
INTRODCUTION
The COVID-19 pandemic, caused by the novel SARS-CoV-2 virus, has emerged as a significant global health crisis [1]. The rapid transmission and severe pathogenic effects of the virus have overwhelmed healthcare systems and economies worldwide, highlighting the urgent need for effective treatments [2,3]. Traditional drug discovery methods are often time-consuming, often taking 10–15 years, and costly, billions of dollars, posing a significant challenge in responding promptly to such a crisis [4]. Despite this investment, the success rate is low, with many drugs failing in clinical trials due to efficacy or safety issues. These methods struggle to fully understand complex biological systems, relying on known chemical compounds, which limits innovation. Predicting safety and side effects is challenging, and the process is slowed by ethical and regulatory hurdles. Additionally, traditional methods may not be effective for complex or rare diseases. These limitations have spurred interest in new technologies like computational drug discovery, high-throughput screening, and AI-driven approaches to improve efficiency and success rates. Therefore, innovative approaches to drug discovery, such as in silico methods, have gained considerable attention for their potential to expedite the development of therapeutic interventions [5]. One of the most promising targets for antiviral drug development within coronaviruses is the main protease (Mpro) [6]. This enzyme plays a critical role in processing the polyproteins translated from the viral RNA, making it essential for viral replication and maturation [7]. Key residues within the active site of Mpro namely, His41, Cys44, Met49, Gly143, Ser144, Cys145, and Glu166 are crucial for its enzymatic function [8,9]. Inhibiting Mpro can effectively block the replication of the virus, thereby halting the progression of the infection [7]. E-pharmacophore virtual screening is a powerful computational technique employed in drug discovery to identify potential drug candidates from extensive chemical libraries or databases [10,11]. This approach leverages the three-dimensional structural information of target proteins and the physicochemical properties of ligands to predict binding affinity and specificity [11,12]. Drug repurposing, also known as drug repositioning, is the process of identifying new therapeutic uses for existing drugs. This approach employs previously approved medications, which can accelerate the drug development process since these drugs have already been tested for safety in humans [13]. In the context of the COVID-19 pandemic, repurposing existing FDA-approved drugs for new therapeutic indications offers a pragmatic and efficient strategy. This approach significantly reduces the time and cost associated with traditional drug development, as these compounds have already undergone extensive testing for safety and efficacy. This manuscript explores the application of e-pharmacophore virtual screening in identifying potential inhibitors of the SARS-CoV-2 main protease from a library of FDA-approved drugs. The findings of the current study provide insights into promising drug candidates that could be repurposed to combat COVID-19, thereby contributing to the global effort to control the pandemic.
MATERIALS AND METHODS
All computational studies, including protein and ligands’ preparation, e-pharmacophore modeling, virtual screening, MM-GBSA calculations, and molecular dynamics (MD) simulations, were executed using Maestro interface incorporated in Schrodinger suite (version 2023.1) (Schrödinger, LLC, New York, NY, 2023). A dataset comprising 2,619 FDA-approved drugs was obtained from the DrugBank database [14] (accessed in January 2024).
Protein and ligands preparations
Using the Maestro interface, all FDA-approved drugs, the reference inhibitor 3WL (Fig. 1), and the target enzyme 6M2N were prepared. Ligands’ preparation was carried out using the LigPrep tool (Ligprep, Schrödinger, LLC, New York, NY, 2023). This process generated and minimized 3D structures, including all possible tautomers and ionization states at pH 7.0 ± 2.0 for the drugs and 3WL, employing the OPLS4 force field. Protein preparation utilized Schrödinger’s multistep Protein Preparation Wizard (PrepWizard, Schrödinger, LLC, New York, NY, 2023). The high-resolution crystal structure of the SARS-CoV-2 main protease (Mpro) (PDB: 6M2N) at 2.2 Å resolution, bound to 3WL, was initially retrieved from the RCSB Protein Data Bank [15]. The process included adding hydrogen atoms, assigning bond orders, creating zero-order bonds to metals, forming disulfide bonds, removing water molecules beyond 5 Å from hetero groups, and capping the termini. Hydrogen bonding networks were optimized, and the orientations of water molecules were sampled. Finally, the structures were refined by minimizing them with the OPLS4 force field, restraining nonhydrogen atoms to a root-mean-square deviation (RMSD) of 0.3 Å. The co-crystallized ligand (3WL) was retained in the active site throughout the preparation process. A 3D cubic grid box was constructed around the co-crystal ligand using the receptor grid generation tool in the Maestro suite.
