
INTRODUCTION
Endophytic fungi are microorganisms capable of colonizing plant tissues asymptomatically, establishing mutualistic relationships without causing harm to their host. These fungi are known to be prolific producers of secondary metabolites exhibiting a wide range of biological activities [1]. Among the various genera of endophytes, Paecilomyces remains relatively underexplored compared to well-documented genera such as Penicillium and Aspergillus [2,3]. Despite this, members of the Paecilomyces genus have been reported to produce diverse bioactive compounds, including sterols, fatty acids, terpenoids, macrocycles, peptides, polyketides, quinones, xanthones, pyrones, pyrenosine, and alkaloids [4,5].
In our previous study, Paecilomyces subglobosus CBK3, one of 19 fungal species isolated from the medicinal tree fern Cyathea contaminans (Hook.) Copel. exhibited potent antibacterial activity in its ethyl acetate (EtOAc) extract and fractions (Table 1) [6]. Further investigation led to the isolation of the dichloromethane (DCM) fraction, from which two major compounds were isolated: compound 1, identified as the isoquinoline derivative O-methylcorypaline (OMC), and compound 2, identified as the phenylacetate derivative phenylglyoxylic acid (PGA). Although neither compound exhibited significant antibacterial activity in vitro, this prompted further evaluation of their pharmacological potential through in silico studies.
 | Table 1. Antibacterial activity of the crude extract and fractions of endophytic fungal Paecylomyces subglobosus CBK3. [Click here to view] |
Isoquinoline alkaloids like OMC are known to exhibit a broad spectrum of biological activities, including neuroprotective, cardioprotective, antitumor, antidepressant, and anti-inflammatory effects [7]. Similarly, phenylacetate derivatives are reported to possess analgesic and anti-inflammatory properties and serve as biosynthetic precursors for various therapeutic agents [8,9]. While OMC has been naturally isolated from several plant species [10,11], its pharmacological profile remains poorly characterized. Limited reports suggest its potential as an antidepressant [12]. Quinapril is an isoquinoline derivative that works as an angiotensin-converting enzyme inhibitor (ACEI). It has been distributed and used to treat hypertension and congestive heart failure [13]. Meanwhile, specific phenylacetate-based nonsteroidal anti-inflammatory drugs (NSAIDs) are known for their potent anti-inflammatory effects and reduced side effects compared to conventional analgesics [14].
This study is the first to report the isolation of OMC and PGA from P. subglobosus CBK3. To explore their potential beyond antibacterial activity, molecular docking simulations were performed targeting several key proteins associated with blood pressure regulation, including angiotensin-converting enzyme (ACE), angiotensin II receptor type 1 ( AT1), and cyclooxygenase-2 (COX-2) [15,16]. Notably, recent studies have identified a role for COX-2 in modulating renal blood flow, linking its activity to the vasodilatory effects of antihypertensive agents [17].
This research aims to isolate and characterize secondary metabolites from P. subglobosus CBK3 and to investigate their potential antihypertensive properties through in silico evaluation against ACE,AT1, and COX-2 receptors. The findings are expected to contribute to the discovery of novel natural compounds with antihypertensive potential and expand the pharmacological relevance of endophytic fungi as sources of drug leads.
MATERIALS AND METHODS
General experimental procedures
Thin-layer chromatography was performed using precoated silica gel 60 F254 plates (layer thickness 0.2 mm; Merck, Darmstadt, Germany). Compound isolation was carried out using a semi-preparative high-pressure liquid chromatography (HPLC) system (Waters 600, USA) equipped with a C18 ODS3 column (10 × 150 mm) and a Waters 2487 UV detector. A gradient elution system consisting of acetonitrile and water containing 0.1% trifluoroacetic acid (TFA) was employed as the mobile phase, with a gradient of the mobile phase against time for 30 minutes, namely 0–15 minutes (100% water 0.1% TFA); 16 minutes (56% Acetonitrile: 44% water 0.1% TFA); 17–24 minutes (100% acetonitrile); 25–30 minutes (100% water 0.1% TFA), with a flow rate of 4.7 ml/min. Sample injections were performed at a volume of 100 µl, and UV detection was monitored at a wavelength of 276 nm. The purity of the isolated compounds was confirmed using an analytical HPLC system (Knauer, Germany) equipped with a C18 ODS3 column (4.6 × 150 mm, 5 µm) and a photodiode array detector. The mobile phase system used in analytical HPLC is the same as the mobile phase used in semipreparative HPLC. The analysis was conducted with an injection volume of 20 µl to ensure the reproducibility and resolution of the target peaks. For compound characterization, liquid chromatography-mass spectrometry analysis was carried out using an ultra performance liquid chromatography (UPLC) system (ACQUITY UPLC® H-Class System, Waters, USA) coupled with an Xevo G2-S Quadrupole Time-of-Flight mass spectrometer (Waters, USA). Chromatographic separation was achieved using an HSS C18 column (2.1 × 100 mm, 1.8 µm; Waters, USA), with a column temperature of 50°C and room temperature of 25°C. The mobile phase used was water +5 mM ammonium formic (A) and acetonitrile + 0.05% formic acid (B), Flow rate: 0.2 ml/min (step gradient), running 23 minutes, injection volume: 5 μl.
Fungal material
The endophytic fungus P. subglobosus CBK3 was previously isolated from the medicinal tree fern C. contaminans (Hook.) Copel., collected in Kerinci, Jambi, Indonesia (geographic coordinates: 2°3’32.1624 “S, 101°21’56.2428’’E). Taxonomic identification was conducted at the ANDA Herbarium, Department of Biology, Faculty of Mathematics and Natural Sciences, Andalas University, Padang, Indonesia, under voucher numbers 51803–51805. Molecular identification was carried out based on sequencing of the 18S rRNA region. The fungal strain is preserved at the Sumatra Biota Laboratory, Andalas University, Padang, Indonesia [6].
