
INTRODUCTION
Prevalence of type 2 diabetes mellitus (T2DM) has been globally increasing together with obesity. Most T2DM patients in Western countries are obese (Eckel et al., 2011). By contrast, Asian populations show an increase in non-obese (lean) T2DM (Kashima et al., 2015) with a high risk of diabetic complications similar to obese T2DM. One morbidity from diabetic complications is skeletal disorders, such as osteopenia or osteoporosis (Schwartz, 2003). Osteoporosis is widely recognized as the most important global age-related metabolic bone disorder, not only in the elderly but also in patients with diabetes mellitus (Hegazy, 2015). T2DM patients were 1.7 times more likely to suffer from hip fractures compared with age-matched non-diabetic controls (Nicodemus and Folsom, 2001; Yaturu et al., 2009). Therefore, more attention should be given to the elevated fracture risk presented by T2DM (Petit et al., 2010).
Hyperglycemia results in an imbalance of bone remodeling, in particular by enhancing osteoclast activity while reducing osteoblast differentiation. High glucose concentrations markedly suppress bone cell growth, mineralization, and expression of various osteoblast-related markers (Kayal et al., 2007). Hyperglycemia also activates osteoclast proliferation and expression of osteoclast genetic mediators (Kayal et al., 2007) which enhance bone resorption while inhibiting bone formation.
Metformin is a first-line oral anti-hyperglycemic drug for the treatment of T2DM by suppressing gluconeogenesis from the liver (Cheng and Fantus, 2005). Previous studies showed that the metformin-induced in vitro osteoblast differentiation and enhance bone matrix production (Cortizo et al., 2006; Kanazawa et al., 2008). In addition, in vivo treatment of metformin in ovariectomized rats prevented bone loss (Mai et al., 2011). Several clinical studies identified the beneficial effect of metformin on preserving bone health (Borges et al., 2011; Kahn et al., 2006; Vestergaard et al., 2005). Metformin controls blood glucose and also has a positive effect on bone (Yan and Li, 2013). Metformin is also involved in microarchitectures, such as improving bone quality (Gao et al., 2010) and osteoblast differentiation (Jang et al., 2011).
The p38 mitogen-activated protein kinase (p38 MAPK) signaling pathway is recognized as essential to positively regulate skeleton development and bone homeostasis (Thouverey and Caverzasio, 2015). Alteration of p38 MAPK signaling pathway affects bone health and pathogenesis. Disruption of the pathway in mice lacking p38 MAPK downstream molecule (MAPKAPK2) increased bone mass and thickness, with lower osteoclast population and lower bone resorption (Braun et al., 2013). In addition, treatment with p38 MAPK inhibitors showed therapeutic potential for osteoporosis (Redman et al., 2001). Hyperglycemia causes metabolic disturbance and also induces cellular injury and death. Hyperglycemic conditions increase basal p38 MAPK activation as one of the signaling pathways that play an important role in cellular injury and apoptosis (Kumphune et al., 2013; Nakagami et al., 2001). Therefore, inhibition of p38 MAPK activation reduces cellular injury and death with therapeutic potential in diabetic-induced osteoporosis.
Both metformin and p38 MAPK inhibition show beneficial effects on bone health; however, the effects of metformin and p38 MAPK inhibitor in non-obese T2DM, as well as combinatorial treatment of both drugs has never been investigated. Here, metformin and p38 MAPK inhibitor treatment, both individually and in combination were investigated on bone cell growth and differentiation as well as bone porosity in non-obese T2DM rats.
MATERIALS AND METHODS
Chemicals and reagents
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum, and trypsin-ethylenediaminetetraacetic acid (EDTA) were sourced from Gibco BRL; Life Technologies Inc. (NY, USA). Other purchases included 2ʹ,7ʹ -dichlorofluorescein diacetate from Merck (Billerica, MA) 3-(4,5-dimethyl-2-thiazol)-2,5-diphenyl-2H-tetrazolium bromide (MTT) from Amreco (Solon, OH), metformin from Merck Serono (Germany) and p38 MAPK inhibitor (SB203580) from Tocris Bio-Techne (USA). Other chemicals were purchased from Sigma Chemical Company (USA).
Experimental animals
Male type 2 diabetic Goto-Kakizaki (GK) rats (n = 40) and age-matched Wistar rats (n = 10) weighing approximately 200–250 g were purchased from Nomura Siam International. All the animals were maintained under environmentally-controlled conditions (22°C ± 1°C, 12 hours light: dark cycle) at the Centre for Animal Research, Naresuan University, Phitsanulok, Thailand. All the protocols were approved by the Committee of the Centre for Animal Research, Naresuan University (NU-AE581023).
Study groups and treatment
All the rats were kept for 4 weeks after purchase and glycemic parameters were performed to confirm their diabetic status, including fasting blood glucose (FBG), haemoglobin A1c level, and oral glucose tolerance test. The rats were divided into two major groups as the control non-diabetic group (Wistar rats; n = 10) and the diabetic group (GK rats; n = 40). Diabetic GK rats were further divided into four subgroups as (i) diabetic group (GK rats; n = 10) receiving deionized water as a control treatment group, (ii) diabetic group treated with metformin (n = 10) receiving metformin 15 mg/kg body weight (BW) twice daily (b.i.d) by oral gavage, (iii) diabetic group treated with SB203580 (n = 10) receiving 2 mg/kg BW of SB203580 by intraperitoneal (IP) injection every 3 days, and (iv) diabetic group treated with both metformin and SB203580, the so-called “combination group” (n = 10) receiving metformin 15 mg/kg BW b.i.d by oral gavage and 2 mg/kg BW of SB203580 IP.
Determination of fasting blood glucose
All the rats fasted for 12–14 hours and then tail vein blood samples were collected to measure blood glucose levels using a glucometer (SD GlucoNavii® GDH, SB Biosensor, Korea). Rat tails were cleaned with 70% (v/v) ethanol and blood was collected with a 1-ml needle, dropped on a glucose strip and measured by a glucometer. Amount of electricity used was proportional to glucose content in the sample. Quality control for blood glucose determination was performed using quality control material provided by SD Biosensor Company.