Generation of e-pharmacophore model and pharmacophore-based virtual screening
The X-ray crystal-bound ligand, 3WL, was utilized to generate energy-optimized pharmacophore hypotheses through the “Develop a Pharmacophore from Receptor-Ligand Complex” option in the Phase module [16] (Phase, Schrödinger, LLC, New York, NY, 2023). The bound ligand was automatically identified and selected, with all default parameters retained. Five pharmacophore sites were predicted, resulting in a final hypothesis consisting of two aromatic moieties, one heteroatom, one hydrogen bond donor, and one hydrogen bond acceptor, as illustrated in Figure 2. The pharmacophore-based screening was conducted against a LigPrep-prepared library of FDA-approved drugs sourced from the DrugBank database to identify drugs with the desired chemical features using the Phase module of Schrodinger. A minimum match of four sites on the generated e-pharmacophore hypothesis was required for the drugs. The phase fitness score, which measures how well the ligands match the chemical features of the pharmacophore sites based on vector alignments, volume terms, and RMSD site matching, was used to rank the final drugs obtained from the virtual screening [17].
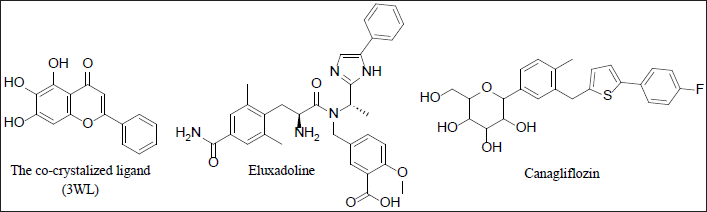 | Figure 1. Chemical structures of the co-crystalized ligand, Eluxadoline, and Canagliflozin. [Click here to view] |
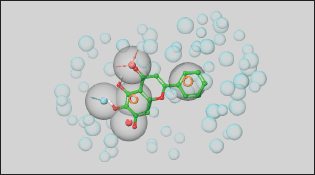 | Figure 2. The generated pharmacophore model. [Click here to view] |
Molecular docking
A grid was generated around the native ligand 3WL to define the binding site, using default settings. The Glide XP (extra precision) module of the Schrödinger Suite (Glide, Schrödinger, LLC, New York, NY, 2023) was then used to perform docking into the active site of the crystal structure [18]. To validate the docking protocol, 3WL was re-docked into the prepared binding site of Mpro. The best-scoring docked pose of 3WL was superimposed on the X-ray crystal conformation of the bound ligand to calculate the RMSD [16,19].
MM-GBSA free binding energy calculations
In this study, the the Molecular Mechanics with Generalized Born and Surface Area (MM-GBSA) calculations were employed as a post-docking validation protocol. The Prime MM-GBSA method, integrated with the Maestro module of the Schrödinger suite (Prime, Schrödinger, LLC, New York, NY, 2023), was used for energy calculations. All calculations were performed using the default parameters [20].
MD simulations
The complexes of the co-crystallized ligand 3WL, Eluxadoline, and Canagliflozin with the Mpro enzyme (PDB: 6M2N) underwent MD simulations using the Desmond Schrödinger program (Desmond, Schrödinger, Schrödinger, LLC, New York, NY, 2023). The system construction wizard was initiated by selecting the SPC solvent model and the OPLS4 force field, creating an orthorhombic box with dimensions of 10 × 10 × 10. The required number of ions was calculated and added to neutralize the system, and a salt concentration of 0.15 M was included to mimic physiological conditions. To prevent steric clashes, the system builder output was employed to perform a minimization step for the entire complex and solvent system for 100 ps using the steepest descent method. Finally, an MD simulation was conducted in an isothermal-isobaric ensemble (NPT mode) with a constant number of particles for 100 ns.