Extraction and isolation
The fungus cultivation medium was prepared by combining rice (100?g) with distilled water (110?ml) in a 1?l Erlenmeyer flask and allowing it to soak overnight. The medium was then sterilized in an autoclave. A rejuvenated endophytic fungal isolate, P. subglobosus, cut into 1–2?cm² pieces, was inoculated into 40 Erlenmeyer flasks, each containing sterile rice medium (totaling 4?kg). The cultures were maintained under static conditions at room temperature, which maintained a neutral pH, and were exposed to natural sunlight for 4–6 weeks or until the fungus mycelium fully colonized the medium. The solid-state fermentation medium of P. subglobosus CBK3 was extracted with EtOAc and sealed for at least 24 hours. The mixture was filtered to separate the liquid phase from the biomass, and this extraction process was repeated three times to ensure maximum yield. The combined EtOAc extracts were concentrated under reduced pressure, yielding a crude extract (90.9 g). This crude extract was then suspended in 90% aqueous methanol (MeOH) and sequentially partitioned with n-hexane and DCM [18–20]. The partitioning process yielded 42?g of the n-hexane fraction, 19.8 g of the DCM fraction, and 26.5?g of the MeOH fraction. Subsequently, 10?g of the DCM fraction was subjected to vacuum liquid chromatography over silica gel 60 using a step-gradient elution with n-hexane, EtOAc, DCM, and MeOH. This procedure resulted in nine major fractions, designated F1 to F9 [20]. Fraction F2 (0.9?g) exhibited promising chemical profiles and was selected for further purification. Purification of F2 was carried out using semi-preparative HPLC with a gradient mobile phase of acetonitrile and water containing 0.1% TFA, operated at a 4.7 ml/min flow rate. Analytical HPLC was used to verify the purity of the isolated compounds. This process successfully led to the isolation of two compounds: compound 1 (24.8 mg/0.25%, tR = 9.3 minutes) and compound 2 (2 mg/0.02%, tR = 11.5 minutes), as shown in Figure 1. The structures of isolated compounds were elucidated using liquid chromatography-tandem mass spectrometry, UV, and IR spectroscopy [21,22]. Mass spectral data were analyzed using MassLynx™ V4.1 software and interpreted by comparison with compound databases, including PubChem, Human Metabolome Database (HMDB), and ChemSpider.
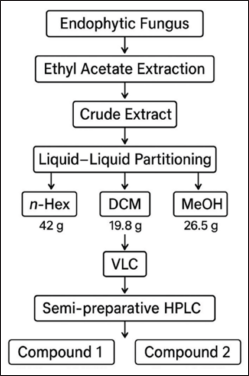 | Figure 1. Scheme of isolation compounds of the endophytic fungus Paecylomyces Subglobosus. [Click here to view] |
Molecular docking
Validation of molecular docking
Before conducting docking simulations of the test compounds, docking protocol validation was performed to ensure the reliability and accuracy of the method. This was achieved by re-docking the native ligands into the binding sites of their respective protein targets: COX-2, ACE, and angiotensin II receptor type 1 (ARB). The re-docked ligand conformations were then compared to their original co-crystallized poses in the protein structures. The accuracy of the docking protocol was assessed by calculating the root mean square deviation (RMSD) between the heavy atoms of the re-docked and original ligand structures. An RMSD value of less than 2.0 Å is generally considered acceptable, indicating that the docking algorithm can reliably reproduce the experimentally determined binding modes [23,24]. A low RMSD value obtained in this study confirmed the validity of the docking parameters and the suitability of the selected scoring function for subsequent ligand–receptor interaction analyses.
Molecular docking simulation
Three receptors were evaluated through in-silico molecular docking, specifically the anti-inflammatory impact on the COX-2 receptor, PDB ID:6COX (https://www.wwpdb.org/pdb?id=pdb_00006cox) with resolution 2.80 Å; the binding site His90, Tyr115, Arg120, Gln192, Tyr355, Leu359, Tyr385, Trp387, Arg513, Phe518, Val523, and Ser530 [25]. Two receptors were utilized for the antihypertensive effect: the ACE receptor, PDB ID: 4AA1 (https://www.wwpdb.org/pdb?id=pdb_00004aa1); resolution 1.99 Å; binding sites: Gln265, Tyr504, and Lys495 [26], and angiotensin II type 1 (AT1), PDB ID: 4ZUD (https://www.wwpdb.org/pdb?id=pdb_00004zud); resolution 2.80 Å; binding sites: Tyr35, Trp84, and Arg167 [27]. The three crystal structures of the X-ray receptors used were downloaded from PDB (https://www.rcsb.org/), while the chemical structures of compounds 1 and 2 were taken from the Pub-Chem database (https://pubchem.ncbi.nlm.nih.gov). The receptors and ligands obtained were interacted using MOE Software. The XYZ coordinates of the active site of the receptor were determined using DS software [28,29].
Absorption, distribution, metabolism, excretion, and toxicity simulation
An in silico assessment of absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties and drug-likeness profiles was conducted to evaluate the pharmacokinetic behavior and drug development potential of the isolated compounds. These predictive simulations provide valuable insights into the physicochemical characteristics and bioavailability of candidate molecules early in the drug discovery process, reducing the need for extensive in vivo testing.
The drug-likeness of the compounds was evaluated using multiple rule-based filters, including Lipinski’s Rule of Five, Ghose filter, Veber rule, Egan rule, and Muegge criteria. These filters assess properties such as molecular weight (MW), lipophilicity (LogP), hydrogen bond donors and acceptors, topological polar surface area (TPSA), and number of rotatable bonds. In addition, the synthetic accessibility score was computed on a scale from 1 (very easy) to 10 (very difficult), indicating the feasibility of compound synthesis. All predictions were performed using the SwissADME web server provided by the Swiss Institute of Bioinformatics (http://www.swissadme.ch/) [30,31], a widely accepted platform for computational pharmacokinetics and medicinal chemistry profiling.
RESULTS AND DISCUSSION
Two compounds, designated as compound 1 and compound 2, were successfully extracted from the fungus P. subglobosus CBK3. Compound 1 is a white solid with limited solubility in MeOH. Its UV spectrum displays a peak absorption at 276 nm. This value is consistent with the UV absorption range (210–288 nm) reported for OMC in studies using HCl or water as solvents, as Menachery et al. [32] noted. The absorption peak observed at 276 nm in MeOH closely aligns with these findings, supporting the identification of compound 1 as an OMC derivative. The FT-IR spectrum of compound 1 reveals characteristic absorption bands corresponding to specific functional groups: a broad band at 3,743.45 cm−1 indicative of O–H stretching (phenol), a peak at 2,900 cm−1 attributed to C–H stretching (alkane), a band at 1,698.67 cm−1 corresponding to C=C stretching (alkene), and a signal at 1,535.82 cm−1 associated with aromatic C=C stretching [33,34].
The mass spectrum of compound 1, obtained using the electrospray ionization mass spectrometry (ESI-MS) technique, revealed a protonated molecular ion [M+H]? with an m/z value of 208.1338, corresponding to the molecular formula C??H??NO?. The CH? molecular ion exhibited characteristic fragmentation, producing fragment ions at m/z 176.1052 (loss of CH?OH), m/z 152.0703 (loss of CCOCH?), m/z 106.0655 (loss of CH? and 2COCH?), and m/z 77.0733 (loss of CH?OH and CH?NCH?). The MS² spectrum showed additional fragment ions at m/z 208.1336, 194.0742, 195, 180.1012, 176.1052, 162.0905, 151.0623, 134.0957, 120.0446, and 106.0655. The fragmentation pattern observed in both TOF MS1 and MS2 spectra of compound 1 is consistent with the known fragmentation scheme of O-methylcorypalline [11]. Furthermore, a comparison of the obtained mass spectra with established databases, including the HMDB (HMDB0029370) and PubChem (CID 27694), supports the identification of compound 1 as O-methylcorypalline. The proposed fragmentation scheme and corresponding mass spectrum are presented in Figure 2.
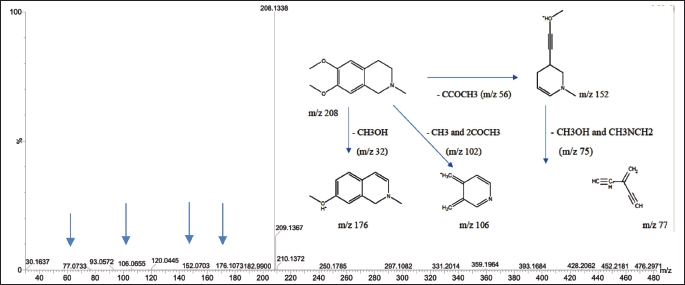 | Figure 2. Mass spectrum with ESI-MS and fragmentation scheme of precursor ions for OMC. [Click here to view] |
Compound 2 is a white solid that exhibits a maximum absorption at 285 nm in the UV spectrum. The FT-IR analysis reveals a broad and intense band at 3,330.92 cm−1, corresponding to O–H stretching vibrations typical of carboxylate groups. A strong absorption at 1,639.87 cm−1 is attributed to C=O stretching (carboxylate), while peaks at 1,457.09 cm−1 and 1,393.50 cm−1 are assigned to aromatic C=C stretching and C–O–H bending of the carboxylate group, respectively [33,34].
The mass spectrum of compound 2, acquired using the ESI-MS technique, revealed a protonated molecular ion [M+H]? at m/z 151.0395, corresponding to the molecular formula C?H?O?. Upon fragmentation, the molecular ion produced characteristic fragment ions at m/z 105.0334, corresponding to the loss of CH?CH? and OH groups, and at m/z 91.0536, associated with the loss of CH? and COOH groups. The observed fragmentation pattern is illustrated in Figure 3. This pattern closely aligns with that of PGA, as previously reported [35]. The structures of compounds 1 and 2 are presented in Figure 4, along with a comparative table summarizing the characterization data for both compounds.
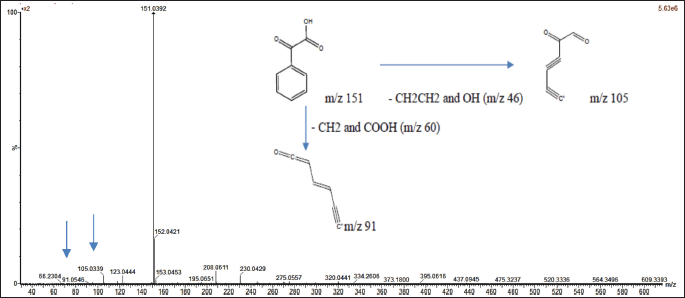 | Figure 3. Mass spectrum with ESI-MS and fragmentation scheme of precursor ions for PGA. [Click here to view] |
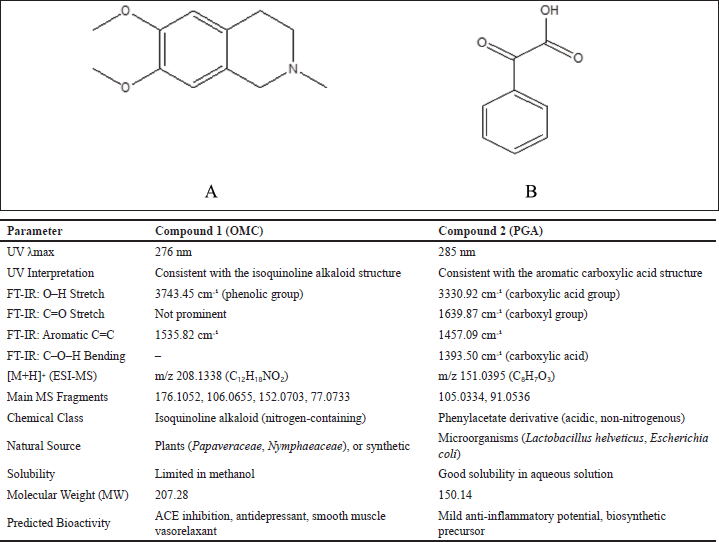 | Figure 4. Structure of OMC (A), PGA (B), and the comparison table of compounds. [Click here to view] |
OMC was characterized and reported in 1976; however, recent publications on this compound remain limited. OMC has been identified in various plant species across different families, including Papaver bracteatum (Papaveraceae), Nelumbo nucifera (Nymphaeaceae), and Pachycereus weberi (Cactaceae), as well as in commercially significant plants such as Coffea arabica and Camellia sinensis. In addition to its natural occurrence, OMC can also be synthesized [10–12].
PGA occurs naturally and has been detected in various foods, organisms, and microorganisms, typically in trace amounts. It is naturally formed through the oxidation of mandelic acid and can also be synthesized. PGA has been identified in microorganisms such as Lactobacillus helveticus and Escherichia coli [9,36].
OMC is categorized as a simple isoquinoline derivative alkaloid, whereas PGA is classified as a phenylacetate derivative. Isoquinoline-based compounds have been reported to exhibit a wide range of bioactivities, including neuroprotective, cardioprotective, antidepressant, antitumor, analgesic, and anti-inflammatory effects [7,12,37]. Phenylacetate derivatives, on the other hand, are known for their analgesic and anti-inflammatory potential, as well as their role as biosynthetic precursors for various other bioactive compounds [8,9].
Literature studies on OMC bioactivity are still relatively few; some have reported OMC activity as an antidepressant [12], but the cardiovascular bioactivity of OMC has not been found, especially related to ACEI. On the other hand, several reports on the bioactivity of isoquinoline derivative alkaloids have been tested in vitro, which is related to cardiovascular effects. DHQ-11 conjugate, the result of molecular hybridization between flavonoid dihydroquercetin and isoquinoline alkaloids, which produces high positive inotropic and vasorelaxant activity in rat cardiac papillary muscles, with EC50 values of flavonoid derivatives, isoquinolines, and their conjugates of 21.2 ± 4.1, 14.6 ± 3.5, and 9.7 ± 4.3 ìM [13]. Other isoquinoline derivatives that produce adverse inotropic effects that can reduce the heart’s workload in the rat heart’s papillary muscles obtained an IC50 value of 12–16.8 µM [38,39]. Several isoquinoline alkaloids have a positive inotropic effect by increasing the strength of cardiac papillary muscle contractions, using the highest concentration of 100 μM, by 75.7% ± 4.7% compared to the control [40,41]. This cardioprotective effect test was carried out on the alkaloid 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline. The structure of this compound is classified as a simple isoquinoline similar to the structure of OMC (6,7-dimethoxy-2-methyl-1,2,3,4-tetrahydroisoquinoline), so this OMC compound has the potential to have activity that affects the cardiovascular system. Furthermore, phenylacetate derivatives from the NSAID class have demonstrated potent anti-inflammatory activity with fewer side effects than conventional analgesics [42].
One limitation of the present study is that although the crude extract, fractions, and subfractions showed antibacterial activity, especially against resistant bacteria [6], the isolated compounds did not exhibit the same antibacterial effect. Due to the limited yield of the isolated compounds, further characterization via nuclear magnetic resonance (NMR) spectroscopy and in vivo antihypertensive testing could not be conducted. Instead, their potential activities were explored through in silico molecular docking studies. The structural similarity of these two compounds (Fig. 4) suggests they could serve as lead compounds for drug development. Future studies can expand on this by evaluating their interactions with various biological targets to design novel therapeutic agents.
Molecular docking analysis
Molecular docking has emerged as a vital tool in the discovery of novel biologically active lead compounds (ligands) with specific affinity for target proteins, typically enzymes or receptors, with known three-dimensional structures. This computational technique aims to predict the optimal conformation, binding orientation, and binding affinity of a ligand within the active site of the target receptor [43].
Molecular docking was performed following the validation of the docking protocol through re-docking of the native ligands with their corresponding target proteins. Re-docking is a critical step to ensure procedural accuracy and reliability, as it assesses the efficiency of the docking method in reproducing the experimentally observed binding poses [44].
The active site coordinates used for docking were determined using DS Software. For the COX-2 receptor (PDB ID: 3LN1), the XYZ coordinates of the binding pockets are as follows: Chain A (−30.988577, −22.283615, and −16.507231 Å), Chain B (77.335538, −14.640769, and −8.091077 Å), and Chain C (65.246500, −45.378923, and 45.422885 Å), each with a radius of 7.00 Å (Fig. 5A). For the ACE receptor (PDB ID: 4AA1), the coordinates are −14.888079, 23.278079, and 22.259579 Å, with a radius of 11.516213 Å (Fig. 5B). The AT1 receptor (PDB ID: 4ZUD) has coordinates −40.873424, 63.309333, and 28.223636 Å, with a radius of 8.138906 Å (Fig. 5C).
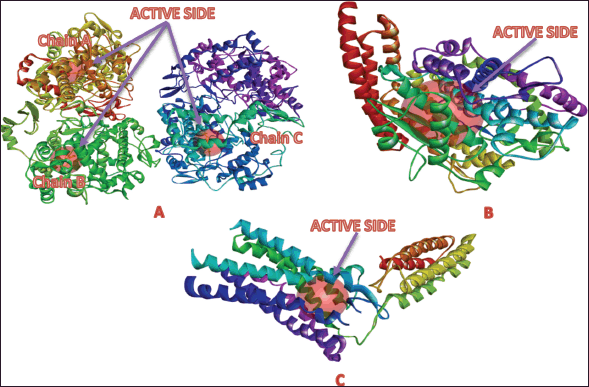 | Figure 5. Active site of the target protein: A) cyclooxygenase-2, B) angiotensin-converting enzyme (ACE), and C) angiotensin II type 1 (AT1). [Click here to view] |
Validation of the docking results was assessed through RMSD analysis, comparing the docked poses of the native ligands with their crystallographic conformations. RMSD analysis from re-docking the native ligands to the COX-2, ACE, and AT1 receptors yielded RMSD values of 0.7786, 1.0474, and 1.7806 Å, respectively (Fig. 6). RMSD reflects the deviation between the docked ligand poses and their original crystallographic coordinates, serving as a key metric for assessing the accuracy of the docking method. An RMSD value below 2.0 Å is generally acceptable, indicating that the docking protocol can reliably reproduce experimentally observed binding modes [38,39]. Based on these results, the molecular docking method used in this study is validated and can be confidently applied for further docking simulations.
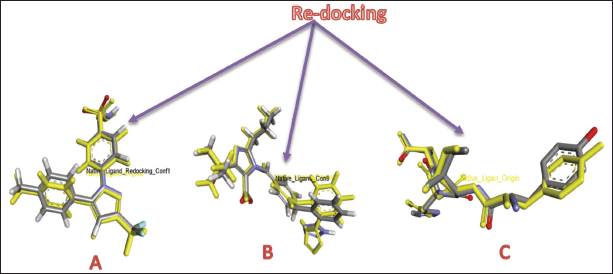 | Figure 6. RMSD of redocking native ligands to receptors: COX-2 (A), ACE (B), and AT1 (C). [Click here to view] |
Tables 2–4 summarize the molecular docking results of the isolated compounds. The binding free energy (BFE) values represent the interactions of OMC, PGA, captopril, candesartan, celecoxib, and native ligands with the target proteins ACE, AT1, and COX-2. The results showed that OMC showed better binding affinity to the ACE receptor compared to PGA, captopril, and the native ligand. In contrast, the interactions of both OMC and PGA with the AT1 and COX-2 receptors demonstrated weak or negligible binding affinities, suggesting limited or no potential activity against these targets (Figs. 9 and 10).
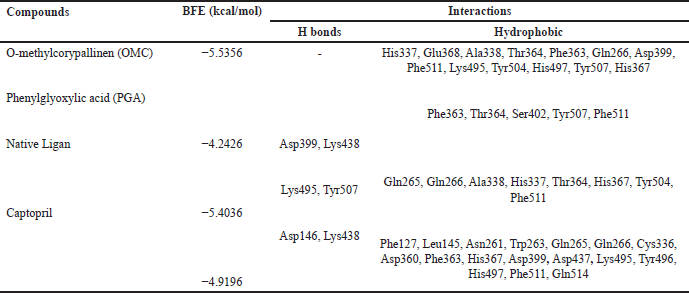 | Table 2. Binding free energies and results of protein-ligand interactions of ACE with OMC, PGA and captopril with MOE (PDB ID: 4AA1). [Click here to view] |
 | Table 3. Binding free energies and results of protein-ligand interactions of AT1 with OMC, PGA, and candesartan with MOE (PDB ID: 4AA1). [Click here to view] |
 | Table 4. Binding free energies and results of protein-ligand interactions of COX-2 with OMC, PGA, and celecoxib with MOE (PDB ID: 4AA1). [Click here to view] |
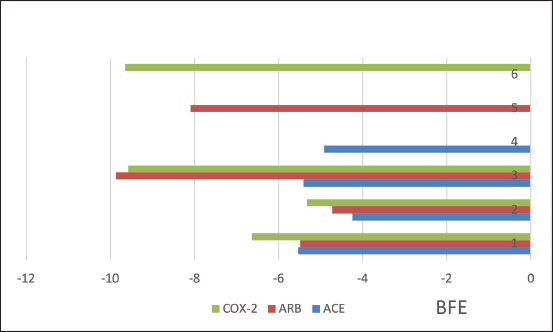 | Figure 7. Binding free energies from ACE, AT1, and COX2 receptors interactions with OMC (1), PGA (2), native ligands (3), captopril (4), candesartan (5), and celecoxib (6) by MOE. [Click here to view] |
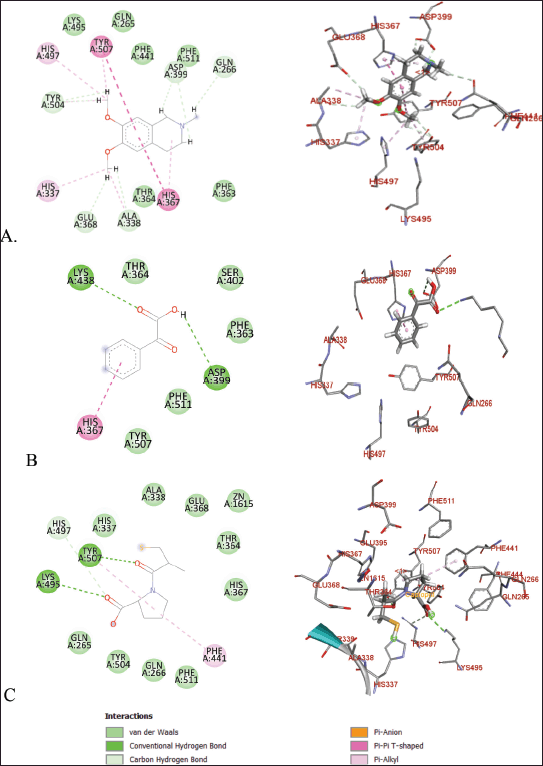 | Figure 8. Molecular docking of 2D and 3D active sites of ACE with interacting ligands OMC (A), PGA (B), and captopril (C). [Click here to view] |
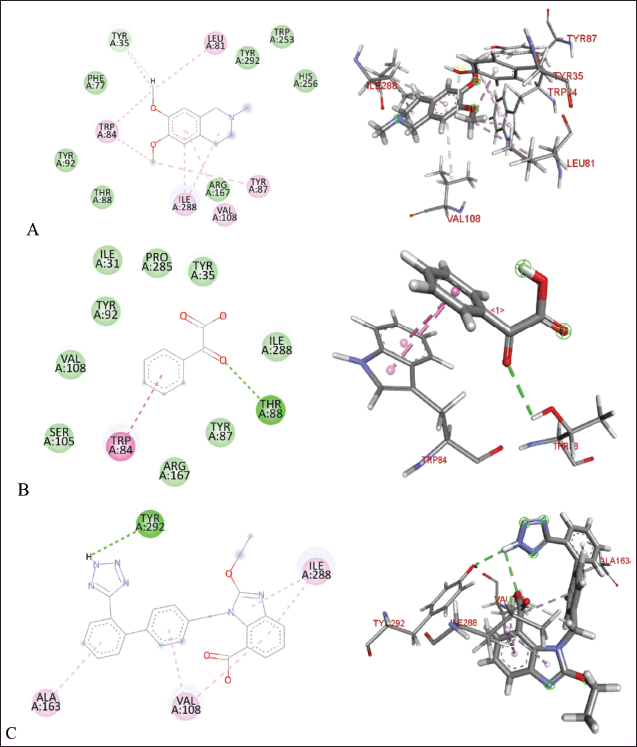 | Figure 9. Molecular docking of 2D and 3D active sites of AT1 with interacting ligands OMC (A), PGA (B), and candesartan (C). [Click here to view] |
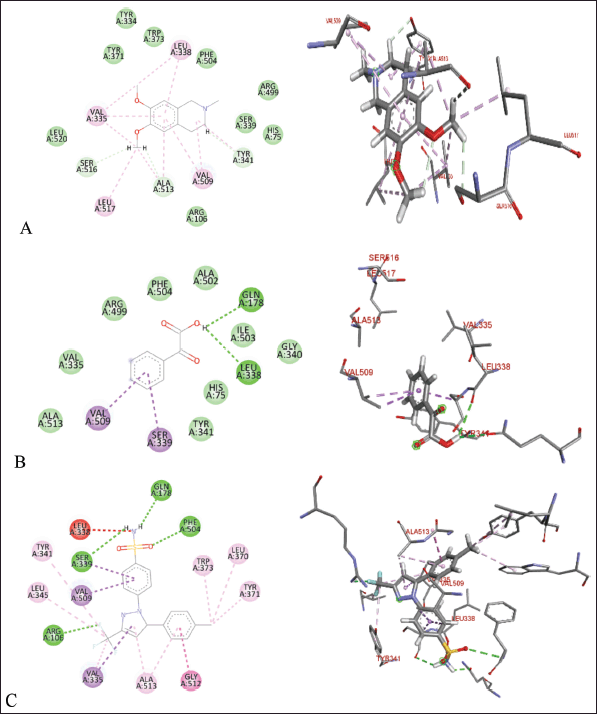 | Figure 10. Molecular docking of 2D and 3D active sites of COX-2 with interacting ligands OMC (A), PGA (B), and celecoxib (C). [Click here to view] |
The molecular docking results reveal the interactions between OMC and PGA with the ACE, AT1, and COX-2 receptors. Among the tested compounds, OMC exhibits the most favorable interaction with the ACE receptor, as indicated by its lower BFE value compared to PGA (Fig. 7). A lower BFE value signifies stronger binding affinity between the ligand and the target protein, highlighting OMC’s potential as a more effective ACE inhibitor.
OMC exhibits interactions with the amino acid residues of the ACE receptor that closely resemble those of the native ligand. In addition to hydrogen bonding, hydrophobic interactions play a key role in stabilizing the protein-ligand complex (Fig. 8). OMC forms four hydrophobic interactions with residues Gln265, Gln266, Phe363, and Lys495, identical to those observed in the native ligand. Similarly, captopril, used as a positive control, forms four hydrophobic bonds with similar amino acid residues. In contrast, PGA forms only two hydrophobic interactions with residues shared by the native ligand (Table 2). These findings suggest that OMC binds more stably to the active site of the ACE receptor, supporting its potential as a promising ACE inhibitor.
ACE is a metalloprotein that contains zinc to interact with ligands at its active site. The results of docking OMC with the ACE receptor resulted in an interaction at the catalytic active site containing zinc ions, which have zinc-binding residues, namely His337, Glu368, and His367 (Table 2), which produces a conserved zinc metalloprotease motif 337HEXXH367. Another study produced an active site involving Zn2+ in the ACE interaction, namely His383, Glu411, and His387, with the motif 383HEXXH387 [15]. Meanwhile, another study resulted in an ACE-Omapatrilat interaction forming a zinc metalloprotease motif with conserved residues His361, Glu389, and His365 361HEXXH365 complementing zinc binding [45].
The amino acid residues interacting with OMC are believed to represent the active site of the ACE receptor, where ligand binding occurs. These residues facilitate the receptor’s biochemical functions [46]. Previous studies have identified a key peptide sequence involved in converting angiotensin I to the vasoconstrictor angiotensin II, which includes the amino acids Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-Leu [47]. Specific interactions observed include GLU forming conventional hydrogen bonds, ALA and VAL involved in alkyl interactions, and PHE, HIS, and TYR participating in π-alkyl interactions [15]. The binding site identified in this study for OMC corresponds closely with the active site reported in earlier ACE receptor studies [15,41], further supporting OMC’s potential role as an ACE inhibitor.
OMC has demonstrated potential as an ACE inhibitor based on in silico predictions. To further evaluate its drug-likeness and feasibility as a therapeutic agent, an ADMET analysis is necessary to assess its pharmacokinetic profile.
ADMET analysis
Physicochemical properties are essential components of a compound’s pharmacokinetic profile and serve as a foundation for evaluating its potential as a drug candidate. Key parameters include MW, the logarithm of the partition coefficient between octanol and water (log?P), the number of rotatable bonds (torsion), hydrogen bond donors (HBDs), hydrogen bond acceptors (HBAs), and polar surface area (PSA). These properties are crucial for predicting the compound’s drug-likeness and oral bioavailability, as outlined in Lipinski’s Rule of Five [48], a widely accepted guideline for assessing compounds’ suitability for development as orally active drugs.
Lipinski’s Rule of Five suggests that compounds are more likely to exhibit good absorption or permeability when they meet the following criteria: no more than 5 hydrogen bond donors (HBDs), no more than 10 hydrogen bond acceptors (HBAs), a MW not exceeding 500 Da, and a calculated Log P (CLog?P) value of 5 or less [48]. Based on the physicochemical property analysis presented in Tables 6 and 8, both OMC and PGA satisfy all parameters of Lipinski’s rule, indicating favorable potential as orally active drug candidates. These findings are further supported by ADMET analysis, which provides additional insight into their pharmacokinetic behavior and safety profile.
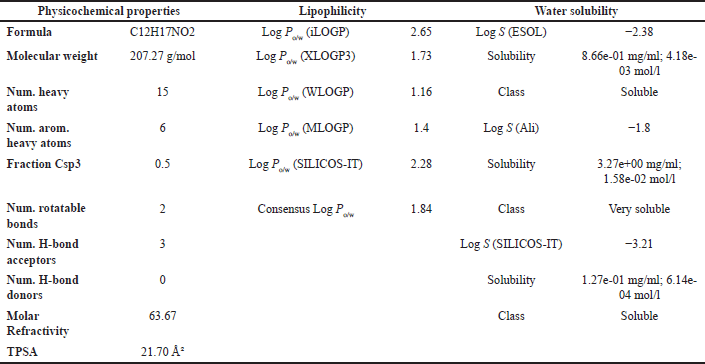 | Table 5. ADMET result of OMC with parameters of psychochemical properties, lipophilicity, and solubility in water. [Click here to view] |
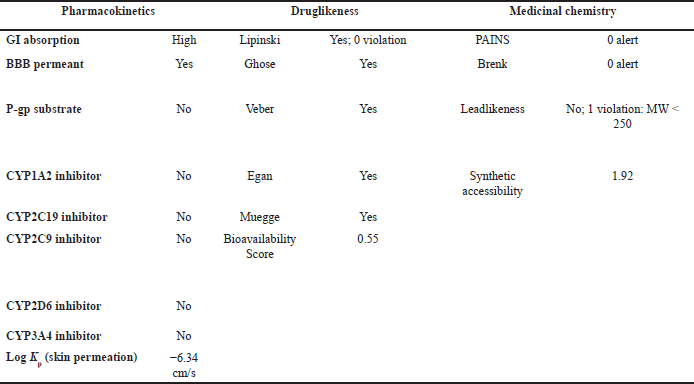 | Table 6. ADMET result of OMC with pharmacokinetic, drug-likeness, and medicinal chemistry parameters. [Click here to view] |
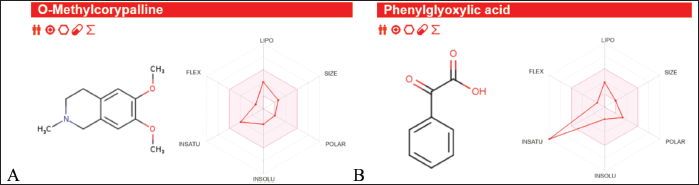 | Figure 11. ADMET result of (A) OMC, and (B) PGA. [Click here to view] |
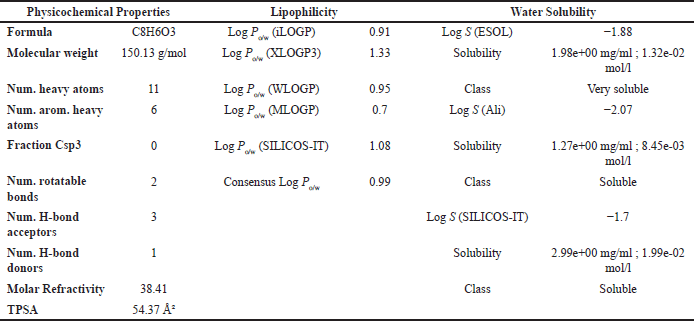 | Table 7. ADMET result of PGA with parameters of psychochemical properties, lipophilicity, and solubility in water. [Click here to view] |
 | Table 8. ADMET result of PGA with pharmacokinetic, drug-likeness, and medicinal chemistry parameters. [Click here to view] |
The ADMET analysis results revealed that OMC (Fig. 11A; Tables 5 and 6) and PGA (Fig. 11 B; Tables 7 and 8) exhibit moderate lipophilicity, good aqueous solubility, and favorable gastrointestinal absorption profiles. Although their skin permeability is low, both compounds are predicted to cross the blood–brain barrier (BBB).
OMC demonstrated superior pharmacokinetics and drug-likeness profiles, fulfilling all criteria under the Lipinski, Ghose, Veber, Egan, and Muegge rules, with a predicted oral bioavailability of 0.55. In contrast, PGA failed to meet several thresholds, particularly those outlined in the Ghose rule, namely MW (<160 Da), molar refractivity (<40), and atom count (<20), as well as the Muegge rule for MW (<200 Da), despite a higher predicted bioavailability of 0.85. Based on these results, it can be concluded that OMC satisfies the requirements for drug-likeness. At the same time, PGA does not fully meet the necessary criteria for development as a drug candidate [49,50].
CONCLUSION
This study demonstrates the potential of P. subglobosus CBK3, an endophytic fungus isolated from C. contaminans, as a promising source of bioactive secondary metabolites with pharmaceutical applications. Chromatographic separation of the DCM fraction yielded two major compounds: OMC (0.25%) and PGA (0.02%). Molecular docking analysis revealed that OMC exhibited a stronger binding affinity toward the ACE compared to the captopril reference inhibitor. Although these findings suggest potential antihypertensive activity, the results remain predictive and require further validation through in vitro and in vivo ACE inhibition assays. OMC also displayed a favorable ADMET profile, reinforcing its potential as a safe and effective lead compound. These results collectively support OMC as a promising candidate for developing novel antihypertensive agents derived from fungal endophytes.