Determination of oral glucose tolerance test
All the rats have fasted for 12–14 hours and blood was collected from the tail vein as a baseline. The rats were then fed 2 g/kg BW of 40% (w/v) glucose solution by the oral gavage. Blood was collected from the tail vein at 30, 60, 90, and 120 minutes after glucose treatment and analyzed for blood glucose using a glucometer.
Determination of hemoglobin A1c
Blood samples from the tail vein were collected and dropped on a test cartridge for measurement of hemoglobin A1c (HbA1c) by a Clover A1c™ Self Analyser. Percentage of HbA1c % in the blood sample was presented on the LCD screen of the Clover A1c™ Self Analyser. Quality control for HbA1c was performed using material provided by EuriMedix Company.
Determination of plasma insulin levels
Plasma insulin levels were determined by Sandwich ELISA (Millipore, MI). Plasma samples were added to a microtiter plate coated with monoclonal mouse anti-rat insulin antibodies. Unbound material was then washed out and immobilized biotinylated antibody was added before 3, 3ʹ, 5, 5ʹ-tetramethylbenzidine substrates. Enzyme activity was measured using a spectrophotometer (BioTek, Winooski, VT) with absorbance at 450 nm and corrected wavelength at 590 nm. Plasma insulin concentrations in unknown samples were derived by interpolation from a reference curve generated in the same assay with the reference standard for a known concentration of rat insulin.
Animal euthanasia and bone sample preparation
Rats were anesthetized with IP injection of pentobarbital (100 mg/kg) and heparin (150 units). Rats were euthanized by heart isolation. After that, the femur, tibia, and iliac bones were collected to determine microarchitecture by micro-CT and calvaria were collected for osteoblast isolation. Surrounding tissues and muscle were removed. The bone samples were then cleaned by washing with phosphate buffer saline (PBS), wrapped in gauze soaked in PBS solution, and stored at −20°C until required for analysis.
Determination of bone porosity by synchrotron computed tomography
Bone porosity was analyzed by synchrotron computed tomography (synchrotron CT) at the Synchrotron Light Research Institute, Thailand. Bone samples were measured from 0° to 180° in 0.2° steps under 5× magnification. The bones were cut into small pieces (less than 6 mm) to measure trabecular and cortical bone porosity and images were normalized using bright and dark fields (n = 100). Raw images were reconstructed via Octopus software and bone porosity analysis was performed in individual regions of interest. 3D images were constructed using Drishti software. Bone porosity was calculated from 3D images using octopus analysis software for selective areas of 200 slices, height 200 pixels, and length 200 pixels (voxel size = 1.4 μm).
Isolation of primary osteoblast cells from non-obese T2DM rats
After animal scarification, calvaria were rapidly removed to prepare the osteoblastic culture. Calvarial bones were cut into small pieces and transferred to a sterile petri dish. The bone pieces were digested with 2 mg/ml of collagenase II (Worthington Biochemical Corporation), dissolved in Hanks’ balanced salt solution (HBSS) buffer, for 30 minutes at 37C in a shaking water bath. Then, collagenase II solution was replaced by trypsin-EDTA (Gibco®) solution and further incubated for 30 minutes. Repeated steps of collagenase II and trypsin-EDTA were performed twice. Then, bone pieces were transferred to 25-cm2 flasks containing 5-ml fresh DMEM (Gibco®) supplemented with 10% (v/v) foetal bovine serum (FBS) (Sigma®), 5,000 units/ml of penicillin and 5,000 g/ml of streptomycin (Gibco®). Culture media were changed every 2 days. All the experiments on osteoblasts were expanded for three cell passages.
Determination of primary osteoblast proliferation
Osteoblast cells were plated in 24-well plates at a density of 2 × 104 cells/well. Cell numbers were counted using Automated Cell Counters (Thermo Fisher Scientific, Waltham, MA) every day for a period of 7 days. A growth curve was plotted and the population doubling time was calculated from the cell count on day 7.
Population doubling time PDT = duration × log(2)log final concentration -loga(initial concentration)
Determination of alkaline phosphatase activity
Osteoblast cells were plated in six-well plates at a density of 5 × 104 cells/well with DMEM supplemented with 50 μg/ml ascorbic acid (Sigma®) and 10 mM β-glycerophosphate (Merck®). Cells were cultured for 7 and 14 days. Culture medium was changed every 3 days until 14 days. For alkaline phosphatase (ALP) staining, cells were treated with Nitro blue tetrazolium chloride/ 5-Bromo-4-chloro-3-indolyl phosphate (NBT/BCIP) solution (Roche®) in dark environment before fixing with 10% (v/v) formaldehyde for 1 minute. Positive ALP staining produced a purple color. Five images were taken at random locations in each well using a microscope. Quantitative ALP was eluted by dimethyl sulfoxide and absorbance was measured at a wavelength of 405 nm.
Determination of mineralization ability
Calcium deposit detection was performed by Alizarin red S staining. Cells were seeded at a density of 5 × 104 cells/well in six-well plates with DMEM supplemented with 50 g/ml ascorbic acid (Sigma®) and 10 mM β-glycerophosphate (Merck®) for 14 and 21 days. Osteoblast cells were washed and stained with 2% (w/v) Alizarin red S, with staining visualized under a microscope after 10 minutes. Five images were taken at random locations in each well.
Statistical analysis
All the values were expressed as mean ± SEM. Comparisons involving more than one group were assessed for significance using one-way analysis of variance followed when appropriate by Tukey’s post hoc test. A value of less than 0.05 was considered as statistically significant.