RESULTS AND DISCUSSION
The SARS-CoV-2 virus, responsible for COVID-19, emerged in late 2019 and swiftly became a global pandemic, causing millions of infections and deaths [21]. Its rapid spread overwhelmed healthcare systems, leading to significant health crises and economic disruptions, including business closures and job losses [22]. Public health measures such as lockdowns and social distancing had profound impacts on daily life and mental health [23]. The pandemic prompted unprecedented global collaboration in vaccine development and therapeutic research, underscoring the importance of robust public health infrastructure and preparedness [24]. The continuous emergence of new variants and the issue of “Long COVID-19” highlight the ongoing need for effective prevention, treatment, and equitable vaccine access [25,26]. Drug targets for COVID-19 are not just essential but pivotal in advancing understanding of the disease and in developing effective therapeutic interventions to combat the pandemic. They provide the critical foundation for targeted treatments that can disrupt viral replication, modulate the immune response, and minimize side effects, ultimately aiming to reduce transmission rates, hospitalizations, and mortality, and alleviate the burden on healthcare systems worldwide [27]. The SARS-CoV-2 main protease (Mpro, also known as 3CLpro) is one of the most well-characterized drug targets among coronaviruses. Inhibiting the activity of this enzyme would effectively block viral replication. Interestingly, no human proteases with similar cleavage specificity are known, reducing the likelihood of toxicity from inhibitors [28]. The literature review revealed that a substantial number of papers have identified the main protease of SARS-CoV-2 as a promising target for in silico drug discovery for COVID-19 [6,7]. Millions of compounds, including known drugs [29], synthetic molecules [30], and natural products [31], were screened using various molecular modeling tools, resulting in numerous candidates demonstrating potential anti-SARS-CoV-2 activity. Additionally, bioinformatics tools were employed to evaluate the ADMET profiles of some of these inhibitors [32]. The analysis of binding affinities and interactions suggested that several FDA-approved drugs could be repurposed as potential treatments for COVID-19 [33]. In this study, Mpro was selected as the target for COVID-19 drug discovery using a computational approach that included E-pharmacophore-based virtual screening, molecular docking, MM-GBSA calculations, and MD simulations’ studies. The crystal structure of Mpro bound to a novel inhibitor (PDB: 6M2N) was downloaded from the Protein Data Bank and prepared using default parameters in the Prime module within the Maestro interface of Schrödinger [20]. Next, Schrödinger’s Phase module [16] was utilized to generate a pharmacophore from the protein-ligand complex, extracting five features: two aromatic moieties, one heteroatom, one hydrogen bond donor group, and one hydrogen bond acceptor group (Fig. 2). The generated pharmacophore model was used to perform virtual screening against a library of FDA-approved drugs [14] with the aid of Schrödinger’s Glide module [18]. Out of a total of 2619 FDA-approved drugs, 308 were identified to match the pharmacophore model generated in the study. These drugs exhibit chemical features and spatial arrangements similar to those specified in the pharmacophore model, suggesting they could potentially interact with the target protein (Mpro) similar to the known inhibitor. This similarity makes them promising candidates for further investigation as potential treatments for COVID-19. Therefore, the next step involved docking each of the 308 drugs into the binding site of Mpro to select the best candidates based on their docking scores. However, before proceeding, it was necessary to validate the docking protocol first. To this end, the co-crystallized ligand was removed from the binding site and re-docked using the Glide module of Schrödinger. The result of the XP docking showed that the docking protocol was valid, with an RMSD found to be as small as 0.47 Å [19] (Fig. 3). Following this validation, XP molecular docking was performed for each of the 308 drugs against the binding site of Mpro. These drugs were then ranked according to the obtained docking scores, which indicated their potential affinity to the target protein. This ranking helped identify the most promising drug candidates to advance to the next step, which involved free-binding energy calculations. In experiments, co-crystallized ligands are molecules known to effectively bind to a target protein’s active site, verified through crystallography. Using their docking scores as benchmarks predicts the binding potential of new ligands. Lower scores indicate stronger binding, identifying promising candidates for further investigation. This approach accelerates drug discovery by prioritizing compounds with potential biological activity and optimizing screening efforts efficiently. Thus, based on initial considerations, the docking score of the redocked co-crystallized ligand is a cutoff point to select the best drug candidates for MM-GBSA calculations. Specifically, all drugs exhibiting a docking score more negative than −7.849 kcal/mol were chosen for the next step, as a more negative docking score typically indicates a stronger predicted binding affinity to the target protein [34]. In this context, only three drugs—Pitavastatin, Canagliflozin (Fig. 1), and Fluvastatin—were found to have a stronger affinity for Mpro compared to the co-crystallized ligand, with docking scores of −8.839, −8.513, −7.859, and −7.849 kcal/mol, respectively (Table 1).