ABBREVIATIONS
ACE angiotensin-converting enzyme
ACEI angiotensin-converting enzyme inhibitor
ARBs angiotensin II receptor blockers
AT1 Angiotensin II receptor type 1
BFE binding free energy
COX-2 cyclooxygenase-2
DCM dichloromethane
EtOAc ethyl acetate
HMDB Human Metabolome Database
HPLC high-pressure liquid chromatography
MeOH methanol
MRSA methicillin-resistant Staphylococcus aureus
NSAID nonsteroidal anti-inflammatory drugs
OMC O-methylcorypalline
PGA phenylglyoxylic acid
RMSD root mean square deviation
TFA trifluoroacetic acid
TPSA topological polar surface area
UPLC ultra performance liquid chromatography
ACKNOWLEDGEMENT
We appreciate the financial support from the Ministry of Education, Culture, Research and Technology Indonesia, Directorate General of Higher Education, Research and Technology: Doctoral Dissertation Research scheme No. 041/E5/PG.02.00.PL/2024.
AUTHORS’ CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVAL
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All of the collected and analyzed data are in this research article.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Wijesekara T, Xu B. Health-promoting effects of bioactive Compounds from plant endophytic fungi. J Fungi. 2023;9(997):1–14.
2. Visagie C, Yilmaz N, Kocsubé S, Frisvad J, Hubka V, Samson R, et al. A review of recently introduced Aspergillus, Penicillium, Talaromyces and other Eurotiales species. Stud Mycol. 2024;10(107):1–66.
3. Li XQ, Xu K, Liu XM, Zhang P. A systematic review on secondary metabolites of Paecilomyces species: chemical diversity and biological activity. Planta Med. 2020;86(12):805–21. CrossRef
4. Dai ZB, Wang X, Li GH. Secondary metabolites and their bioactivities produced by Paecilomyces. Molecules. 2020;25(21):1–18.
5. Ramos G da C, Silva-Silva JV, Watanabe LA, Siqueira JE de S, Almeida-Souza F, Calabrese KS, et al. Phomoxanthone A, compound of endophytic fungi Paecilomyces sp. and its potential antimicrobial and antiparasitic. Antibiotics 2022;11(10):1332.
6. Yulianis, Rustini, Supriyono A, Sandrawati N, Handayani D. Ethyl acetate extracts endophytic fungi from the medicinal tree fern Cyathea Contaminans (Hook) Copel with antimicrobial activity. TRENDS Sci. 2024;21(10):1–10.
7. Yang X, Miao X, Dai L, Guo X, Jenis J, Zhang J, et al. Isolation, biological activity, and synthesis of isoquinoline alkaloids. Nat Prod Rep. 2024;41:1652–722.
8. Mustafa YF, Zain Al-Abdeen SH, Khalil RR, Mohammed ET. Novel functionalized phenyl acetate derivatives of benzo [e]-bispyrone fused hybrids: synthesis and biological activities. Results Chem. 2023;5:100942. CrossRef
9. Tang CD, Shi HL, Xu JH, Jiao ZJ, Liu F, Ding PJ, et al. Biosynthesis of phenylglyoxylic acid by LhDMDH, a novel d -mandelate dehydrogenase with high catalytic activity. J Agric Food Chem. 2018;66(11):2805–11.
10. Wei X, Zhang M, Yang M, Ogutu C, Li J, Deng X. Lotus (Nelumbo nucifera) benzylisoquinoline alkaloids: advances in chemical profiling, extraction methods, pharmacological activities, and biosynthetic elucidation. Veg Res. 2024;4:e005.
11. Wu WN, Huang CH. Structural elucidation of isoquinoline, isoquinolone, benzylisoquinoline, aporphine, and phenanthrene alkaloids using API-ion spray tandem mass spectrometry. Chinese Pharm J. 2006;58(1):41–55.
12. Crestey F, Jensen AA, Borch M, Andreasen JT, Andersen J, Balle T, et al. Design, synthesis, and biological evaluation of Erythrina alkaloid analogues as neuronal nicotinic acetylcholine receptor antagonists. J Med Chem. 2013;56(23):9673–82. CrossRef
13. Usmanov PB, Jumayev IZ, Rustamov SY, Zaripov AA, Esimbetov AT, Zhurakulov SN, et al. The combined inotropic and vasorelaxant effect of DHQ-11, a conjugate of flavonoid dihydroquercetin with isoquinoline alkaloid 1-aryl-6,7-dimethoxy-1,2.3,4-tetrahydroisoquinoline. Biomed Pharmacol J. 2021;14(2):651–61.
14. Amir M, Shikha K. Synthesis and anti-inflammatory, analgesic, ulcerogenic and lipid peroxidation activities of some new 2-[(2,6-dichloroanilino) phenyl]acetic acid derivatives. Eur J Med Chem. 2004;39(6):535–45.
15. Ko SC, Kim JY, Lee JM, Yim MJ, Kim HS, Oh GW, et al. Angiotensin I-converting enzyme (ACE) inhibition and molecular docking study of meroterpenoids isolated from brown alga, Sargassum macrocarpum. Int J Mol Sci. 2023;24(13):11065. CrossRef
16. Agostini L da C, Silva NNT, Belo V de A, Luizon MR, Lima AA, da Silva GN. Pharmacogenetics of angiotensin-converting enzyme inhibitors (ACEI) and angiotensin II receptor blockers (ARB) in cardiovascular diseases. Eur J Pharmacol [Internet]. 2024;981:176907. CrossRef
17. Kirkby NS, Sampaio W, Etelvino G, Alves DT, Anders KL, Temponi R, et al. Cyclooxygenase-2 selectively controls renal blood flow through a novel PPARβ/δ-dependent vasodilator Pathway. Hypertension 2018;71:297–305. CrossRef
18. Kjer J, Debbab A, Aly AH, Proksch P. Methods for isolation of marine-derived endophytic fungi and their bioactive secondary products. Nat Protoc. 2010;5(3):479–90.
19. Handayani D, Muslim RI, Syafni N, Artasasta MA, Riga R. Endophytic fungi from medicinal plant Garcinia cowa Roxb. ex Choisy and their antibacterial activity. J Appl Pharm Sci. 2024;14(9):182–8. CrossRef
20. Handayani D, Dwinatrana K, Rustini R. Antibacterial compound from marine sponge derived fungus Aspergillus sydowii DC08. Rasayan J Chem. 2022;15(4):2485–92.
21. Handayani D, Aminah I, Pontana Putra P, Eka Putra A, Arbain D, Satriawan H, et al. The depsidones from marine sponge-derived fungus Aspergillus unguis IB151 as an anti-MRSA agent: molecular docking, pharmacokinetics analysis, and molecular dynamic simulation studies. Saudi Pharm J. 2023;31(9):101744. CrossRef
22. Kilicaslan OS, Cretton S, Quir L, Bella MA, Kaiser M, Mäser P, et al. Isolation and structural elucidation of compounds from Pleiocarpa bicarpellata and their in vitro antiprotozoal activity. Molecules. 2022;27(7):2200.
23. Weng G, Gao J, Wang Z, Wang E, Hu X, Yao X, et al. Comprehensive evaluation of fourteen docking programs on protein-peptide complexes. J Chem Theory Comput. 2020;16(6):3959–69. CrossRef
24. Khachatryan H, Matevosyan M, Harutyunyan V, Gevorgyan S, Shavina A, Tirosyan I, et al. Computational evaluation and benchmark study of 342 crystallographic holo-structures of SARS-CoV-2 Mpro enzyme. Sci Rep. 2024;14(1):1–15. CrossRef
25. Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, et al. Structure basis for selective inhibitionof cyclooxygenase-2 by anti-inflammatory agents. Nat Publ Gr. 1996;384(19):644–8.
26. Akif M, Masuyer G, Bingham RJ, Sturrock ED, Isaac RE, Acharya KR. Structural basis of peptide recognition by the angiotensin-1 converting enzyme homologue AnCE from Drosophila melanogaster. FEBS J. 2012;279:4525–34. CrossRef
27. Zhang H, Unal H, Desnoyer R, Han GW, Patel N, Katritch V, et al. Structural basis for ligand recognition and functional selectivity at angiotensin receptor. JBS Pap. 2015;90(49):29127–39.
28. Alnajjar R, Mostafa A, Kandeil A, Al-Karmalawy AA. Molecular docking, molecular dynamics, and in vitro studies reveal the potential of angiotensin II receptor blockers to inhibit the COVID-19 main protease. Heliyon 2020;6(12):e05641.
29. Ahmed MZ, Hameed S, Ali M, Zaheer A. Computational analysis of the interaction of limonene with the fat mass and obesity-associated protein. Sci J Informatics. 2021;8(1):154–60. CrossRef
30. Daina A, Michielin O, Zoete V. SwissADME?: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017 3:7:42717. CrossRef
31. Hadni H, Elhallaoui M. Heliyon 3D-QSAR, docking and ADMET properties of aurone analogues as antimalarial agents. HLY. 2020;6(4):e03580. CrossRef
32. Menachery MD, Lavanier GL, Wetherly ML, Guinaudeau H, Shamma M. Simple isoquinoline alkaloids. J Natl Prod. 1986;49(5):745-78.
33. Silverstein RM, Webster FX, Kiemle DJ. Spectrometric identification of organic compounds. 7th ed. New York, NY: United States of America; 2005. 1–502 pp.
34. Rouessac F, Rouessac A. Chemical analysis: modern instrumentation methods and techniques. 2nd ed. Willey, UK: John Wiley & Sons Ltd; 2007. 1–574 pp.
35. Ohashi Y, Mamiya T, Mitani K, Wang B, Takigawa T, Kira S, et al. Simultaneous determination of urinary hippuric acid, o-, m- and p-methylhippuric acids, mandelic acid and phenylglyoxylic acid for biomonitoring of volatile organic compounds by gas chromatography-mass spectrometry. Anal Chim Acta. 2006;566(2):167–71.
36. Tang C, Ding P, Shi H, Jia Y, Zhou M, Yu H, et al. One-pot synthesis of phenylglyoxylic acid from racemic mandelic acids via cascade biocatalysis. J Agric Food Chem. 2019;67:2946–53. CrossRef
37. Aalinezhad S, Dabaghian F, Namdari A, Akaberi M, Emami SA. Phytochemistry and pharmacology of alkaloids from Papaver spp.: a structure–activity based study. Phytochem Rev. 2025;24:585–657. CrossRef
38. Jumayev I, Usmanov P, Rustamov S, Zhurakulov S. Comparative inotropic effects of the some isoquinoline alkaloids. Biomed Pharmacol J. 2020;13(1):325–333.
39. Jumayev IZ, Usmanov PB, Rustamov SY, Boboev SN, Ibragimov EB, Esimbetov AT. Evaluation of negative inotropic effects of an isoquinoline alkaloid N-14. Biomed Res Clin Rev. 2021;4(3):1–5.
40. Boboev SN, Zhumaev IZ, Usmanov PB, Shakhnoza B, Zhurakulov SN, Author C, et al. Effects of some isoquinoline alkaloids on cardiac muscle. West Eur J Med Med Sci. 2024;2(11):30–6.
41. Boboev SN, Zhumaev IZ, Usmanov PB, Yusubovich RS, Zhuraqulov SN. Effects of 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride alkaloid on cardiomyocyte Na+/Ca2+ exchange under normoxia and hypoxia conditions. Asian J Pharm Biol Res. 2023;12(3):1–10.
42. Li G, Kim T, Huang R, Hu D, Shi S, Wang H. Synthesis, anti-inflammatory and analgesia activities of diclofenac and their derivatives. Mater Express. 2022;1:865–70.
43. Paggi JM, Pandit A, Dror RO. The art and science of molecular docking. Annu Rev Biochem. 2024;93(April):389–410.
44. C S, S. DK, Ragunathanc V, Tiwari P, Sumitha A, P BD. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J Biomol Struct Dyn. 2020;40(2):585–611. CrossRef
45. Sharma U, Cozier GE, Sturrock ED, Acharya KR. Molecular basis for omapatrilat and sampatrilat binding to neprilysin - implications for dual inhibitor design with angiotensin-converting enzyme. J Med Chem. 2020;63:5488−500.
46. Chen J, Chen C, Zhang Z, Zeng F, Zhang S. Exploring the key amino acid residues surrounding the active center of lactate dehydrogenase a for the development of ideal inhibitors. Molecules. 2024;29(9):2029.
47. Daskaya-Dikmen C, Yucetepe A, Karbancioglu-Guler F, Daskaya H, Ozcelik B. Angiotensin-I-converting enzyme (ACE)-inhibitory peptides from plants. Nutrients. 2017;9(4):316. CrossRef
48. Benet LZ, Hosey CM, Ursu O, Oprea TI. BDDCS, the rule of 5 and drugability. Adv Drug Deliv Rev. 2016;101:89–98.
49. Girbane VM, Kedari RR, Munde RA. In silico docking and ADMET evaluation of bioactive compounds from Phyllanthus niruri and captopril as angiotensin-converting enzyme (ACE) inhibitors for hypertension management. Int J Sci Res Arch. 2025;14(01):423–33.
50. Guan L, Yang H, Cai Y, Sun L, Di P, Li W, et al. ADMET-score – a comprehensive scoring function for evaluation of chemical drug-likeness. R Soc Chem Med Chem Commun. 2018;10(1):148–57. CrossRef