
INTRODUCTION
Chalcones are one of the most important classes of natural products existing in many plant species, such as compound 1 in Macaranga denticulata (Lei et al., 2016), 2 in Uvaria siamensis roots (Salae et al., 2017), 3 in Stevia lucida (Morales et al., 2018), and 4 in Pongamia pinnata (L.) Pierre roots (Wen et al., 2018). Recently, some new hydroxychalcones bearing sugar functionalities (5–7) have been isolated and identified from the flowers of Coreopsis lanceolata by Kim et al. (2019) (Fig. 1). In basic structure, they are 1,3-diphenyl-2-propen-1-ones (two aromatic rings connected with α,β-unsaturated carbonyl moiety). In nature, chalcones serve as precursors for flavonoids and isoflavonoids biosynthesis (Ahmadi et al., 2019). Scientific study has demonstrated that chalcone derivatives display a wide variety of attractive biological activities, such as anti-inflammatory (Rücker et al., 2015), anticancer (Bonakdar et al., 2017), antidiabetic (Shukla et al., 2017), antiprotozoal (Hayat et al., 2011), antibacterial (Osorio et al., 2012), antiviral (Pradip et al., 2016), and antioxidant (Díaz-Carillo et al., 2018). They have also been reported to show antihypertension and antitumor effects (Avila-Villarreal et al., 2013; Bandgar et al., 2012). Thus, their fascinating biological properties has made chalcone derivatives as a main target for the development of novel molecular diversity in drug design and discovery projects (Espinoza-Hicks et al., 2019; Tajuddeen et al., 2018; Zhuang et al., 2017).
Nowadays, nitrogen, oxygen, and sulfur containing heterocyclic compounds have been widely studied due to their interesting applications as bioactive molecules (Kalaria et al., 2018; Saleh et al., 2019). For instance, compounds containing some nitrogen heterocycles, such as pyrazole, benzimidazole, and triazole have been reported to possess antiproliferative, anti-inflammatory, kinase inhibitory, antimicrobial, and anticancer properties (Celik et al., 2018; El-Gamal et al., 2017; Pinto et al., 2014 ; Sashidhara et al., 2015). Meanwhile, oxygen as well as sulfur containing heterocycles, such as benzofuran, benzopyran, and benzothiophene derivatives, have attracted medicinal chemist’s interest because of their therapeutics potential as antitubercular, antibacterial, antioxidant, cytotoxic, and anticancer (Baldisserotto et al., 2018; Romero-Parra et al., 2016; Singh et al., 2016; Xu et al., 2019). With the widespread of benefit of organic compounds bearing heterocyclic moieties, many researchers have focused their study in the synthesis of chalcone derivatives containing heterocyclic scaffolds. A large number of bioactive chalcones are synthesized together with the existence of heterocyclic ring in the structure (Fig. 2). This review focuses on the recent development of these compounds with significance in the field of medicinal chemistry due to their notable biological actions.
 | Figure 1. Basic structure of chalcone (middle) and some naturally occurring chalcones. [Click here to view] |
BIOLOGICAL ACTIVITIES OF CHALCONE-HETEROCYCLIC HYBRIDS
Chalcones containing N-heterocycles
5-membered ring
Imidazole and Benzimidazole. Several previously reported chalcone derivatives containing 5-membered ring of N-heterocycles are depicted in Figure 3. Sasidharan et al. (2018) synthesized 11 derivatives of (2E)-1-[4-(1H-imidazol-1-yl) substituted phenyl]-3-phenylprop-2-en-1-one and evaluated their biological action as inhibitors of recombinant human monoamine oxidase (MAO) A and B. Compound (1) was denoted to be the most active in the series, a nonselective and reversible competitive inhibitor of MAO-A and MAO-B having IC50 values of 0.30 ± 0.010 and 0.40 ± 0.017 μM, respectively. Meanwhile, compound (2) exhibited appreciable activity with IC50 values of 1.06 ± 0.090 and 0.32 ± 0.021 μM, respectively. These compounds have better inhibitory activities on MAO-A than Toloxatone (IC50 = 1.10 ± 0.0085 μM) but lower activities on MAO-B inhibition than Pargyline (IC50 = 0.082 ± 0.010 μM) as standard. Several substituted styryl 2-benzimidazole chalcone derivatives have been prepared by fly-ash:H2SO4 catalyst and tested for their insect anti-feedant activities using Dethler’s method (Janaki et al., 2016). It was found that the compound containing p-bromo substituted to the phenyl ring (3) was found to be the most active as antifeedant at 150 ppm concentration. Some N-benzyl substituted benzimidazole chalcones have been synthesized via multistep reaction sequences (Padhy et al., 2016). The synthesized compounds were screened for their antibacterial performance against some selective Gram-positive and Gram-negative bacteria. Amongst tested benzimidazole-chalcone hybrids, compound (4) showed good activity against Staphylococcus aureus and Bacillus subtilis with MIC value of 62.5 μg/ml on both cases. However, this compound possessed less activity against Escherichia coli and Pseudomonas aeruginosa as Gram-negative bacteria with MIC values of 125 and 500 μg/ml, respectively.
Carbazole. Considering that topoisomerase II is one of promising targets for anticancer drugs, some chalcone derivatives containing carbazole moiety have been synthesized and evaluated as non-intercalative topoisomerase II inhibitors and apoptosis-inducing agents (Li et al., 2018). Although compound (5) possessed moderate topoisomerase II inhibitory activity at 50 μM, it is noteworthy that this derivative displayed high growth inhibition against four human cancer cell lines with IC50 values of 0.36 μM (on HeLa), 2.16 μM (on A549), 0.62 μM (on PC-3), and 0.22 μM (on HL-60). Meanwhile, compound (6) displayed high activity as topoisomerase II inhibitor but showed less inhibitory activity against tested cancer cell lines with IC50 values of 5.48, 9.57, 7.12, and 2.85 μM, respectively. Compound (5) and (6) can arrest the HL-60 cells in sub G1 phase by induction of apoptosis.
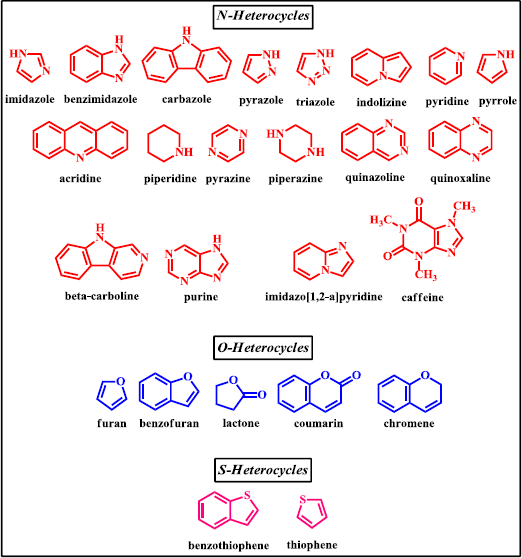 | Figure 2. Various heterocyclic systems integrated in chalcone derivatives. [Click here to view] |
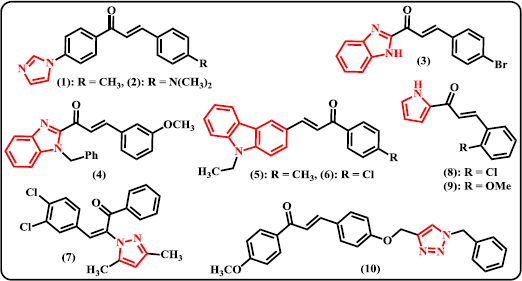 | Figure 3. Chalcone derivatives containing 5-membered N-heterocycles. [Click here to view] |
Pyrazole. A series of pyrazole-based chalcone derivatives were synthesized by Kumari et al. (2018) and subjected to antimicrobial and antioxidant screening. Some of the tested compounds exhibited significant activity against bacterial strains. Amongst the pyrazole-chalcone hybrids, compound (7) displayed the most potent activity against B. subtilis, P. aeruginosa, S. aureus, and E. coli with inhibition zone inhibition of 16 ± 0.82, 14 ± 1.24, 13 ± 0.60, and 14 ± 0.83 mm, respectively. The in vitro antioxidant evaluation of the synthesized compounds was performed using 1,1-diphenyl-2-picryl hydrazide (DPPH) radical scavenging method. Compound (7) possessed promising activity with IC50 value of 88.04 μg/ml.