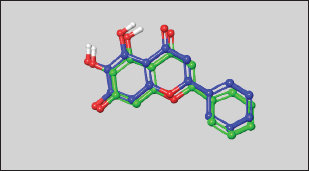 | Figure 3. The superposition of the docked ligand (blue) on the co-crystalized ligand (green). [Click here to view] |
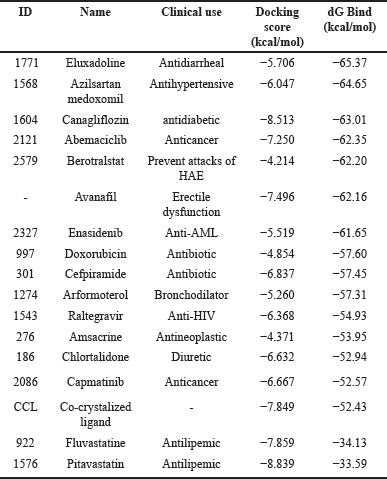 | Table 1. XP docking scores and MM-GBSA binding free energies of the co-crystalized ligand (3WL) and the top-ranked FDA-approved drugs with SARS-CoV-2 main protease (Mpro) (PDB: 6N2N). [Click here to view] |
MM-GBSA calculations on the complexes of these drugs with Mpro revealed that only Canagliflozin had better binding energy compared to the co-crystallized ligand. Specifically, Pitavastatin and Fluvastatin demonstrated weaker binding to the active site of Mpro, with binding energies of −33.59 and −34.13 kcal/mol, respectively, compared to the co-crystallized ligand’s −52.43 kcal/mol. In contrast, Canagliflozin exhibited a binding energy of −63.01 kcal/mol, indicating it bound the target protein more strongly than that of the co-crystallized ligand [35]. The discrepancy between the docking scores of Pitavastatin and Fluvastatin and their MM-GBSA binding free energies strengthens the assumption that molecular docking may yield false-positive results [36]. Thus, it became apparent that relying solely on docking scores to select the best drugs for MM-GBSA calculations was not advisable. Therefore, MM-GBSA calculations were conducted, using the Prime module of Schrödinger, on all 308 drugs retrieved through e-pharmacophore-based virtual screening. By this time, it was found that the initial cutoff point (docking score of the co-crystallized ligand) was not suitable. Consequently, the binding free energy exhibited by the co-crystallized ligand was chosen as another cutoff point. The MM-GBSA energy of the co-crystallized ligand plays a critical role in drug discovery as it quantitatively measures the binding affinity between a specific ligand and its target protein through computational simulations. This provides a comprehensive estimation of the ligand’s stability within the protein’s active site. Establishing the MM-GBSA energy of the co-crystallized ligand as a cutoff allows researchers to efficiently screen potential drug candidates. Ligands with energies comparable to or lower than that of the co-crystallized ligand are prioritized, indicating stronger and more specific binding to the target protein. In this regard, 14 drugs exhibited more negative binding energies than the co-crystallized ligand, with Eluxadoline (Fig. 1) being the top-ranked (−65.37 kcal/mole) (Table 1). Following this, Schrödinger’s Desmond module was used to conduct MD simulations for 100 nanoseconds on the complexes of Eluxadoline and the co-crystallized ligand with Mpro. These simulations allowed for the exploration of protein-ligand interactions, stability, and conformational changes [37]. The findings from the MD simulations revealed Eluxadoline’s potential as a SARS-CoV-2 main protease inhibitor, demonstrating parameters almost comparable to those of the co-crystallized ligand and superior to those of Canagliflozin. The Mpro complexed with Eluxadoline demonstrated RMSD values with a maximum of 3.2Å, a minimum of 1.0Å, and an average of 2.4Å. In comparison, the 3WL-Mpro complex revealed RMSD values with a maximum of 3.2Å, a minimum of 1.2Å, and an average of 1.9Å. Both Eluxadoline and 3WL (the co-crystallized ligand) appeared to have a stabilizing effect on the Mpro structure, whereas the Apo (unbound protein) form of Mpro exhibited higher fluctuation with a maximum of 4.4Å, a minimum of 1.1Å, and an average of 2.6Å (Fig. 4) (Table 2).