RESULTS
Characterization of a lean hyperglycemic rat model
To ensure that the GK rats gave a valid phenotype of lean type 2 diabetes, several parameters were explored. We first confirmed the non-obese or lean animal model by measuring body weight. Body weights of the diabetic group were significantly lower than Wistar rats (301.6 ± 5.9 g vs. 458 ± 19.0 g, *p < 0.001). Mean FBG of the diabetic group was significantly higher than Wistar rats (control group) (176.6 ± 14.01 mg/dl vs. 75.71 ± 2.5 mg/dl, *p < 0.05). Mean percentage HbA1c level of the diabetic group was also higher compared with the control group (6.43% ± 0.12% vs. 4.11% ± 0.05%, *p < 0.001, Table 1). Results indicated the diabetic-like status of GK rats in this study.
Effect of metformin and SB203580 on blood glucose and plasma insulin
GK rats were treated with metformin, SB203580, or a combination of metformin and SB203580 for 4 weeks. Results showed no significant difference in body weight before and after treatment in all sub-groups. FBG and HbA1c were also measured. Results indicated significantly decreased FBG in the diabetic group treated with metformin before and after treatment (126.1 ± 5.08 mg/dl vs. 180.7 ± 15.48 mg/dl, p < 0.05) and HbA1c (5.19% ± 0.18% vs. 6.93% ± 0.18%, p < 0.01, Table 1). Treatment with SB203580 significantly reduced FBG when compared to the level before treatment (134.5 ± 2.8 mg/dl vs. 170.6 ± 10.64 mg/dl, #p < 0.05) but there was no difference in HbA1c level (6.23% ± 0.19% vs. 6.53% ± 0.19%, p > 0.05). Combination of metformin and SB203580 reduced FBS (126 ± 5.62 mg/dl vs. 186.9 ± 9.03 mg/dl, #p < 0.05) and HbA1c (5.04% ± 0.13% vs. 6.76% ± 0.31%, #p < 0.01, Table 1) compared to the diabetic group but did not synergistically reduce FBS and HbA1c compared to the two single drug treatment sub-groups.
Treatment with metformin significantly lowered plasma insulin level compared to the diabetic group (3.210 ± 1.338 ng/ml vs. 9.233 ± 0.6731 ng/ml, #p < 0.05). By contrast, plasma insulin level in the diabetic group treated with SB203580 alone (11.73 ± 3.075 ng/ml, p < 0.05) was significantly higher than in the metformin-treated group. Combination of both drugs did not significantly affect plasma insulin level compared to single drug diabetic groups (4.77 ± 1.18 ng/ml vs. 9.233 ± 0.67 ng/ml, p > 0.05, Table 2).
Serum biochemical parameters in normal and hyperglycemic rats
Serum mineral levels and ALP activity were measured between control and hyperglycemic rats. Treatment with metformin significantly increased serum phosphorous (5.199 ± 0.5 mg/dl vs. 4.174 ± 0.4 mg/dl) and magnesium (3.654 ± 0.34 mg/dl vs. 3.209 ± 0.2 mg/dl) compared to the diabetic group (Table 2). Moreover, serum ALP in the SB203580 treated group was significantly higher than the diabetic group (10.67 ± 1.67 mU/I vs. 5.619 ± 1.05 mU/I, *p < 0.05, Table 2). However, the combination of metformin and SB203580 did not affect serum mineral levels after treatment.
Combination of metformin and SB203580 improved femur density
Femur bones were isolated from all the rat groups to determine cortical and trabecular bone porosity. Results showed that femur cortical bone porosity of the diabetic group was significantly higher than control rats (26.50% ± 2.42% vs. 5.363% ± 0.76%, p < 0.0001). Treatment with metformin, SB203580, or a combination of these two drugs significantly reduced femur cortical bone porosity compared with the diabetic group (6.394% ± 1.72%, 8.721% ± 1.59%, 7.967% ± 2.36% vs. 26.50% ± 2.42%, #p < 0.0001, Fig. 1A, respectively).
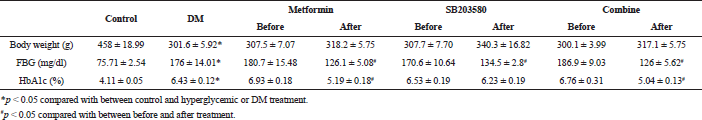 | Table 1. Body weight and blood glucose parameter of Wistar rats and GK rats. [Click here to view] |
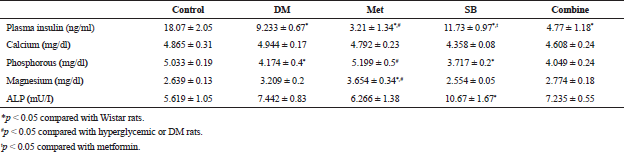 | Table 2. Mineral blood parameters of Wistar rats and GK rats. [Click here to view] |
Results also indicated that isolated femur trabecular bone porosity of diabetic rats was significantly higher than control rats (71.07% ± 2.61% vs. 38.16% ± 1.84%, *p < 0.0001), while a combination of metformin and SB203580 significantly reduced femur trabecular bone porosity compared with the diabetic group (60.14% ± 2.40% vs. 71.07% ± 2.61%, #p < 0.0001, Fig. 1B).
Metformin and SB203580 reduced bone porosity in tibia bones
The individual effects of metformin and SB203580 and a combination of these two drugs on tibial bone porosity was investigated. Results showed that percentages of tibial cortical bone porosity for the diabetic group were significantly higher than the control group (33.37% ± 2.87% vs. 6.054% ± 0.87%, *p < 0.001). Treatment with metformin, SB203580, or a combination of the two drugs significantly reduced tibial cortical bone porosity compared to diabetic group (8.847% ± 1.15%, 8.459% ± 0.93%, 6.296% ± 1.61% vs. 33.37% ± 2.87%, #p < 0.0001, Fig. 2A, respectively).
Results showed that isolated tibial trabecular bone porosity of diabetic rats was significantly higher than control rats (68.88% ± 2.43% vs. 44.47% ± 2.35%, *p < 0.001). However, all the treatment groups showed a reduction in tibial trabecular bone porosity but no significant differences when compared to the diabetic group (58.69% ± 2.89%, 63.92% ± 2.35%, 62.98% ± 2.84% vs. 68.88% ± 2.43%, Fig. 2B, respectively).