Pyrrole. Synthesis of a series of pyrrole-based chalcone derivatives and biological evaluation of their CYP1 enzyme inhibitory activity were performed by Williams et al. (2017). From the assay, compound (8) was denoted as the most active agent that selectively inhibited CYP1B1 isoform with IC50 of ~0.2 μM. However, compound (9) has shown to inhibit both CYP1A1 and CYP1B1 isoforms with IC50 value of ~0.9 μM in the same conditions. Further study revealed that this compound totally protected human cells from benzo[a]pyrene toxicity and reversed cisplatin resistance.
Triazole. Some chalcone-1,2,3-triazole conjugates have been prepared by using copper nanoparticles supported on cellulose in aqueous medium (Yadav et al., 2017). The synthesized compounds were evaluated for their anticancer potential using 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay against four human cancer cell lines (HepG2, MIA-Pa-Ca-2, MCF-7, and A549). The most active compound (10) (IC50 against abovementioned cell lines = 9, 4, 5 and 11 μM, respectively) displayed better or comparable performance to the reference drugs. Furthermore, this lead compound not only induced apoptosis and G2/S arrest but also triggered mitochondrial potential loss in MIA-Pa-Ca-2 cells.
.png) | Figure 4. Chalcone derivatives containing 6-membered N-heterocycles. [Click here to view] |
6-membered ring
Pyridine. The structure of chalcone compounds bearing 6-membered N-heterocycles are presented in Figure 4. Several substituted pyridine-containing chalcone derivatives were designed, synthesized, and biologically screened for their antiviral activity (Chen et al., 2015). Results demonstrated that at 500 μg/ml, most of the synthesized compounds exhibited appreciable antiviral activity against cucumber mosaic virus (CMV). Particularly, compound (11) displayed an enhanced antiviral activity with curative, protection, and inactivation activities of 69.8 ± 1.8%, 39.1 ± 2.6%, and 88.1 ± 1.5% respectively, while compound (12) also exhibited high activity against CMV with above parameter values of 65.0 ± 3.9%, 54.9 ± 1.4%, and 88.6 ± 1.2%, respectively. These compounds were denoted as the most active compounds in the series.
Acridine. The significant role of acridine moiety contributes to molecular diversity of chalcone derivatives, which is essential for potent antimalarial activity. Tomar et al. (2010) designed new chalcone derivatives containing acridinyl scaffold from the reaction between various 3ʹ- or 4ʹ-aminochalcones and 9-chloroacridine via catalyst-free nucleophilic aromatic substitution. The synthesized compounds have been elucidated and in vitro evaluated for their antimalarial properties against Plasmodium falciparum NF-54. All the chalcone derivatives demonstrated 100% inhibition at concentration of 10 μg/ml and above. Compounds (13)–(15) showed >71% inhibition at 2 μg/ml. Although at low concentration (0.4 μg/ml), compound (13) exhibited 71.4% inhibition of parasite.
Piperidine. Some piperidine tethered chalcone derivatives were synthesized and tested as inhibitors of normal human basophil degranulation and anti-PI3Kδ activities (Dumontet et al., 2018). Compound (16) actively demonstrated to reduce respiratory pressure and inhibit normal human basophil degranulation in a dose-dependent manner. This compound has comparable activity with reference drug betamethasone.
Pyrazine. Synthesis and biological assay of chalcone derivatives containing pyrazine nucleus were conducted by Kucerova-Chlupacova et al. (2015). Compounds (17) and (18) bearing nitro substituent showed good potency against some fungal strains. For instance, on Trychophyton mentagrophytes, they have MIC values of 7.81 and 3.90 μM, respectively. They displayed comparable activity to that of fluconazole, a reference mycoses drug which is occasionally used after voriconazole and terbinafine. The synthesized compounds were also tested for their antimycobacterial activity. Not only in antifungal screening, these two compounds were also denoted as the most potent agents against Mycobacterium tuberculosis H37Rv (ATCC 27294) with MIC90 value of 6.25 μg/ml for both compounds.
Piperazine. A series of new chalcone-based dithiocarbamate derivatives containing piperazine and morpholine moieties were synthesized, structurally elucidated and evaluated for their antibacterial activity against multidrug-resistant Gram-negative bacteria (Ayman et al., 2019). In chalcone-piperazine series, compound (19) has proved to inhibit P. aeruginosa (Ps12), and Klebsiella pneumoniae (K4) with inhibition zone diameter of 24 and 25 mm, respectively. However, compound (20) with inhibition diameter of 23 and 20 mm, respectively, showed better DNA binding affinity than reference drug doxorubicin with IC50 value of 30.97 μg/ml. This result indicating that compound (20) could have a role in high antibacterial effect. Molecular docking study revealed that aromatic planar moiety of this compound intercalates toward the minor groove between the double strands of DNA.
Quinazoline. Inspired by the availability of some anticancer drugs containing quinazoline moiety, such as Gefitinib and Erlotinib (Herbst, 2003; Murphy and Stordal, 2011; Sun et al., 2011), a new series of 10 quinazoline-based chalcone derivatives were designed and synthesized via four steps reaction starting from 2-aminobenzoic acid (Madhavi et al., 2017). Amongst the synthesized derivatives, compounds (21)–(24) exhibited higher or comparable anticancer potential than the reference drug Combretastatin-A4 against four human cancer cell lines. These compounds have IC50 values of 2.90, 0.10, 0.10, and 2.10 μM, respectively, against A549 human alveolar adenocarcinoma cell line. On HT-29 human colorectal adenocarcinoma cell line, these compounds have IC50 of 0.18, 0.13, 1.56, and 2.89 μM, respectively. Against A375 melanoma cell line, these compounds displayed IC50 of 1.89, 1.34, 0.19, and 1.37 μM, respectively. On MCF-7 human breast adenocarcinoma cell line inhibition, compound (22)–(24) were checked with IC50 values of 0.17, 0.14, and 0.16 μM, respectively. Meanwhile, control drug has IC50 values of 0.11, 0.93, 0.18, and 0.21 μM against A549, HT-29, MCF-7, and A375 cancer cell lines, respectively. Recently, Han et al. (2019) synthesized chalcone analogues containing a 4-oxoquinazolin-2-yl scaffold and evaluated for their antitumor effects. From 38 compounds synthesized, (25) was denoted as the most potent agent that exhibit cytotoxic activities against HCT-116 and MCF-7 cells with IC50 of 3.56 and 4.08 μM, respectively. Compound (25) induced apoptosis in the sub-G1 phase and mitochondrial death pathway.