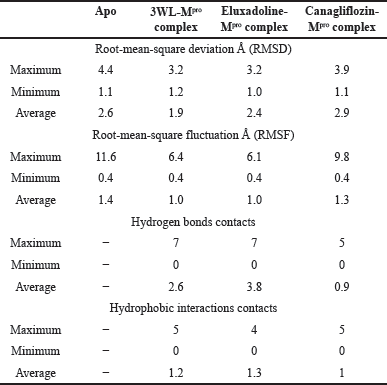 | Table 2. The maximum, minimum, and average values of different parameters, RMSD, RMSF, hydrogen bonds contacts, and hydrophobic interactions contacts of the co-crystalized ligand (3WL), Eluxadoline, and Canagliflozin complexed with SARS-CoV-2 main protease (Mpro) (PDB: 6N2N). [Click here to view] |
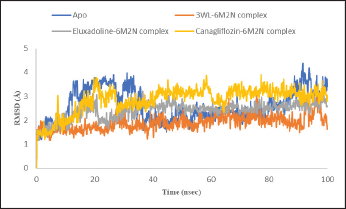 | Figure 4. RMSD plot of Mpro Apo protein (PDB: 6M2N) and Mpro (PDB: 6M2N) bound with the co-crystalized ligand (3WL), Eluxadoline and Canagliflozin versus time of the simulation. [Click here to view] |
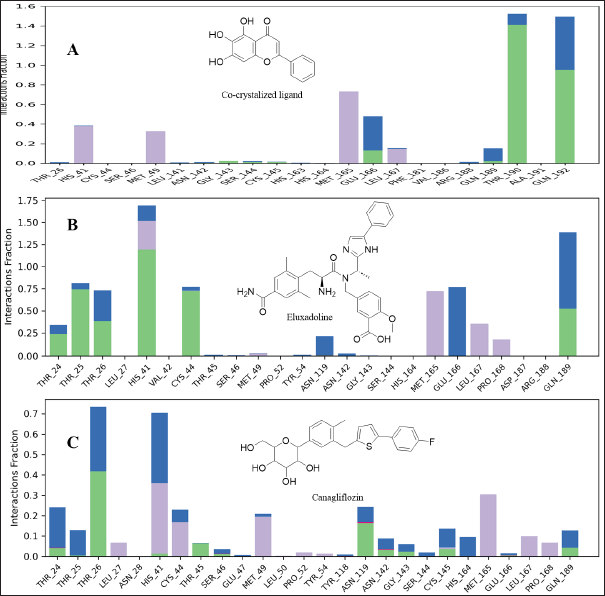 | Figure 5. Interaction Mpro residues with the co-crystalized ligand (A), Eluxadoline (B), and Canagliflozin (C). Different interactions were observed: hydrogen bonds (green); Water bridges (blue), hydrophobic interactions (grey); and ionic interactions (red). [Click here to view] |
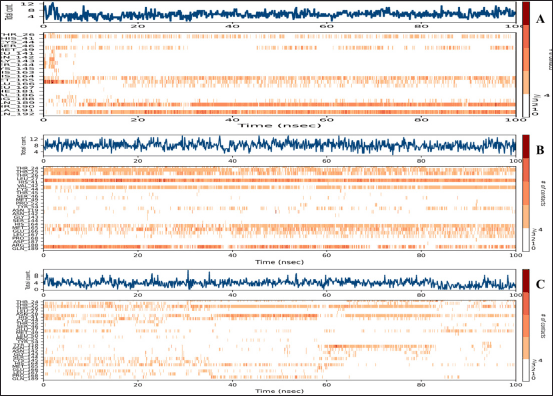 | Figure 6. A timeline representation of the interactions and contacts (H-bonds, Hydrophobic, Ionic, Water bridges) of Mpro residues with the co-crystalized ligand (A), Eluxadoline (B), and Canagliflozin (C). The top panel shows the total number of specific contacts the protein makes with the ligand over the course of the trajectory. The bottom panel shows which residues interact with the ligand in each trajectory frame. Some residues make more than one specific contact with the ligand, which is represented by a darker shade of orange, according to the scale to the right of the plot. [Click here to view] |
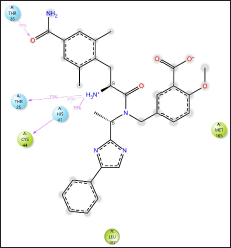 | Figure 7. Eluxadoline interactions with Mpro residues. Only interactions that occur more than 30.0% of the simulation time in the selected trajectory (0.00 through 100.00 nsec), are shown. [Click here to view] |
The maximum deviation in the Eluxadoline-Mpro complex is the same as in the 3WL-Mpro complex (3.2Å). However, the minimum deviation is slightly lower (1.0Å vs. 1.2Å), and the average deviation is slightly higher (2.4Å vs. 1.9Å). These findings suggest that Eluxadoline, similar to the co-crystallized ligand 3WL, stabilizes the Mpro structure, reducing its fluctuations. In contrast, the unbound Mpro protein (Apo) exhibits greater structural variability. This stabilizing effect of the ligands is demonstrated through lower and more consistent RMSD values compared to the higher RMSD values of the Apo form. Data presented in Table 2 clearly indicate that the Canagliflozin-Mpro complex exhibited lower stability compared to both the co-crystallized ligand and Eluxadoline. The maximum RMSD of 3.9 shows the largest deviation observed, the minimum RMSD of 1.1 shows the smallest deviation, and the average RMSD of 2.9 shows the typical deviation across the simulation. The differences between the maximum and minimum RMSD values for the complexes suggest varying degrees of stability and flexibility in the binding of Eluxadoline and Canagliflozin to the SARS-CoV-2 main protease. Eluxadoline showed a narrower range (2.2Å), indicating more consistent binding, whereas Canagliflozin exhibited a wider range (2.8Å), potentially suggesting less stable interactions [38]. As mentioned earlier, RMSD is a critical metric in MD simulations, assessing the stability and accuracy of molecular structures over time by measuring the average distance between simulated and reference structures. It indicates structural stability, detects conformational changes, validates simulation quality, compares molecular states, and provides insights into biomolecular dynamics and behavior. The lower RMSD values observed in the Eluxadoline-Mpro complex suggest stable structure maintenance throughout the simulation, indicating effective binding of Eluxadoline to the target protein with minimal deviations. This stability reflects a stronger and more stable interaction between Eluxadoline and Mpro, implying potential inhibitory properties of Eluxadoline against Mpro. Throughout the simulation, Eluxadoline, Canagliflozin, and the co-crystallized ligand remained bound to Mpro (PDB: 6M2N) binding site, forming contacts with 24, 26, and 22 residues, respectively (Fig. 5A-C). Eluxadoline consistently maintained more than four persistent contacts with key residues His41, Cys44, and Glu166. In contrast, the interactions of Canagliflozin and the co-crystallized ligand with these residues were less stable (Fig. 6A-C). The stability of the Eluxadoline-Mpro complex observed during MD simulations can be attributed in part to the formation of hydrogen bonds with residues His41 and Cys44, which persisted for up to 79% of the simulation time (Fig. 7). On average, Eluxadoline formed 3.8 hydrogen bonds with different residues in the Mpro binding site (Fig. 8) (Table 2). In contrast, neither the co-crystallized ligand nor Canagliflozin exhibited stable hydrogen bonds lasting for more than 25% of the simulation time with any residues in the Mpro active site. Their overall averages were 2.6 and 0.9 hydrogen bonds, respectively (Table 2). Hydrophobic interactions played a crucial role in the relative stability of the Eluxadoline-Mpro complex compared to both the co-crystallized ligand and Canagliflozin, with average values of 1.3, 1.2, and 1, respectively [39] (Fig. 9) (Table 2). Additionally, water-mediated hydrogen bonds, known as water bridges, were observed between Eluxadoline and several residues in the Mpro binding site, including the key residue Glu166 (Fig. 5). The stability of interactions between Eluxadoline and Mpro during the simulation was reflected in reduced RMSF values observed for Mpro amino acid residues [40]. The overall average RMSF value for the Eluxadoline-Mpro complex was comparable to that of the co-crystallized ligand-Mpro complex, approximately 1Å (Table 2) (Figure 10). In contrast, RMSF values for Apo protein residues averaged 1.4Å, similar to those observed for the Canagliflozin-Mpro complex (1.3Å). Reduced RMSF values indicate that Eluxadoline binds tightly to the protein binding site, reducing the atomic displacements of residues from their average positions and reflecting a stabilized and less flexible protein-ligand complex. As noted earlier, MM-GBSA calculations revealed that the Eluxadoline-Mpro complex exhibited a binding free energy of −65.37 kcal/mol, compared to −63.01 kcal/mol exhibited by the Canagliflozin-Mpro complex (Table 1). These findings, supported by both MD simulations and MM-GBSA calculations, demonstrate the strong binding affinity of Eluxadoline to the SARS-CoV-2 main protease (Mpro). Based on this comprehensive analysis, Eluxadoline emerges as a promising candidate for repurposing as an Mpro inhibitor. While computational studies offer valuable insights into molecular interactions and dynamics, they are limited by factors such as the accuracy of force fields, simulation parameters, and the simplification of complex biological environments, all of which can impact results. Therefore, it is essential to complement computational findings with experimental validations conducted in vitro (in controlled laboratory settings) and in vivo (within living organisms). In vitro experiments enable detailed analysis of molecular interactions under controlled conditions, while in vivo studies provide crucial insights into biological systems, including metabolism, toxicity, and efficacy. These combined experimental approaches provide robust validation of computational predictions, bolstering confidence in the potential therapeutic applications of molecules like Eluxadoline against targets such as Mpro. Interestingly, Eluxadoline has been used to treat abdominal cramps associated with COVID-19 and has shown good efficacy in alleviating abdominal disturbances [41,42]. The findings of the current study suggest that Eluxadoline might also play a role in eradicating the virus.
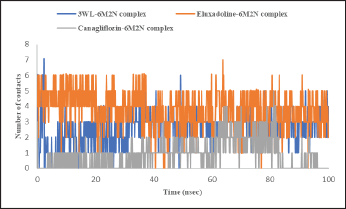 | Figure 8. Time-dependent hydrogen bond analysis of Mpro (PDB: 6M2N) bound with the co-crystalized ligand (3WL), Eluxadoline, and Canagliflozin calculated from MD simulations trajectories. [Click here to view] |
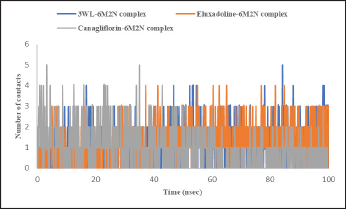 | Figure 9. Time-dependent hydrophobic interactions analysis of Mpro (PDB: 6M2N) bound with the co-crystalized ligand (3WL), Eluxadoline, and Canagliflozin calculated from MD simulations trajectories. [Click here to view] |
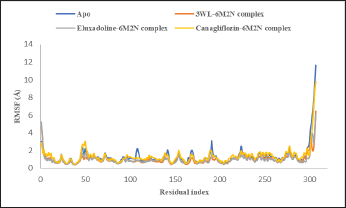 | Figure 10. RMSF plot of Mpro Apo protein (PDB: 6M2N) and Mpro (PDB: 6M2N) bound with the co-crystalized ligand (3WL), Eluxadoline and Canagliflozin versus time of the simulation. [Click here to view] |
CONCLUSION
In conclusion, a computational approach, combining e-pharmacophore-based virtual screening, molecular docking, MM-GBSA calculations, and MD simulations’ studies, has successfully identified Eluxadoline as a potential inhibitor of the SARS-CoV-2 main protease. These findings shed light on the molecular interactions driving Eluxadoline’s inhibitory activity and highlight the promise of repurposing existing drugs to combat emerging viral threats such as SARS-CoV-2.
ACKNOLWEDGMENT
T.E. acknowledges Karthi Niranjan from Schrödinger, LLC. for the one-month free trial.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Atzrodt CL, Maknojia I, McCarthy RDP, Oldfield TM, Po J, Ta KTL, et al. A guide to COVID-19: a global pandemic caused by the novel coronavirus SARS-CoV-2. FEBS J. 2020;287(17):3633-50. CrossRef
2. Sindhuja T, Kumari R, Kumar A. Epidemiology, transmission and pathogenesis of SARS-CoV-2. In: Arpana Parihar, Raju Khan, Ashok Kumar, Ajeet Kumar Kaushik, Hardik Gohel editors. Computational Approaches for Novel Therapeutic and Diagnostic Designing to Mitigate SARS-CoV-2 Infection. 2022. 1st ed. Amsterdam, The Netherlands: Elsevier Inc; 2022.
3. Batista C, Shoham S, Ergonul O, Hotez P, Bottazzi ME, Figueroa JP, et al. Urgent needs to accelerate the race for COVID-19 therapeutics. EClinicalMedicine. 2021;36:100911. CrossRef
4. Vora LK, Gholap AD, Jetha K, Thakur RRS, Solanki HK, Chavda VP. Artificial intelligence in pharmaceutical technology and drug delivery design. Pharmaceutics. 2023;15(7):1916. CrossRef
5. Shaker B, Ahmad S, Lee J, Jung C, Na D. In Silico methods and tools for drug discovery. Comput Biol Med. 2021;137:104851. CrossRef
6. Eissa IH, Saleh AM, Al-Rashood ST, El-Attar A-AMM, Metwaly AM, Penoni A. Multistaged in silico discovery of the best SARS-CoV-2 main protease inhibitors amongst 3009 clinical and FDA-approved compounds. J Chem. 2024;2024:1-19. CrossRef
7. Pang X, Xu W, Liu Y, Li H, Chen L. The research progress of SARS-CoV-2 main protease inhibitors from 2020 to 2022. Eur J Med Chem. 2023;257:115491. CrossRef
8. Antonopoulou I, Sapountzaki E, Rova U, Christakopoulos P. Inhibition of the main protease of SARS-CoV-2 (Mpro) by repurposing/designing drug-like substances and utilizing nature’s toolbox of bioactive compounds. Comput Struct Biotechnol J. 2022;20:1306-44. CrossRef
9. Tumskiy RS, Tumskaia AV, Klochkova IN, Richardson RJ. SARS-CoV-2 proteases Mpro and PLpro: design of inhibitors with predicted high potency and low mammalian toxicity using artificial neural networks, ligand-protein docking, molecular dynamics simulations, and ADMET calculations. Comput Biol Med. 2023;153:106449. CrossRef
10. Giordano D, Biancaniello C, Argenio MA, Facchiano A. Drug design by pharmacophore and virtual screening approach. Pharmaceuticals. 2022;15(5):646. CrossRef
11. Leach AR, Gillet VJ, Lewis RA, