of AZT-Ac-A showed that quadratic fitting (i.e., second-order polynomial) gave an excellent regression (R2 > 0.9995). Thus, to estimate the IDR (J) of AZT-Ac-A, linear regression analysis was performed on data points in the initial linear region of the dissolution curve (i.e., up to 10 minutes, inset of Fig. 6) as recommended by the United States Pharmocopeia (USP, 2012), which provided, and IDR of 0.397 mg/min/cm2. It is worth mentioning that after the dissolution experiments, the dies were patted dried and visually examined under a magnifying glass to evaluate if changes had occurred on the surface of discs. No changes in the surface of the compacts of AZT-Ac-C were detected after dissolution. In contrast, the compacts of AZT-Ac-A showed holes and lines, indicating that the surface area of the discs did not remain constant during the assay, and this makes an estimate of its IDR speculative at best.
 | Figure 6. Disc intrinsic dissolution profiles (50 rpm, 250 ml of degassed SGF, 37°C ± 0.5°C) of (a) AZT-Ac-C and (b) AZT-Ac-A. Inset: linear portion of the intrinsic dissolution profile of AZT-Ac-A. [Click here to view] |
According to Kaplan’s classification (Du et al., 2013; Kaplan, 1972; Yao et al., 2014), AZT-Ac belonged to the category of borderline NCE as the J values of AZT-Ac-C and AZT-Ac-A were between 0.1 and 1.0 mg/min/cm2. Hence, additional information may be required to ascertain the effect of dissolution on the absorption rate of AZT-Ac.
CONCLUSION
This investigation contributes to the preformulation assessment of AZT-Ac by exploring some aspects of its solid-state properties and inherent stability. Two solid forms of AZT-Ac were prepared and physically characterized by PXRD, SSNMR, DSC, TG, and HSM. Although the enzymatic and stability of AZT-Ac in solution it was found good, especially in acidic conditions (Ribone et al., 2016), our research studies demonstrated that under hot and humid climate conditions (40°C/75% RH), the solid-state stability of AZT-Ac is not good as the two solid forms partly hydrolyzed to the parent compound AZT after 14 days of storage. In addition, AZT-Ac-A devitrified partially to the crystalline form after 7 days of storage, suggesting that the amorphous form was not optimal to use for further development. The ID rates of AZT-Ac-C and AZT-Ac-A were determined to fall in the range 0.1–1.0 mg/cm2/min, which suggested that this prodrug could exhibit dissolution rate-limited absorption. Therefore, approaches to enhance the solid-state stability and dissolution rate of AZT-Ac-C should be considered in a further development.
ACKNOWLEDGMENTS
This work was supported by SECyT-UNC and FONCyT of Argentina. Flavia P. Bruno and Diego E. Kassuha are grateful for financial support from the CONICET Type II Doctoral fellowships. Diego E. Kassuha also thanks SeCiTI de San Juan (Argentina) for financial support. We thank Dr. P. Hobson, native speaker, for revision of the manuscript.
DECLARATION OF INTEREST
None.
REFERENCES
Barbier O, Turgeon D, Girard C, Green MD, Tephly TR, Hum DW, Bélanger A. 3′-Azido-3′-deoxythymidine (AZT) is glucuronidated by human UDP-glucuronosyltransferase 2B7 (UGT2B7). Drug Metab Dispos 2000; 28:497–502.