Quinoxaline. Organic compounds containing chalcone-quinoxaline hybrid was first ever synthesized by Desai et al. (2017) and subjected to antimicrobial and antitubercular assays. Through biological evaluation, it was noted that compound (26) and (27) with hydroxyl functionality in the ring exhibited significant antitubercular activity toward MTbH37RV with minimum inhibition concentration value of 3.12 μg/ml, comparable to the control drugs Pyrazinamide and Ciprofloxacin. From evaluation of antimicrobial effects, compound (26) displayed antibacterial activity against both Gram-positive and Gram-negative bacteria, but no activity against fungal strains. It has MIC of 5 μg/ml against both S. aureus and E. coli. Meanwhile, compound (27) showed antibacterial as well as antifungal effects with MIC values of 10, 5, and 10 μg/ml toward S. aureus, E. coli and Candida albicans, respectively.
Other N-heterocyclic rings
Caffeine. A novel series of caffeine-based chalcone derivates was synthesized and biologically tested as potential antitrypanosomal, antileishmanial, and antimalarial candidates (Insuasty et al., 2015). Amongst the designed compounds, (28) and (29) showed notable performance against Leishmania panamensis with growth inhibition values of 88.3 ± 1.5% and 82.6 ± 2.2%, respectively, at the concentration of 20 μg/ml (Fig. 5). Meanwhile, compound (30) that contains trimethoxy substituent exhibited remarkable inhibition against Trypanozoma cruzi even at very low concentration (9.7 ± 1.5% at 1.0 μg/ml; EC50 5.9 ± 1.4 μg/ml). Investigation on cytotoxic effects demonstrated that compound (30) was highly cytotoxic against promonocytic human U-937 cell with LC50 of 3.2 ± 0.5 μg/ml. However, none of these compounds exhibited strong antimalarial activity.
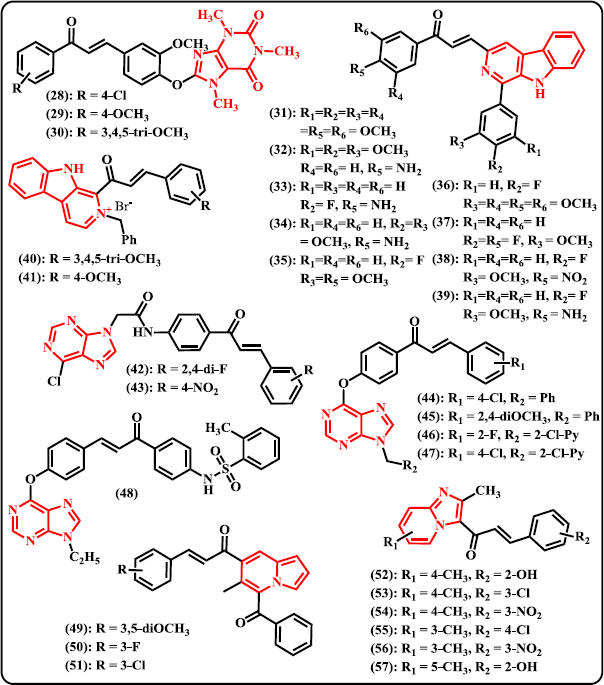 | Figure 5. Chalcone derivatives containing some other ring type of N-heterocycles. [Click here to view] |
Carboline. A new series of tricyclic pyrido[3,4-b] indole (β-carboline) tethered chalcone derivatives were designed, synthesized, and biologically evaluated for their anticancer activity and DNA-binding affinity (Shankaraiah et al., 2015). Most of the synthesized derivatives possessed promising cytotoxic properties against A549 lung adenocarcinoma cancer cell lines with IC50 less than 10 μM. Compound (31) with trimethoxy substituent on the aromatic ring at chalcone skeleton as well as C-1 position of β-carboline was observed to possess remarkable activity against all the tested cancer cell lines with IC50 values of 5.30 (A549 lung adenocarcinoma), 6.37 (B-16 melanoma), 19.59 (PC-3 human prostate), 23.08 (HT-29 human colon colorectal), and 44.26 (HeLa cervical) μM. However, compound (32)–(39) showed significant elevation in ΔTm of DNA compared to reference drug Adriamycin that indicating significant interaction and notable DNA stabilization. Another novel series of chalcone derivates connected to β-carboline via C-1 position were also synthesized and investigated for their activities as potent anticancer and antibacterial agents (Venkataramana Reddy et al., 2018). Off the synthesized β-carboline-chalcone conjugates, compound (40) that contains trimethoxy substituent and the molecule in the form of bromide salt was denoted as the most active compound against a panel of cancer and non-cancerous cell lines with IC50 values of 20 ± 2.1 (BxPC-3), 22.1 ± 3.23 (HeLa), 16.13 ± 4.2 (C4-2), 22.02 ± 3.25 (PC-3), 17.18 ± 2.98 (HEK293T), 15.95 ± 3.41 (MDA-MB-231), and 55.23 ± 5.8 (NIH3T3) μM. Meanwhile, compound (41) with p-methoxy group was found to be the most active analogue in the series to inhibit Gram-positive as well as Gram-negative bacterial strains. The best performance of this compound occurred when it can inhibit S. aureus with 15 mm of zone inhibition diameter and MIC value of 440 μg/ml.
Purine. Purine is N-heterocyclic aromatic compound consisting an imidazole ring fused to a pyrimidine system. Novel chalcone derivatives containing a purine scaffold were designed and synthesized by combining biologically active substructures (Gan et al., 2017). Chalcone and purine moieties were connected with an amide bridge. The synthesized compounds were subjected to antiviral evaluation against tobacco mosaic virus (TMV) and CMV. Amongst them, compounds (42) and (43) demonstrated superior curative, protective, and inactivation ability against TMV, which were better than those of reference drug ribavirin. Compounds (42) and (43) displayed appreciable curative and protective activities on CMV. Particularly, compound (42) exhibited a moderate affinity to TMV coat protein which is in tune with the inactivation ability. Still come from same research group, hydroxy chalcones and 6-chloro-9H-purine were adducted to give chalcone-purine hybrids and tested in vivo for their antiviral activity (Wang et al., 2018). The result revealed that amongst the designed derivatives, compounds (44)–(47) showed superior curative activity toward CMV with EC50 values of 301.1, 315.7, 282.3, and 230.5 μg/ml, respectively. These compounds were more active than that of control drugs ribavirin (726.3 μg/ml) and dufulin (373.7 μg/ml). Furthermore, from fluorescence spectroscopy study demonstrated that compound (44) displayed strong combining capacity to TMC coat protein. In another research paper, Zhou et al. (2018) synthesized a new series of chalcone derivatives bearing a purine and benzenesulfonamide moieties. Through in vivo antiviral assays, some of the derivatives have been reported to exhibit excellent anti-TMV and anti-CMV. With EC50 value of 51.65 μg/ml, compound (48) was denoted as the most potent hybrid that showed significant inactivating activity toward TMV. This derivative also displayed strong binding capacity to TMV coat protein.
Indolizine. An accessible protocol for the synthesis of a new series of chalcone-indolizine hybrids via base-mediated Aldol condensation has been presented by Park et al. (2018). The synthesized hybrids were then subjected to anticancer assessment against lymphoma cells (U937, Raji and JeKo-1 cells). Among them, compound (49) was denoted as the most potent agent that resulting cell viability less than 60% at 1 μM against U937 cells. The data in hand also indicated the importance of 3,5-dimethoxy moiety in the phenyl ring toward anticancer properties. Compound (50) and (51) with halogen substituent at meta position displayed remarkable inhibition of cell activity better than that of the para position. Moreover, compound (49) demonstrated better apoptotic inducing activity than reference drug doxorubicin but lower than that of cisplatin.
Imidazopyridine. A series of imidazo[1,2-a]pyridine-chalcone conjugates were designed, synthesized, and evaluated as potent inhibitors against A549 cell line (Kuthyala et al., 2019). Screening using MTT assay suggested that compounds (52)–(57) displayed significant inhibitory activity with IC50 values in the range of 7.0–42.2 μg/ml. Among the synthesized molecules, compound (54) was denoted as the most active agent having IC50 of 7.0 ± 2.1 μg/ml. Through single crystal XRD study, compounds (53), (55), and (57) showed well-defined crystal structures.
Chalcones containing O-heterocycles
5-membered ring
Furan. Zheng et al. (2011) synthesized 36 new heterocyclic chalcone compounds containing furan, thiofuran, and quinoline systems and investigated for their activity against Gram-positive and Gram-negative bacteria. Some derivatives selectively inhibited Gram-positive bacteria, including the multidrug-resistant isolates. Furan-containing chalcones were found to be more active than two other series. The synthesized compounds displayed high activity against Streptococcus mutans. In particular, compound (58) was noted as the most potent derivative with MIC value of 2 μg/ml, it was as active as positive control norfloxacin but less active than oxacillin (Fig. 6). On the contrary, none of the designed compounds exhibited growth inhibition against Gram-negative bacteria at concentration of 64 μg/ml. The authors noted that no significant difference was observed between electron-donating and electron-withdrawing groups on the aromatic ring for contribution to antibacterial performance. Another research result within the same laboratory, Sun et al. (2013) constructed furan-chalcone derivatives as protein tyrosine phosphatase inhibitors. Amongst the compounds reported, (59) and (60) were selected as potent protein tyrosine phosphatase1B (PTP1B) inhibitors with IC50 values of 2.90 ± 0.12 and 2.49 ± 0.23 μM, respectively. The study suggested that, in general, compounds tethered with electron-withdrawing or dihydroxy groups exhibited promising activity against the PTP1B.
Benzofuran. A number of benzofuran-chalcone hybrids were synthesized and in vivo studied with transgenic Caenorhabditis elegans (Sashidhara et al. 2014). The results showed that compound (61)–(63) significantly reduced Aβ aggregation, acetylcholinesterase (AChE) level, and oxidative stress in the worms. These hybrids were found to increase acetylcholine (ACh) level and provide protection toward chemically-induced cholinergic neurodegeneration. In order to find a novel anticancer candidate containing chalcone-benzofuran system, Coskun et al. (2017) developed a new series of 1-(7-ethoxy-1-benzofuran-2-yl) substituted chalcone derivates by the base-catalyzed Claisen-Schmidt reaction of 1-(7-ethoxy-1-benzofuran-2-yl) with various aromatic aldehydes. It is noteworthy to remember the compound that contain dimethyl amino or trimethoxy substituents showed promising activity against tested cancer cell lines. Compound (24) was denoted as the most potent derivative with IC50 values of 9, 2, and 10 μM against A549, MCF-7, and PC-3, respectively. At 20 μM concentration, compound (64) was in the late apoptotic stage around more that 90% of the cells in MCF-7 and A549, while in PC-3 cell, 6.45% and 59.70% were in the early and late apoptotic stage, respectively. Still at same sample concentration, compound (65) displayed the most growth-inhibitory effect on PC-3 cell line for 72 hours. In a work by Mphahlele et al. (2018), a series of 2-arylbenzo[c]furan-chalcone hybrids were designed and synthesized via Claisen–Schmidt reaction to produce chalcone and subsequently reacted with some aryl alkynes via Sonogashira cross-coupling reaction to construct the benzofuran moiety. The organic derivatives were then investigated for their in vitro antiproliferative effects against MCF-7 cancer cell line and for potential anti-tubulin polymerization and/or EGFR-Tyrosine Kinase phosphorylation. Most of the synthesized compounds displayed medium to superior activity against MCF-7 compared to the reference drug actinomycin D. Amongst them, compound (66) and (67) exhibited significant inhibition with IC50 of 0.55 ± 0.24 and 3.55 × 10−4 ± 0.07 μM, respectively. These lead compounds also demonstrated promising inhibitory activity against EGFR-TK phosphorylation with corresponding IC50 values of 0.17 ± 0.03 and 0.09 ± 0.03 μM, respectively. The values are comparable with standards actinomycin D and gefitinib. The title compounds were found to possess anti-tubulin effect.
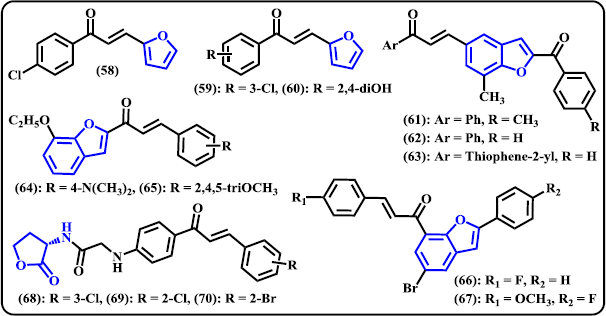 | Figure 6. Chalcone derivatives containing 5-membered O-heterocycles. [Click here to view] |
Homoserine Lactone. Recently, chalcone derivatives bearing homoserine lactone scaffold were synthesized and investigated for their antiproliferative activity against four human cancer cell lines MCF-7, MGC-803 DU145, and PC-3 (Yu et al. 2019). Following in vitro evaluation, several derivatives displayed selective and potent inhibitory activity against two prostate cancer cell lines DU145 and PC-3. Compounds (68)–(70) were found to have the best activity with IC50 values below 5.0 μM, much more active than that of natural OdDHL as reference. Compound (69) showed its ability to inhibit cell migration and colony formation of DU145 cells in dose-dependent manner.
6-membered ring
Coumarin. A novel series of chalcone-coumarin hybrids were synthesized by Sashidhara et al. (2010) and subsequently in vitro evaluated as potential anticancer agents (Fig. 7). Some of these derivatives showed promising activities with broad spectrum against four human cancer cell lines KB (oral squamous carcinoma), C33A (cervical carcinoma), MCF-7 (breast adenocarcinoma), A549 (lung carcinoma), and mouse embryo fibroblast (NIH3T3). Compound (71)–(73) exhibited remarkable performance with IC50 values in the range of 3.59–8.12 μM. Compound (73) was denoted as the most potent derivative in the series that shows 30-fold more selective against C33A than normal fibroblast NIH3T3 cell line with IC50 of 3.59 μM. In order to develop a new class of DNA oxidation inhibitors and radical scavengers, Xi and Liu (2014) have synthesized chalcones containing coumarin framework. Compound (74)–(77) were found to have significant inhibitory activities. Following in vitro investigation, it was found that the antioxidant capacity of compound having hydroxyl group attaching to coumarin skeleton can be increased by the presence of hydroxyl group attaching to the aromatic part of chalcone structure. Coumarin-clubbed chalcone derivatives promoted stronger antioxidant effects with only one or two phenolic OH group in the structure. Moreover, double OH group at adjacent location demonstrated high effectiveness to inhibit Cu(II)/glutathione-induced DNA oxidation and to scavenge ABTS as well as DPPH radicals. Vazquez-Rodriguez et al. (2015) designed and synthesized chalcone-coumarin hybrids as selective antibacterial agent against fish pathogens tenacibaculosis. The hybrid (78)–(80) presented appreciable activities toward fourteen strains of Tenacibaculosis maritimus. Compound (80) was denoted as the most potent derivative exhibiting MIC values 20-fold more than reference drug enrofloxacin against two strains of Tenacibaculosis maritimus LL01 8.3.1 and LL01 8.3.8. It is noteworthy to keep in mind that compounds that bear an amino group at ortho or para position in benzoyl group demonstrated high antibacterial activities. A new series of chalcone-coumarin fibrates were designed, synthesized, and evaluated as PPARα/γ agonists with potent antioxidant ability (Niu et al. 2017). Compounds (81)–(83) exhibited potent dual PPARα and γ agonists. In PPARα agonist activity, they showed to be more active than that of fenofibrate. Investigation of antioxidant capacity revealed that compounds (82) and (84)–(87) had stronger effects than Trolox with IC50 in the range of 9.40–18.63 μM.