Only metformin reduced iliac bone porosity
Effects of drugs on bone porosity were also determined in flat bone. Here, the iliac bone was isolated as a representative of flat bone. Results showed that percentage porosity of iliac bones from the diabetic group was significantly higher than the control group (74.28% ± 2.51% vs. 50.67% ± 3.80%, *p < 0.001). Metformin treatment reduced iliac bone porosity (58.66% ± 3.04% vs. 74.28% ± 2.51%, #p < 0.0001) but combinatorial treatment did not provide a synergistic effect to reduce iliac bone porosity (66.43% ± 2.18%, Fig. 3).
Determination of biological properties of isolated primary osteoblasts from diabetic and drug-treated rats
To determine the effects of hyperglycemia from metformin, SB203580, or a combination of metformin and SB203580 at cellular bone level, osteoblasts from each group of animals were isolated and cultured. Then, the biological properties of the osteoblasts, including cell proliferation, population doubling time, expression of ALP, and mineralization ability were determined.
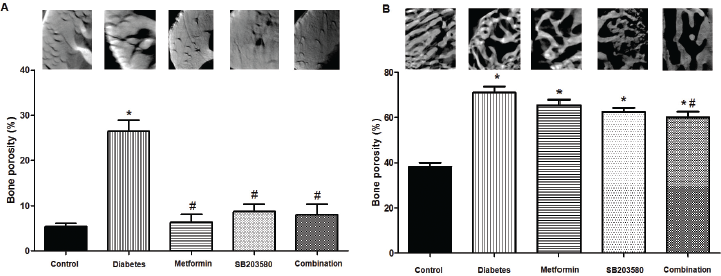 | Figure 1. Femur bones porosity from synchrotron radiation microtomography. (A) Cortical bone (B) Trabecular bone porosity (%) base on the 3D calculation of femur in each group and present with 2D cortical bone of Z-direction images was detected by using octopus visualize software (1.4 μm voxel sizes) (p < 0.0001, *compared with control groups, #compared with DM groups). [Click here to view] |
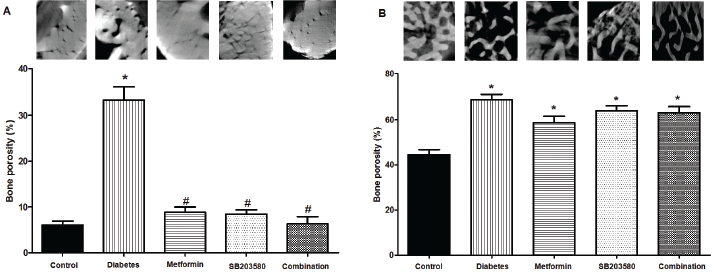 | Figure 2. Tibia bone porosity from synchrotron radiation microtomography. (A) Cortical bone (B) Trabecular bone porosity (%) base on the 3D calculation of femur in each group and present with 2D cortical bone of Z-direction images was detected by using octopus visualize software (1.4 μm voxel sizes) (p < 0.0001, *compared with control groups, #compared with DM groups). [Click here to view] |
Results of cell proliferation ability represented as a growth curve (Fig. 4A) showed that isolated osteoblasts from the diabetic group had a slower growth rate compared to the control group. Treatment with metformin significantly enhanced cell proliferation. Similarly, results from calculated population doubling time (PDT) showed that isolated osteoblasts from the diabetic group did not significantly prolong PDT (104 ± 2.22 hours vs. 109.4 ± 3.25 hours, p > 0.05). By contrast, treatment with metformin gave significantly lower results than the diabetic group (95.46 ± 4.21 hours vs. 109.4 ± 3.25 hours, *p < 0.05, Fig. 4B). There were no significant differences in growth rate and PDT between diabetic sub-groups treated with SB203580 or a combination of metformin and SB203580 (109.4 ± 3.25 hours vs. 96.53 ± 3.54 hours, 102.1 ± 4.0 hours, p > 0.05, Fig. 4B).
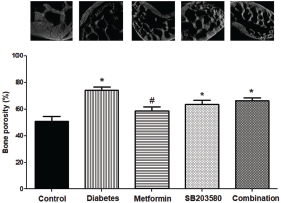 | Figure 3 Iliac bones porosity from synchrotron radiation micrograph. Iliac bones porosity (%) base on the 3D calculation of femur in each group and present with 2D cortical bone of Z-direction images were detected by using octopus visualize software (1.4 μm voxel sizes) (p < 0.0001, *compared with control groups, #compared with DM groups). [Click here to view] |
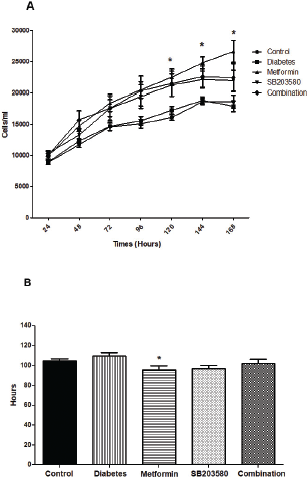 | Figure 4. Cell proliferations in isolations in isolated primary osteoblast from control rats, GK rats represent as a hyperglycemic rat, and GK with treatment rats (metformin, SB203580, and metformin combine with SB203580). (A) The growth curve was measured for 7 days in every group. (B) Population doubling times were calculated from 7 days (*p < 0.05 compared with DM group). [Click here to view] |
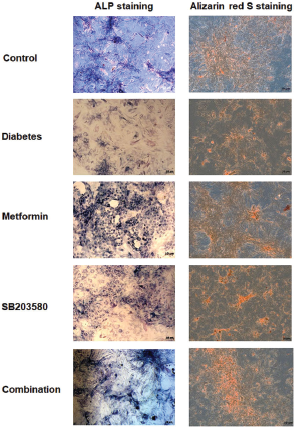 | Figure 5. Characterization of primary osteoblast isolated from calvaria of Control rats, hyperglycemic rats, Metformin, SB203580 and Combine treatment groups. Cells were cultured in osteogenic medium. (Left) ALP staining was determined on 14 days. (Right) Alizarin red S staining was positive in all isolated osteoblast groups on 14 days. [Click here to view] |
The expression of ALP was determined in primary osteoblast cells by ALP staining. Results showed that osteoblasts isolated from the diabetic group expressed fewer ALP stained cells. Treatment with metformin, SB203580, or a combination of metformin and SB203580 gave higher ALP staining compared to osteoblasts isolated from the diabetic group (Fig. 5). However, a combination of metformin and SB203580 showed no synergistic effect to increase ALP activity.
Mineralization ability for measuring calcium deposition was determined in primary osteoblast cells by Alizarin red staining. Results showed that osteoblasts isolated from the diabetic group had less Alizarin red positive cells. Treatment with metformin or a combination of metformin and SB203580 but not SB203580 on its own gave higher Alizarin red positive cells compared to the diabetic group (Fig. 5).
DISCUSSION
This study demonstrated the effect of anti-diabetic drugs metformin, p38 MAPK inhibitor SB203580 and the combinatorial effect of metformin and SB203580 on bone porosity in non-obese diabetic Goto-Kakizaki rats. Major findings showed that non-obese diabetes caused a significant loss of both trabecular and cortical long bone mass as well as flat bone. Treatment with anti-diabetic drugs metformin and p38 MAPK inhibitor SB203580 significantly preserved bone mass, predominantly in cortical bone. Treatment with metformin combined with p38 MAPK inhibitor only synergistically reduced porosity of the femur trabecular bone. Similarly, in bone tissue, osteoblasts isolated from diabetic bone were less proliferate and expressed less osteoblastic characteristics. However, metformin had a dominant effect on osteoblastic properties by significantly reducing population doubling time, expression of ALP and mineralization.
Most previous studies determined that hyperglycemia and diabetes affected bone quality by decreasing whole body bone mineral density (BMD) (Wong et al., 2018) and jaw bone density (Pramojanee et al., 2013). However, some studies revealed no change or slightly increased bone mass but enhanced risk of bone fracture (de et al., 2005; Moseley, 2012; Napoli et al., 2017). Similar to our findings, several previous studies in experimental animal models (Kerckhofs et al., 2016; Sampayo et al., 2017) as well as humans (Kayal et al., 2007; Vestergaard, 2007) demonstrated that hyperglycemia or diabetes caused detrimental effects on bone mass by decreasing bone mass, strength, and bone formation processes while increasing osteoclast activity and bone resorption.
Among the different types of diabetes, type 1 and type 2 showed diverse impacts on the structural and mechanical properties of bone (Sampayo et al., 2017). A greater hyperglycemic condition in T1DM could be a major factor involved in bone alterations, whereas body weight change is considered as one of the key factors in T2DM which impacts bone health. T2DM is the most common type of diabetes in older age-groups (Kalyani et al., 2017) who have an increased risk of bone defects. Globally, the vast majority of patients with T2DM are overweight or obese but more than half of diabetic patients in Asian countries are lean or non-obese (Kashima et al., 2015), they show a high risk for diabetic complications with less awareness.
Different types of diabetes present diverse outcomes on bone porosity. Previous studies demonstrated that T1DM patients had lower BMD which depends on age and gender (Mastrandrea et al., 2008), while body weight was a major influencing factor on BMD for T2DM patients (Kao et al., 2003). BMD in T2DM patients increased, possibly due to poor glycaemia control (Dede et al., 2014; Ma et al., 2012). Therefore, both glycemic level and body weight are predominant factors for bone porosity in T2DM. However, bone porosity in non-obese T2DM might be different from obese T2DM since body weight seems to have less influence. In this study, GK rats were used as representatives of a non-obese diabetic model to demonstrate the effect of hyperglycemic condition on bone remodeling without body weight as a physical stimulant. Our results confirmed the diabetic-like phenotype with mild hyperglycemia and showed that non-obese diabetic GK rats had higher bone porosity compared to control rats. This finding was supported by a previous study confirming that GK rats had low BMD in the femur and lumbar bones as well as low bone volume/total volume (BV/TV) in trabecular bones (Zhang et al., 2009). A recent study showed that GK rats had reduced trabecular and cortical BMD in both tibia and femur bones. BMD was determined by peripheral quantitative computerized tomography (pQCT) (Ahmad et al., 2003). Therefore, non-obese T2DM had higher risk for osteoporosis than obese T2DM. Preventive attention should also be focused on non-obese T2DM.
Our study demonstrated that the treatment with metformin significantly reduced trabecular and cortical bone porosity of tibia, femur, and iliac bones. Furthermore, primary osteoblast cells isolated from the calvarial bones of GK rats treated with metformin improved primary osteoblast proliferation. Moreover, we also showed that metformin promoted osteoblast differentiation by enhancing ALP expression and increasing calcium deposits. Our results suggested the beneficial effect of metformin on bone health for non-obese T2DM. Similar results for metformin enhancement of in vitro osteoblastic differentiation were shown in mouse osteoblast precursor cell line MC3T3-E1 cells treated with 100 μM of metformin (Jang et al., 2011). In an in vivo study, metformin protected bone loss in ovariectomized rats via reduced osteoclastogenesis (Mai et al., 2011). Moreover, metformin treatment delayed bone repair and healing in Zucker diabetic fatty rats (ZDF) (Akash et al., 2013). Beneficial effects of metformin on bone health were reported in a preclinical study and also in patients. Information from the clinical study showed that metformin decreased bone fracture in diabetic patients (Vestergaard et al., 2005).
Metformin improved bone health in diabetes; therefore, lowering blood sugar levels could be a key factor to protect bone loss. Unlike metformin, other anti-diabetic drugs failed to provide beneficial effects on bone density. For example, treatment with anti-diabetic thiazolidinediones, such as rosiglitazone in ovariectomized ZDF rats increased cortical bone porosity (Sardone et al., 2011). Similarly, diabetic patients receiving thiazolidinedione showed enhanced non-spine bone fractures (Schwartz et al., 2015), reduced levels of parameters for osteoblast activity and increased osteoclast activity (Zinman et al., 2010). Other anti-diabetic drugs in the DPP-4 inhibitors family, such as vildagliptin, showed no improvement in BMD in post-menopausal women with T2DM (Vianna et al., 2017). Therefore, the benefits of metformin on bone health might not be due to the sugar lowering effect of the drug but to other mechanisms that require further investigation.
One signaling pathway involved in bone homeostasis, especially in osteoblast activity is p38 MAPK. Phosphorylation of p38 MAPK stimulated runt-related transcription factors (Runx2) and the downstream Wnt/β-catenin signaling pathway that play major roles in osteoblast proliferation and differentiation (Rodríguez-Carballo et al., 2016; Yavropoulou and Yovos, 2007). Therefore, it could be speculated that p38 MAPK signaling plays an important role in osteoblast cells. Hyperglycemic conditions increased basal p38 MAPK activation as a signaling pathway that plays a role in cellular injury and apoptosis (Kumphune et al., 2013; Nakagami et al., 2001). Thus, inhibition of p38 MAPK by means of an inhibitor could benefit osteoblast cells. Our results indicated that osteoblasts isolated from GK rats treated with SB203580 showed no significant difference in cell proliferation compared to osteoblasts isolated from diabetic rats. In addition, treatment with SB203580 improved ALP expression but failed to enhance osteoblast mineralization. A combination of p38 MAPK inhibitor and metformin did not provide synergistic effects to further enhance osteoblast growth and differentiation. This result may be because p38 MAPK positively regulates osteoblast growth and differentiation. Inhibition of p38 MAPK activity could be detrimental to osteoblasts. Loss of p38 MAPK by gene knockout could reduce trabecular and cortical bone density as well as impair osteoblast differentiation (Rodríguez-Carballo et al., 2014). In addition, inhibition of p38 MAPK by inhibitors showed a reduction in ALP activity and a decrease in trabecular bone loss in long bones (Caverzasio et al., 2008; Suzuki et al., 2002). Unlike results from an in vivo study and bone scanning by micro-CT, our results showed that p38 MAPK inhibition significantly preserved bone mass, predominantly on the trabecular bone. On the other hand, treatment with metformin combined with p38 MAPK inhibitor only synergistically reduced the porosity of cortical bone from the femur and tibia. A combination of p38 MAPK inhibitor and metformin significantly reduced only femur trabecular bone porosity. This discrepancy could be due to the effect of p38 MAPK inhibitor on other cell types in the bone such as osteoclasts and osteoprogenitor cells. Therefore, the effect of p38 MAPK inhibitor on these cell types was not examined and is considered as one of the limitation of this study.
To the best of our knowledge, this is the first study to hypothesize the combinatorial effect of metformin and p38 MAPK inhibitor. However, the outcomes do not clearly show the benefits of co-treatment on bone health. This may be, in part, due to the counteracting effects of these two drugs with interfering growth and differentiation of osteoblast or osteoclast cells also considered as another factor of the failure.
CONCLUSION
In conclusion, results showed that non-obese type 2 diabetes aggravated bone porosity, while a single treatment of metformin or SB203580 provided beneficial effects to bone health for non-obese type 2 diabetes patients.
ACKNOWLEDGMENT
The authors would like to thank the Centre for Animal Research, Naresuan University for their excellent technical assistance. The authors also would like to thank BL1.2W: X-ray imaging and X-ray Tomographic Microtomography, Synchrotron light research institute, Thailand for funding the studies and excellent technical assistance. The authors would like to thank the ProofRead4Sure service for English proofreading and editing.
CONFLICT OF INTEREST
The authors have declared that they have no conflict of interest.
FUNDING SOURCES
This thesis is sponsored and supported by the National Research Council of Thailand. Higher Education Research Promotion (HERP) Grant ID number R2559A017. Naresuan University Research endowment fund Grant ID number R2560C138. We would like to thank Ph.D. scholarship from Naresuan University for Punyanuch Adulyarithikul, Royal Golden Jubilee Ph.D. Program-Thailand Research Fund (TRF) (No. PHD/0087/2556) for Jantira Sanit and (No. PHD/0125/2558) for Kantapitch Kongpol.
AUTHORS’ CONTRIBUTIONS
Punyanuch Adulyaritthikul and Sarawut Kumphune conceived and designed the experiments; Punyanuch Adulyaritthikul, Jantira Sanit, Nuttikarn Nokkaew, Kantapich Kongpol, and Podsawee Mongkolpathumrat performed animal treatment and collection of data. Punyanuch Adulyaritthikul and Catleya Rojviriya performed and analyzed the microCT data; Sarawut Kumphune, Catleya Rojviriya, and Sakarat na Lampang contributed reagents/materials/analysis tools; Punyanuch Adulyaritthikul and Sarawut Kumphune wrote and prepared the manuscript.
REFERENCES
Ahmad T, Ohlsson C, Saaf M, Ostenson CG, Kreicbergs A. Skeletal changes in type-2 diabetic Goto-Kakizaki rats. J Endocrinol, 2003; 178:111–6. CrossRef
Akash MS, Rehman K, Chen S. Goto-Kakizaki rats: its suitability as non-obese diabetic animal model for spontaneous type 2 diabetes mellitus. Curr Diabetes Rev, 2013; 9:387–96. CrossRef
Borges JL, Bilezikian JP, Jones-Leone AR, Acusta AP, Ambery PD, Nino AJ, Grosse M, Fitzpatrick LA, Cobitz AR. A randomized, parallel group, double-blind, multicentre study comparing the efficacy and safety of Avandamet (rosiglitazone/metformin) and metformin on long-term glycaemic control and bone mineral density after 80 weeks of treatment in drug-naive type 2 diabetes mellitus patients. Diabetes Obes Metab, 2011; 13:1036–46. CrossRef
Braun T, Lepper J, Ruiz HG, Hofstetter W, Siegrist M, Lezuo P, Gaestel M, Rumpler M, Thaler R, Klaushofer K, Distler JH, Schett G, Zwerina J. Mitogen-activated protein kinase 2 regulates physiological and pathological bone turnover. J Bone Miner Res, 2013; 28:936–47. CrossRef
Caverzasio J, Higgins L, Ammann P. Prevention of trabecular bone loss induced by estrogen deficiency by a selective p38alpha inhibitor. J Bone Miner Res, 2008; 23:1389–97. CrossRef
Cheng AY, Fantus IG. Oral antihyperglycemic therapy for type 2 diabetes mellitus. CMAJ, 2005; 172:213–26. CrossRef
Cortizo AM, Sedlinsky C, McCarthy AD, Blanco A, Schurman L. Osteogenic actions of the anti-diabetic drug metformin on osteoblasts in culture. Eur J Pharmacol, 2006; 536:38–46. CrossRef
de L II, van der Klift M, de Laet CE, van Daele PL, Hofman A, Pols HA. Bone mineral density and fracture risk in type-2 diabetes mellitus: the Rotterdam study. Osteoporos Int, 2005; 16:1713–20. CrossRef
Dede AD, Tournis S, Dontas I, Trovas G. Type 2 diabetes mellitus and fracture risk. Metabolism, 2014; 63:1480–90. CrossRef
Eckel RH, Kahn SE, Ferrannini E, Goldfine AB, Nathan DM, Schwartz MW, Smith RJ, Smith SR. Obesity and type 2 diabetes: what can be unified and what needs to be individualized? J Clin Endocrinol Metab, 2011; 96:1654–63. CrossRef
Gao Y, Li Y, Xue J, Jia Y, Hu J. Effect of the anti-diabetic drug metformin on bone mass in ovariectomized rats. Eur J Pharmacol, 2010; 635:231–6. CrossRef
Hegazy SK. Evaluation of the anti-osteoporotic effects of metformin and sitagliptin in postmenopausal diabetic women. J Bone Miner Metab, 2015; 33:207–12. CrossRef
Jang WG, Kim EJ, Bae IH, Lee KN, Kim YD, Kim DK, Kim SH, Lee CH, Franceschi RT, Choi HS, Koh JT. Metformin induces osteoblast differentiation via orphan nuclear receptor SHP-mediated transactivation of Runx2. Bone, 2011; 48:885–93. CrossRef
Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, Kravitz BG, Lachin JM, O'Neill MC, Zinman B, Viberti G. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med, 2006; 355:2427–43. CrossRef
Kalyani RR, Golden SH, Cefalu WT. Diabetes and aging: unique considerations and goals of care. Diabetes Care, 2017; 40:440–3. CrossRef
Kanazawa I, Yamaguchi T, Yano S, Yamauchi M, Sugimoto T. Metformin enhances the differentiation and mineralization of osteoblastic MC3T3-E1 cells via AMP kinase activation as well as eNOS and BMP-2 expression. Biochem Biophys Res Commun, 2008; 375:414–9. CrossRef
Kao WH, Kammerer CM, Schneider JL, Bauer RL, Mitchell BD. Type 2 diabetes is associated with increased bone mineral density in Mexican-American women. Arch Med Res, 2003; 34:399–406. CrossRef
Kashima S, Inoue K, Matsumoto M, Akimoto K. Prevalence and characteristics of non-obese diabetes in Japanese men and women: the Yuport Medical Checkup Center Study. J Diabetes, 2015; 7:523–30. CrossRef
Kayal RA, Tsatsas D, Bauer MA, Allen B, Al-Sebaei MO, Kakar S, Leone CW, Morgan EF, Gerstenfeld LC, Einhorn TA, Graves DT. Diminished bone formation during diabetic fracture healing is related to the premature resorption of cartilage associated with increased osteoclast activity. J Bone Miner Res, 2007; 22:560–8. CrossRef
Kerckhofs G, Durand M, Vangoitsenhoven R, Marin C, Van der Schueren B, Carmeliet G, Luyten F P, Geris L, Vandamme K. Changes in bone macro- and microstructure in diabetic obese mice revealed by high resolution microfocus X-ray computed tomography. Sci Rep, 2016; 6:35517. CrossRef
Kumphune S, Chattipakorn S, Chattipakorn N. Roles of p38-MAPK in insulin resistant heart: evidence from bench to future bedside application. Curr Pharm Des, 2013; 19:5742–54. CrossRef
Ma L, Oei L, Jiang L, Estrada K, Chen H, Wang Z, Yu Q, Zillikens MC, Gao X, Rivadeneira F. Association between bone mineral density and type 2 diabetes mellitus: a meta-analysis of observational studies. Eur J Epidemiol, 2012; 27:319–32. CrossRef
Mai QG, Zhang ZM, Xu S, Lu M, Zhou RP, Zhao L, Jia CH, Wen ZH, Jin DD, Bai XC. Metformin stimulates osteoprotegerin and reduces RANKL expression in osteoblasts and ovariectomized rats. J Cell Biochem, 2011; 112:2902–9. CrossRef
Mastrandrea LD, Wactawski-Wende J, Donahue RP, Hovey KM, Clark A, Quattrin T. Young women with type 1 diabetes have lower bone mineral density that persists over time. Diabetes Care, 2008; 31:1729–35. CrossRef
Moseley KF. Type 2 diabetes and bone fractures. Curr Opin Endocrinol Diabetes Obes, 2012; 19:128–35. CrossRef
Nakagami H, Morishita R, Yamamoto K, Yoshimura SI, Taniyama Y, Aoki M, Matsubara H, Kim S, Kaneda Y, Ogihara T. Phosphorylation of p38 mitogen-activated protein kinase downstream of Bax-caspase-3 pathway leads to cell death induced by high D-glucose in human endothelial cells. Diabetes, 2001; 50:1472–81. CrossRef
Napoli N, Chandran M, Pierroz DD, Abrahamsen B, Schwartz AV, Ferrari SL. Mechanisms of diabetes mellitus-induced bone fragility. Nat Rev Endocrinol, 2017; 13:208–19. CrossRef
Nicodemus KK, Folsom AR. Type 1 and type 2 diabetes and incident hip fractures in postmenopausal women. Diabetes Care, 2001; 24:1192–7. CrossRef
Petit MA, Paudel ML, Taylor BC, Hughes JM, Strotmeyer ES, Schwartz AV, Cauley JA, Zmuda JM, Hoffman AR, Ensrud KE. Bone mass and strength in older men with type 2 diabetes: the Osteoporotic Fractures in Men Study. J Bone Miner Res, 2010; 25:285–91. CrossRef
Pramojanee SN, Phimphilai M, Kumphune S, Chattipakorn N, Chattipakorn SC. Decreased jaw bone density and osteoblastic insulin signaling in a model of obesity. J Dent Res, 2013; 92:560–5. CrossRef
Redman AM, Johnson JS, Dally R, Swartz S, Wild H, Paulsen H, Caringal Y, Gunn D, Renick J, Osterhout M, Kingery-Wood J, Smith RA, Lee W, Dumas J, Wilhelm SM, Housley TJ, Bhargava A, Ranges GE, Shrikhande A, Young D, Bombara M, Scott WJ. p38 kinase inhibitors for the treatment of arthritis and osteoporosis: thienyl, furyl, and pyrrolyl ureas. Bioorg Med Chem Lett, 2001; 11:9–12. CrossRef
Rodríguez-Carballo E, Gámez B, Sedó-Cabezón L, Sánchez-Feutrie M, Zorzano A, Manzanares-Céspedes C, Rosa JL, Ventura F. The p38α MAPK function in osteoprecursors is required for bone formation and bone homeostasis in adult mice. PLoS One, 2014; 9:e102032. CrossRef
Rodríguez-Carballo E, Gámez B, Ventura F. p38 MAPK signaling in osteoblast differentiation. Front Cell Dev Biol, 2016; 4:40. CrossRef
Sampayo C, Alves Agripino A, Stilwell D, Vidal B, Fernando A, Silva Lima B, Vaz MF, Canhão H, Cristina Marques M. Chronic hyperglycemia modulates rat osteoporotic cortical bone microarchitecture into less fragile structures. Int J Endocrincol, 2017; 2017:9. CrossRef
Sardone LD, Renlund R, Willett TL, Fantus IG, Grynpas MD. Effect of rosiglitazone on bone quality in a rat model of insulin resistance and osteoporosis. Diabetes, 2011; 60:3271–8. CrossRef
Schwartz AV. Diabetes mellitus: does it affect bone? Calcif Tissue Int, 2003; 73:515–9. CrossRef
Schwartz AV, Chen H, Ambrosius WT, Sood A, Josse RG, Bonds DE, Schnall AM, Vittinghoff E, Bauer DC, Banerji MA, Cohen RM, Hamilton BP, Isakova T, Sellmeyer DE, Simmons DL, Shibli-Rahhal A, Williamson JD, Margolis KL. Effects of TZD use and discontinuation on fracture rates in ACCORD bone study. J Clin Endocrinol Metab, 2015; 100:4059–66. CrossRef
Suzuki A, Guicheux J, Palmer G, Miura Y, Oiso Y, Bonjour JP, Caverzasio J. Evidence for a role of p38 MAP kinase in expression of alkaline phosphatase during osteoblastic cell differentiation. Bone, 2002; 30:91–8. CrossRef
Thouverey C, Caverzasio J. Focus on the p38 MAPK signaling pathway in bone development and maintenance. Bonekey Rep, 2015; 4:711. CrossRef
Vestergaard P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes—a meta-analysis. Osteoporos Int, 2007; 18:427–44. CrossRef
Vestergaard P, Rejnmark L, Mosekilde L. Relative fracture risk in patients with diabetes mellitus, and the impact of insulin and oral antidiabetic medication on relative fracture risk. Diabetologia, 2005; 48:1292–9. CrossRef
Vianna AGD, de Lacerda CS, Pechmann LM, Polesel MG, Marino EC, Borba VZC, Barreto FC. Vildagliptin has the same safety profile as a sulfonylurea on bone metabolism and bone mineral density in post-menopausal women with type 2 diabetes: a randomized controlled trial. Diabetol Metab Syndr, 2017; 9:35. CrossRef
Wong SK, Chin KY, Suhaimi FH, Ahmad F, Ima-Nirwana S. Effects of metabolic syndrome on bone mineral density, histomorphometry and remodelling markers in male rats. PLoS One, 2018; 13:e0192416. CrossRef
Yan W, Li X. Impact of diabetes and its treatments on skeletal diseases. Front Med, 2013; 7:81–90. CrossRef
Yaturu S, Humphrey S, Landry C, Jain SK. Decreased bone mineral density in men with metabolic syndrome alone and with type 2 diabetes. Med Sci Monit, 2009; 15:Cr5–9.
Yavropoulou MP, Yovos JG. The role of the Wnt signaling pathway in osteoblast commitment and differentiation. Hormones (Athens), 2007; 6:279–94. CrossRef
Zhang L, Liu Y, Wang D, Zhao X, Qiu Z, Ji H, Rong H. Bone biomechanical and histomorphometrical investment in type 2 diabetic Goto-Kakizaki rats. Acta Diabetol, 2009; 46:119–26. CrossRef
Zinman B, Haffner SM, Herman WH, Holman RR, Lachin JM, Kravitz BG, Paul G, Jones N P, Aftring R P, Viberti G, Kahn S E. Effect of rosiglitazone, metformin, and glyburide on bone biomarkers in patients with type 2 diabetes. J Clin Endocrinol Metab, 2010; 95:134–42. CrossRef