Bruno FP, Caira MR, Monti GA, Kassuha DE, Sperandeo NR. Spectroscopic, thermal and X-ray structural study of the antiparasitic and antiviral drug nitazoxanide. J Mol Struct 2010; 984:51–7. CrossRef
Cuffini S, Ayala A, Mendes Filho AJ, Ellena J, Mascarenhas Y, Sielser HW, Monti GA, Aiassa V, Sperandeo NR. Physicochemical characterization of deflazacort: thermal analysis, crystallographic and spectroscopic study. Steroids 2007; 72:61–9. CrossRef
Cunha S. Métodos Simples de Formação de Monocristal de Substância Orgânica para Estudo Estrutural por Difração de Raios X. Quim Nova 2008; 31:906–9. CrossRef
D’Andrea G, Brisdelli F, Bozzi A. AZT: an old drug with new perspectives. Curr Clin Pharmacol 2008; 3:20–37. CrossRef
Dalpiaz A, Paganetto G, Pavan B, Fogagnolo M, Medici A, Beggiato S, Perrone D. Zidovudine and ursodeoxycholic acid conjugation: design of a new prodrug potentially able to bypass the active efflux transport systems of the central nervous system. Mol Pharmaceut 2012; 9:957–68. CrossRef
De Clercq E. The design of drugs for HIV and HCV. Nat Rev Drug Discov 2007; 6(12):1001–18. CrossRef
Du W, Zhou Y, Gong Y, Zhao C. Investigation of physicochemical properties and in-vitro in-vivo evaluation of agomelatine polymorphs. Asian J Pharm Sci 2013; 8:181–90. CrossRef
Eilers M, Roy U, Mondal D. MRP (ABCC) transporters-mediated efflux of anti-HIV drugs, saquinavir and zidovudine, from human endothelial cells. Exp Biol Med 2008; 233:1149–60. CrossRef
European Pharmacopoeia (EP). Council of Europe, Strasbourg, France, Intrinsic dissolution, 8th edition, pp. 331–332, 2013.
Gandhi RB, Bogardus JB, Bugay DE, Perrone RK, Kaplan MA. Pharmaceutical relationships of three solid state forms of stavudine. Int J Pharm 2000; 201:221–37. CrossRef
Kaplan SA. Biopharmaceutical considerations in drug formulation design and evaluation. Drug Metab Rev 1972; 1:15–33. CrossRef
Kassuha D, Aiassa V, Bruno F, Cuadra G, Sperandeo N. Preparation and characterization of polymorphs of the glucocorticoid deflazacort. Pharm Dev Technol 2015; 20(4):401–9. CrossRef
Khandazhinskaya AL, Shirokova EA. AZT 5’-phosphonates: achievements and trends in the treatment and prevention of HIV infection. Acta Naturae 2013; 5(3): 54–61. CrossRef
Kolodziejski W, Klinowski J. 13C CP/MAS NMR study of anti-HIV-1 agent AZT. Chem Phys Letters 1999; 303:183–6. CrossRef
Löbmann K, Flouda K, Qiu D, Tsolakou T, Wang W, Rades T. The influence of pressure on the intrinsic dissolution rate of amorphous indomethacin. Pharmaceutics 2014; 6:481–93. CrossRef
Monti GA, Chattah AK, Garro Linck Y. Solid state nuclear magnetic resonance in pharmaceutical compounds. Annu Rep NMR Spectro 2014; 83:221–69. CrossRef
Moroni GN, Bogdanov PM, Briñón MC. Synthesis and in vitro antibacterial activity of novel 5-O-analog derivatives of zidovudine as potential prodrugs. Nucleos Nucleot Nucl 2002; 21:231–41. CrossRef
Narciso P, Bolli A, Fasano M, Ascenzi P. Binding of anti-HIV drugs to human serum albumin. IUBMB Life 2014; 56(10):609–14. CrossRef
Olivieri AC, Frydman L, Grasselli M, Días LE. Analysis of 13C, 14N residual dipolar coupling in the 13C CP/MAS NMR spectra of ribonucleosides. Magn Res Chem 1988; 26:281–6. CrossRef
Parang K, Wiebe LI, Knaus EE. Novel approaches for designing 5-O-ester prodrugs of 3-azido-2, 3-dideoxythymidine (AZT). Curr Med Chem 2000; 7:995–1039. CrossRef
Pecharsky VK, Zavalij PY. Fundamentals of powder diffraction and structural characterization of materials. Springer Science + Business Media, Inc., p. 389, 2003. ISBN 978-0-387-09579-0
Price CP, Grzesiak AL, Matzger AJ. Crystalline polymorph selection and discovery with polymer heteronuclei. J Am Chem Soc 2005; 127:5512–7. CrossRef
Quevedo MA, Briñón MC. In vitro and in vivo pharmacokinetic characterization of two novel prodrugs of zidovudine. Antiviral Res 2009; 1:103–1. CrossRef
Quevedo MA, Ribone SR, Moroni GR, Briñón MC. Binding to human serum albumin of zidovudine (AZT) and novel AZT derivatives. Experimental and theoretical analyses. Bioorg Med Chem 2008; 16:2779–90. CrossRef
Raviolo MA, Trinchero-Hernández JS, Turk G, Briñón MC. Synthesis and antiretroviral evaluation of novel derivatives of Zidovudine. J Braz Chem Soc 2009; 20:1870–7. CrossRef
Ribone, SR, Schenfeld, EM, Madrid M, Pierini AB, Quevedo MA. Evaluation and synthesis of AZT prodrugs with optimized chemical stabilities: experimental and theoretical analyses. New J Chem 2016; 40:2383–92. CrossRef
Sehic S, Betz G, Hadzidedi S, Kocova El-Arini S, Leuenberger H. Investigation of intrinsic dissolution behavior of different carbamazepine samples. Int J Pharm 2010; 386:77–90. CrossRef
Shey MS, Kongnyuy EJ, Alobwede SM, Wiysonge CS. Co-formulated abacavir-lamivudine-zidovudine for initial treatment of HIV infection and AIDS. Cochrane Database Syst Rev 2013; 28(3):1–38. CrossRef
Soares KC, Rediquieri CF, Souza J, Serra CH, Abrahamsson B, Groot DW, Kopp S, Langguth P, Polli JE, Shah VP, Dressman J. Biowaiver monographs for immediate-release solid oral dosage forms: Zidovudine (azidothymidine). J Pharm Sci 2013; 102:2409–23. CrossRef
Solyev PN, Shipitsin AB, Karpenko IL, Nosik DN. Synthesis and anti-HIV properties of new carbamate prodrugs of AZT. Chem Biol Drug Des 2012; 80:947–52. CrossRef
Sperandeo NR, de Bertorello MM. Solid state characterization of potential protozoocidal agents-aminoisoxazolyl napthoquinones. Thermochim Acta 2001; 378:67–71. CrossRef
Sperandeo NR, Karlsson A, Cuffini S, Pagola S, Stephens PW. The crystal structure and physicochemical characteristics of 2-Hydroxy-N- [3(5)-pyrazolyl]-1,4-naphthoquinone-4-imine, a new antitrypanosomal compound. AAPS Pharm Sci Tech 2005; 6(4):E655–63. CrossRef
Teagarden DL, Baker DS. Practical aspects of lyophilization using non-aqueous co-solvent systems. Eur J Pharm Sci 2002; 15:115–33. CrossRef
United States Pharmacopeia (USP) General Information chapter <1087> Apparent Intrinsic Dissolution-Dissolution Testing Procedures for Rotating Disc and Stationary Disc. In The United States Pharmacopeia and National Formulary USP 35–NF 30; The United States Pharmacopeial Convention, Inc., Rockville, MD, pp 660–3, 2012.
Yao Y, Lin G, Xie Y, Ma P, Li G, Meng Q, Wu T. Preformulation studies of myricetin: a natural antioxidant flavonoid. Pharmazie 2014; 69:19–26.
SUPPLEMENTARY MATERIAL
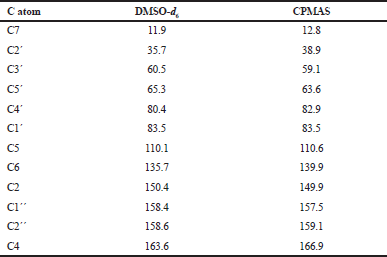 | Table S1. 13C NMR chemical shifts (in ppm) of AZT-Ac-A in solution (DMSO-d6) and in the solid state (CPMAS spectrum). [Click here to view] |
 | Figure S1. Images of AZT-Ac-C (a) and AZT-Ac-A (b) taken through a stereomicroscope (StereoZoom® Leica S8 APO, magnification of 10×). [Click here to view] |