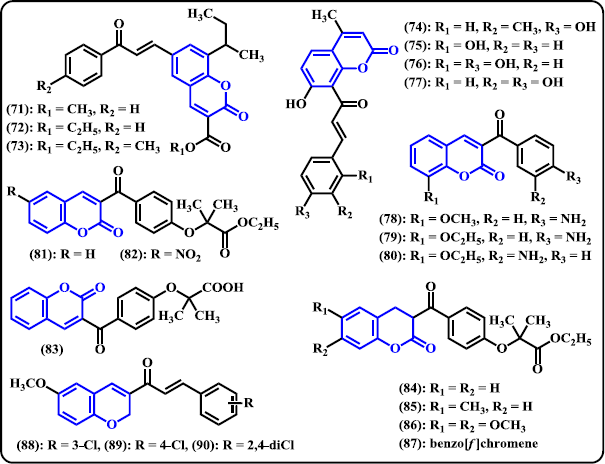 | Figure 7. Chalcone derivatives containing 6-membered O-heterocycles. [Click here to view] |
Chromene. Foroumadi et al. (2010) have synthesized two series of chalcone derivatives tethering chromene moiety, namely, 1-(6-methoxy-2H-chromen-3-yl)-3-phenylpropen-1-ones and 3-(6-methoxy-2H-chromen-3-yl)-1-phenylpropen-1-ones. Following in vitro biological evaluation as antileishmanial agents, chloro-substituted of the 1-(6-methoxy-2H-chromen-3-yl)-3-phenylpropen-1-ones showed superior activity toward Leishmania major at non-cytotoxic concentrations. Compounds (88)–(90) were found to be the most powerful antileishmanial agents with IC50 values less than 1 μM.
Chalcones containing S-heterocycles
5-membered ring
Thiophene. Mathew et al. (2016) reported the synthesis of thiophene-based chalcone derivatives from 2-acetyl thiophene and substituted aromatic aldehydes via Claisen-Schmidt aldol condensation reaction (Fig. 8). Following ADME studies, in silico toxicity prediction and exploration of molecular recognition, compound (91) demonstrated a docking score of –8.46 kcal/mol with calculated inhibition constant toward the active site of MAO-B of about 0.64 μM. Synthesis and anticancer studies of the novel aryl/heteroaryl chalcones derived from 3-aryl thiophene-2-carbaldehydes were developed by Venkataramireddy et al. (2016). Amongst the compounds evaluated, (92) was denoted to be the best anticancer candidate with IC50 of 21 μg/ml against HCT-15 human colon cell line, slightly better than that of doxorubicin as control drug. Meanwhile, compound (93) showed remarkable activity with IC50 of 22.8 μg/ml. Anticancer effects of chalcone derivatives of 2-acetyl thiophene were studied by Fogaça et al. (2017). After in vitro assessment against human breast cancer cells MCF-7 and MDA-MB-231 for 48-hour treatment, all the chalcone derivatives significantly reduced cells viability in a dose-dependent manner. In particular, compound (94) displayed the best cytotoxic performance with IC50 values of 11.76 ± 4.87 and 5.52 ± 4.26 μM against MCF-7 and MDA-MB-231 cell lines, respectively. In a work by Lokesh et al. (2017), new series of 2,5-dichloro-3-acetylthiophene chalcone hybrids were synthesized and in vitro investigated for their antifungal, antitubercular, and cytotoxic activity against DU145 prostate cancer cell line. Among the synthesized derivatives, compound (95) displayed comparable antifungal activity to the reference Fluconazole against Aspergillus niger and Candida tropicalis with MIC value of 4 μg/ml. This compound also demonstrated the most potent cytotoxic property toward DU145 with IC50 value of 5 ± 1 μg/ml, same ability compared to the control Methotrexate (MTX). Compound (96) showed the best performance against M. tuberculosis H37Rv with MIC of 3.12 μg/ml, same activity when compared to the Pyrazinamide as general medication for tuberculosis. Still in the same year, thiophene-based heterocyclic chalcones were synthesized, characterized, and biologically evaluated as antifungal agents (Ming et al. 2017). Out of the tested compounds, (97) was found to exhibit appreciable inhibition against C. albicans and A. niger with MIC values of 128 and 64 μg/mL, respectively. Chandra Sekhar et al. (2018) reported the synthesis and biological assessment of the chalcone derivatives linked to a thiophene scaffold, 5-aryl-thieno[3,2-b] thiophene-chalcones. Compounds (98)–(101) showed promising antibacterial activity against S. aureus (ZOI = 24, 23, 22, and 22 mm, respectively), more active than that of Ampicillin (ZOI = 20 mm) as reference antibacterial drug. Meanwhile, compounds (102)–(104) displayed excellent activity toward both A549 and SKNSH cancer cell lines with IC50 values of 52.40, 51.00, and 47.17 μM, respectively. Ritter et al. (2015) reported the synthesis and antimicrobial evaluation of thiophene-chalcone hybrids. Amongst the prepared derivatives, compound (105) demonstrated activity against broad spectrum of bacteria with the IC50 values of 219.1 μg/ml (S. aureus), 441.9 μg/ml (P. aeruginosa), 338.5 μg/ml (Enterococcus faecalis). However, none of these values are lower than that of reference drugs.
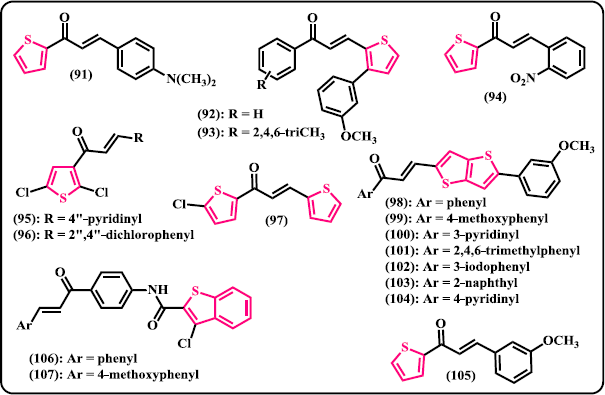 | Figure 8. Chalcone derivatives containing 5-membered S-heterocycles. [Click here to view] |
Benzothiophene. Benzfused thiophene, also called benzothiophene, linked to the chalcone structures via amide bond were synthesized and in vitro investigated for their antimicrobial potential (Naganagowda et al. 2012). In this series, compounds (106) and (107) showed moderate activity against bacterial strains and significant activity against fungal strains.
CONCLUSION AND PROSPECTIVE
Chalcone, a diaromatic connected by ketovinyl chain, can be traced in a number of biologically active agents. The biological potential of chalcone derivatives may be increased by the presence of nitrogen, oxygen, or sulfur-containing heterocyclic scaffolds. They demonstrate promising activities compared to the control drugs to inhibit or destroy several cancer cell lines, fungus, bacteria, and other specific target of diseases.
FINANCIAL SUPPORT
None.
CONFLICT OF INTEREST
The author declares that he has no conflict of interest.
REFERENCES
Ahmadi S, Mardinia F, Azimi N, Qomi M, Balali E. Prediction of chalcone derivative cytotoxicity activity against MCF-7 human breast cancer cell by Monte Carlo method. J Mol Struct, 2019; 1181:305–11. CrossRef
Avila-Villarreal G, Hernández-Abreu O, Hidalgo-Figueroa S, Navarrete-Vázquez G, Escalante-Erosa F, Pena-Rodríguez LM, Villalobos-Molina R, Estrada-Soto S.Antihypertensive and vasorelaxant effects of dihydrospinochalcone-A isolated from Lonchocarpus xuul Lundell by NO production: computational and ex vivo approaches. Phytomedicine, 2013; 20(14):1241–6. CrossRef
Ayman M, El-Messery SM, Habib EE, Al-Rashood ST, Almehizia AA, Alkahtani HM, Hassan GS. Targeting microbial resistance: synthesis, antibacterial evaluation, DNA binding and modeling study of new chalcone-based dithiocarbamate derivatives. Bioorg Chem, 2019; 85: 282–92. CrossRef
Baldisserotto A, Demurtas M, Lampronti I, Moi D, Balboni G, Vertuani S, Manfredini S, Onnis V. Benzofuran hydrazones as potential scaffold in the development of multifunctional drugs: synthesis and evaluation of antioxidant, photoprotective and antiproliferative activity. Eur J Med Chem, 2018; 156:118–25. CrossRef
Bandgar BP, Jalde SS, Adsul LK, Shringare SN, Lonikar SV, Gacche RN, Dhole NA, Nile SH, Shirfule AL. Synthesis of new olefin chalcone derivatives as antitumor, antioxidant and antimicrobial agents. Med Chem Res, 2012; 21(12):4512–22. CrossRef
Bonakdar APS, Vafaei F, Farokhpour M, Esfahani MHN, Massah AR. Synthesis and anticancer activity assay of novel chalcone-sulfonamide derivatives. Iran J Pharm Res, 2017; 16(2):565–8.
Celik F, Unver Y, Barut B, Ozel A, Sancak K. Synthesis, characterization and biological activities of new symmetric bis-1,2,3-triazoles with click chemistry. Med Chem, 2018; 14(3):230–41. CrossRef
Chandra Sekhar D, Tejeswara Rao A, Venkata Rao DV, Lav Kumar U, Anjali J. Synthesis and anticancer/antibacterial activity of compounds containing thiophene ring linked to a chalcone derivatives. Chem Biol Interface, 2018; 8(2):94–105.
Chen Z, Li P, Hu D, Dong L, Pan J, Luo L, Zhang W, Xue W, Jin L, Song B. Synthesis, antiviral activity, and 3D-QSAR study of novel chalcone derivatives containing malonate and pyridine moieties. Arab J Chem, 2015; Article in press DOI of the published paper: http://dx.doi.org/10.1016/j.arabjc.2015.05.003 CrossRef
Coskun D, Erkisa M, Ulukaya E, Coskun MF, Ari F. Novel 1-(7-ethoxy-1-benzofuran-2-yl) substituted chalcone derivatives: synthesis, characterization and anticancer activity. 2017; 136: 212–222. CrossRef
Desai V, Desai S, Gaonkar SN, Palyekar U, Joshi SD, Dixit SK. Novel quinoxalinyl chalcone hybrid scaffolds as enoyl ACP reductase inhibitors: synthesis, molecular docking and biological evaluation. Bioorg Med Chem Lett, 2017; 27:2174–80. CrossRef
Díaz-Carillo JT, Díaz-Camacho SP, Delgado-Vargas F, Rivero IA, López-Angulo G, Sarmiento-Sánchez JI, Montes-Avila J. Synthesis of leading chalcones with high antiparasitic, against Hymenolepis nana, and antioxidant activities. Braz J Pharm Sci, 2018; 54(3):1–13. CrossRef
Dumontet C, Beck G, Gardebien F, Haudecoeur R, Mathé D, Matera E-L, Tourette A, Mattei E, Esmenjaud J, Boyère C, Nurisso A, Peuchmaur M, Pérès B, Bouchaud G, Magnan A, Monneret G, Boumendjel A. Piperidinyl-embedded chalcones possessing anti PI3Kδ inhibitory properties exhibit anti-atopic properties in preclinical models. Eur J Med Chem, 2018; 158:405–13. CrossRef
El-Gamal MI, Abdel-Maksoud MS, Gamal El-Din MM, Shin JS, Lee KT, Yoo KH, Oh CH. Synthesis, in vitro antiproliferative and antiinflammatory activities, and kinase inhibitory effects of new 1,3,4-triarylpyrazole derivatives. Anticancer Agents Med Chem, 2017; 17(1):75–84. CrossRef
Espinoza-Hicks JC, Chacon-Vargas KF, Hernandez-Rivera JL, Nogueda-Torres B, Tamariz J, Shancez-Torres LE, Camacho-Dávila A. Novel prenyloxy chalcones as potential leishmanicidal and trypanocidal agents: design, synthesis and evaluation. Eur J Med Chem, 2019; 167:402–13. CrossRef
Fogaça TB, Martins RM, Begnini KR, Carapina C, Ritter M, de Pereira CMP, Seixas FK, Collares T. Apoptotic effect of chalcone derivatives of 2-acetylthiophene in human breast cancer cells. Pharmacol Rep, 2017; 69:156–61. CrossRef
Foroumadi A, Emami S, Sorkhi M, Nakhjiri M, Nazarian Z, Heydari S, Ardestani SK, Poorrajab F, Shafiee A. Chromene-based synthetic chalcones as potent antileishmanial agents: synthesis and biological activity. Chem Biol Drug Des, 2010; 75:590–6. CrossRef
Gan X, Wang Y, Hu D, Song B. Design, synthesis, and antiviral activity of novel chalcone derivatives containing a purine moiety. Chin J Chem, 2017; 35:665–72. CrossRef
Han X, Peng B, Xiao B-B, Cao S-L, Yang C-R, Wang W-Z, Wang FC, Li HY, Yuan XL, Shi R, Liao J, Wang H, Li J, Xu X. Synthesis and evaluation of chalcone analogues containing a 4-oxoquinazolin-2-yl group as potential anti-tumor agents. Eur J Med Chem, 2019; 162:586–601. CrossRef
Hayat F, Moseley E, Salahuddin A, Van Zyl RL, Azam A. Antiprotozoal activity of chloroquinoline based chalcones. Eur J Med Chem, 2011; 46(5):1897–905. CrossRef
Herbst, RS. Erlotinib (Tarceva): an update on the clinical trial program. Semin Oncol, 2003; 30:34–46. CrossRef
Insuasty B, Ramírez J, Becerra D, Echeverry C, Quiroga J, Abonia R, Robledo SM, Vélez ID, Upegui Y, Muñoz JA, Ospina V, Nogueras M, Cobo J. An efficient synthesis of new caffeine-based chalcones, pyrazolines and pyrazolo[3,4-b][1,4]diazepines as potential antimalarial, antitrypanosomal and antileishmanial agents. Eur J Med Chem, 2015; 93:401–13. CrossRef
Janaki P, Sekar KG, Thirunarayanan G. Synthesis, spectral correlation and insect antifeedant activities of some 2-benzimidazole chalcones. J Saudi Chem Soc, 2016; 20:58–68. CrossRef
Kalaria PN, Karad SC, Raval DK. A review on diverse heterocyclic compounds as the privileged scaffolds in antimalarial drug discovery. Eur J Med Chem, 2018; 158:917–36. CrossRef
Kim H-G, Oh H-J, Ko J-H, Song HS, Lee Y-G, Kang SC, Lee DY, Baek NI. Lanceoleins A-G, hydroxychalcones, from the flowers of Coreopsis lanceolata and their chemopreventive effects against human colon cancer cells. Bioorg Chem, 2019; 85:274–81. CrossRef
Kucerova-Chlupacova M, Kunes J, Buchta V, Vejsova M, Opletalova V. Novel pyrazine analogs of chalcones: synthesis and evaluation of their antifungal and antimycobacterial activity. Molecules, 2015; 20:1104–17. CrossRef
Kumari S, Paliwal SK, Chauhan R. An improved protocol for the synthesis of chalcones containing pyrazole with potential antimicrobial and antioxidant activity. Curr Bioactive Comp, 2018; 14:39–47. CrossRef
Kuthyala S, Nagaraja GK, Sheik S, Hanumanthappa M, Kumar SM. Synthesis of imidazo [1,2-a]pyridine-chalcones as potent inhibitors against A549 cell line and their crystal studies. J Mol Struct, 2019; 1177:381–90. CrossRef
Lei C, Zhang L-B, Yang J, Gao L-X, Li J-Y, Li J, Hou A-J. Macdentichalcone, a unique polycyclic dimeric chalcone from Macaranga denticulata. Tetrahedron Lett, 2016; 57(49):5475–8. CrossRef
Li P-H, Jiang H, Zhang W-J, Li Y-L, Zhao M-C, Zhou W, Zhang LY, Tang YD, Dong CZ, Huang ZS, Chen HX, Du ZY. Synthesis of carbazole derivatives containing chalcone analogs as non-intercalative topoisomerase II catalytic inhibitors and apoptosis inducers. Eur J Med Chem, 2018; 145:498–510. CrossRef
Lokesh BVS, Prasad YR, Shaik AB. Synthesis and biological activity of novel 2,5-dichloro-3-acetylthiophene chalcone derivatives. Indian J Pharm Ed Res, 2017; 51(4S):S679–90. CrossRef
Madhavi S, Sreenivasulu R, Yazala JP, Raju RR. Synthesis of chalcone incorporated quinazoline derivatives as anticancer agents. Saudi Pharm J, 2017; 25:275–9. CrossRef
Mathew B, Suresh J, Mathew GE, Haridas A, Suresh G, Sabreena P. Synthesis, ADME studies, toxicity estimation, and exploration of molecular recognition of thiophene based chalcones towards monoamine oxidase-A and B. Beni-Suef Univ J Basic Appl Sci, 2016; 5:396–401. CrossRef
Ming LS, Jamalis J, Al-Maqtari HM, Rosli MM, Sankaranarayanan M, Chander S, Fun H-K. Synthesis, characterization, antifungal activities and crystal structure of thiophene-based heterocyclic chalcones. Chem Data Collections, 2017; 9–10:104–13. CrossRef
Morales PAC, Dugarte CS, Amaro Luis JM. 2’,3,4-trihydroxychalcone, phloretin and calomelanone from Stevia lucida. The first chalcones reported in Stevia genus. Biochem Syst Ecol, 2018; 77:57–60. CrossRef
Mphahlele MJ, Maluleka MM, Parbhoo N, Malindisa ST. Synthesis, evaluation for cytotoxicity and molecular docking studies of benzo[c]furan-chalcones for potential to inhibit tubulin polymerization and/or EGFR-tyrosine kinase phosphorylation. Int J Mol Sci, 2018; 19:2552. CrossRef
Murphy M, Stordal B. Erlotinib or gefitinib for the treatment of relapsed platinum pretreated non-small cell lung cancer and ovarian cancer: a systematic review. Drug Resist Update, 2011; 14:177–90. CrossRef
Naganagowda G, Thamyongkit P, Petsom A. Synthesis and antimicrobial activities of benzothiophene derivatives. J Chil Chem Soc, 2012; 57(1):1043–7. CrossRef
Niu H, Wang W, Li J, Lei Y, Zhao Y, Yang W, Zhao C, Lin B, Song S, Wang S. A novel structural class of coumarin-chalcone fibrates as PPARα/γ agonists with potent antioxidant activities: design, synthesis, biological evaluation and molecular docking studies. Eur J Med Chem, 2017; 138:212–20. CrossRef
Osorio TM, Monache FD, Chiaradia LD, Mascarello A, Stumpf TR, Zanetti CR, Silveira, DB, Barardi CRM, Smânia EFA, Viancelli A, Garcia LAT, Yunes RA, Nunes RJ, Smânia A. Antibacterial activity of chalcones, hydrazones and oxadiazoles against methicillin-resistant Staphylococcus aureus. Bioorg Med Chem Lett, 2012; 22(1):225–30. CrossRef
Padhy GK, Panda J, Behera AK. Synthesis and characterization of novel benzimidazole chalcones as antibacterial agents. Der Pharma Chemica, 2016; 8(13):235–41.
Park S, Kim EH, Kim J, Kim SH, Kim I. Biological evaluation of indolizine-chalcone hybrids as new anticancer agents. Eur J Med Chem, 2018; 144:435–43. CrossRef
Pinto E, Neves H, Hrimpeng K, Silva A-F, Begouin A, Lopes G, Queiroz MJ. Antimicrobial activity and mechanism of action of new N-heteroaryl-1H-(benz)imidazoles. Mini-Rev Med Chem, 2014; 14(11):941–52. CrossRef
Pradip S, Khushboo M, Anand C, Devanshi G, Sudha S, Sweta K, Abhay C, Meena K. Virucidal activity of newly synthesized chalcone derivatives against H1N1 virus supported by molecular docking and membrane interaction studies. J Antivir Antiretrovir, 2016; 8(2):79–89. CrossRef
Ritter M, Martins RM, Rosa SA, Malavolta JL, Lund RG, Flores AFC, Pereira CMP. Green synthesis of chalcones and microbiological evaluation. J Braz Chem Soc, 2015; 26(6):1201–10. CrossRef
Romero-Parra J, Mella-Raipán J, Palmieri V, Allarà M, Torres MJ, Pessoa-Mahana H, Iturriaga-Vásquez P, Escobar R, Faúndez M, Di Marzo V, Pessoa-Mahana CD. Synthesis, binding assays, cytotoxic activity and docking tudies of benzimidazole and benzothiophene derivatives with selective affinitiy for the CB2 cannabinoid receptor. Eur J Med Chem, 2016; 124:17–35. CrossRef
Rücker H, Al-Rifai N, Rascle A, Gottfried E, Brodziak-Jarosz L, Gerhäuser C, Dick TP, Amslinger S. Enhancing anti-inflammatory activity of chalcones by tuning the Michael acceptor site. Org Biomol Chem, 2015; 13(10):3040–7. CrossRef
Salae A-W, Chairerk O, Sukkoet P, Chairat T, Prawat U, Tuntiwachwuttikul P, Chalermglin P, Ruchirawat S. Antiplasmodial dimeric chalcone derivatives from the roots of Uvaria siamensis. Phytochem, 2017; 135:135–43. CrossRef
Saleh SS, AL-Salihi SS, Mohammed IA. Biological activity study for some heterocyclic compounds and their impact on the gram positive and negative bacteria. Energy Procedia, 2019; 157:296–306. CrossRef
Sashidhara KV, Avula SR, Doharey PK, Singh LR, Balaramnavar VM, Gupta J, Misra-Bhattacharya S, Rathaur S, Saxena AK, Saxena JK. Designing, synthesis of selective and high-affinity chalcone-benzothiazole hybrids as Brugia malayi thymidylate kinase inhibitors: in vitro validation and docking studies. Eur J Med Chem, 2015; 103:418–28. CrossRef
Sashidhara KV, Kumar A, Kumar M, Sarkar J, Sinha S. Synthesis and in vitro evaluation of novel coumarin-chalcone hybrids as potential anticancer agents. Bioorg Med Chem Lett, 2010; 20:7205–11. CrossRef
Sashidhara KV, Modukuri RK, Jadiya P, Dodda RP, Kumar M, Sridhar B, Kumar V, Haque R, Siddiqi MI, Nazir A. Benzofuran-chalcone hybrids as potential multifunctional agents against Alzheimer’s disease: synthesis and in vivo studies with transgenic Caenorhabditis elegans. ChemMedChem, 2014; 9:2671–84. CrossRef
Sasidharan R, Baek SC, Manju SL, Kim H, Mathew B. Imidazole bearing chalcones as a new class of monoamine oxidase inhibitors. Biomed Pharmacother, 2018; 106:8–13. CrossRef
Shankaraiah N, Siraj KP, Nekkanti S, Srinivasulu V, Sharma P, Senwar KR, Sathish M, Vishnuvardhan MVPS, Ramakrishna S, Jadala C, Nagesh N, Kamal A. DNA-binding affinity and anticancer activity of β-carboline-chalcone conjugates as potential DNA intercalators: molecular modelling and synthesis. Bioorg Chem, 2015; 59:130–9. CrossRef
Shukla P, Satyanarayana M, Verma PC, Tiwari J, Dwivedi AP, Srivastava R, Rehuja N, Tamrakar AK, Dwivedi AK, Kushwaha HN, Gautam N, Singh S, Srivastava M, Nath C, Raghubir R, Srivastava AK, Pratap R. Chalcone-based aryloxypropanolamine as a potential antidiabetic and antidyslipidaemic agent. Curr Sci, 2017; 112(8):1675–89. CrossRef
Singh S, Ahmad A, Raghuvanshi DS, Hasanain M, Agarwal K, Dubey V, Fatima K, Alam S, Sarkar J, Luqman S, Khan F, Tandon S, Gupta A. Synthesis of 3,5-dihydroxy-7,8-dimethoxy-2-(4-methoxyphenyl) benzopyran-4-one derivatives as anticancer agents. Bioorg Med Chem Lett, 2016; 26(21):5322–7. CrossRef
Sun L-P, Jiang Z, Gao L-X, Sheng L, Quan Y-C, Li J, Piao H-R. Synthesis and biological evaluation of furan-chalcone derivatives as protein tyrosine phosphatase inhibitors. Bull Korean Chem Soc, 2013; 34(4):1023–4. CrossRef
Sun M, Zhang YM, Zhang J, Wang S-C, He L-C. A high expression EGFR/cell membrane chromatography and online high performance liquid chromatography/ mass spectrometry method for screening EGFR antagonists from Rhizoma Polygoni Cuspidati. Acta Pharm Sin B, 2011; 1:115–20. CrossRef
Tajuddeen N, Isah MB, Suleiman MA, van Heerden FR, Ibrahim MA. The chemotherapeutic potential of chalcones against leishmaniases: a review. Int J Antimicrob Agents, 2018; 51(3):311–18. CrossRef
Tomar V, Bhattacharjee G, Kamaluddin, Rajakumar S, Srivastava K, Puri SK. Synthesis of new chalcone derivatives containing acridinyl moiety with potential antimalarial activity. Eur J Med Chem, 2010; 45:745–51. CrossRef
Vazquez-Rodriguez S, López RL, Matos MJ, Armesto-Quintas G, Serra S, Uriarte E, Santana L, Borges F, Crego AM, Santos Y. Design, synthesis and antibacterial study of new potent and selective coumarin-chalcone derivatives for the treatment of tenacibaculosis. Bioorg Med Chem, 2015; 23:7045–52. CrossRef
Venkataramireddy V, Shankaraiah M, Rao AT, Kalyani Ch, Narasu ML, Varala R, Jayashree A. Synthesis and anti-cancer activity of novel 3-aryl thiophene-2-carbaldehydes and their aryl/heteroaryl chalcone derivatives. Rasayan J Chem, 2016; 9(1):31–9.
Venkataramana Reddy PO, Hridhay M, Nikhil K, Khan S, Jha PN, Shah K, Kumar D. Synthesis and investigation into the anticancer and antibacterial activity studies of β-carboline chalcones and their bromide salts. Bioorg Med Chem Lett, 2018; 28:1278–82. CrossRef
Wang Y-J, Zhou D-G, He F-C, Chen J-X, Chen Y-Z, Gan X-H, Hu D-Y, Song B-A. Synthesis and antiviral bioactivity of novel chalcone derivatives containing purine moiety. Chin Chem Lett, 2018; 29:127–30. CrossRef
Wen R, Lv H-N, Jiang Y, Tu P-F. Anti-inflammatory flavone and chalcone derivatives from the roots of Pongamia pinnata (L.) Pierre. Phytochem, 2018; 149:56–63. CrossRef
Williams IS, Joshi P, Gatchie L, Sharma M, Satti NK, Vishwakarma RA, Chaudhuri B, Bharate SB. Synthesis and biological evaluation of pyrrole-based chalcones as CYP1 enzyme inhibitors, for possible prevention of cancer and overcoming cisplatine resistance. Bioorg Med Chem Lett, 2017; 27:3683–7. CrossRef
Xi G-L, Liu Z-Q. Coumarin moiety can enhance abilities of chalcones to inhibit DNA oxidation and to scavenge radicals. Tetrahedron, 2014; 70:8397–404. CrossRef
Xu Z, Zhao S, Lv Z, Feng L, Wang Y, Zhang F, Bai L, Deng J. Benzofuran derivatives and their anti-tubercular and anti-bacterial activities. Eur J Med Chem, 2019; 162:266–76. CrossRef
Yadav P, Lal K, Kumar A, Guru SK, Jaglan S, Bhushan S. Green synthesis and anticancer potential of chalcone linked-1,2,3-triazoles. Eur J Med Chem, 2017; 126:944–53. CrossRef
Yu B, Liu H, Kong X, Chen X, Wu C. Synthesis of new chalcone-based homoserine lactones and their antiproliferative activity evaluation. Eur J Med Chem, 2019; 163:500–11. CrossRef
Zheng C-J, Jiang S-M, Chen Z-H, Ye B-J, Piao H-R. Synthesis and anti-bacterial activity of some heterocyclic chalcone derivatives bearing thiofuran, furan and quinoline moieties. Arch Pharm Chem Life Sci, 2011; 344:689–95. CrossRef
Zhou D, Xie D, He F, Song B, Hu D. Antiviral properties an interaction of novel chalcone derivatives containing a purine and benzenesulfonamide moiety. Bioorg Med Chem Lett, 2018; 28: 2091–7. CrossRef
Zhuang C, Zhang W, Sheng C, Zhang W, Xing C, Miao Z. Chalcone: a privileged structure in medicinal chemistry. Chem Rev, 2017; 117(12):7762–810. CrossRef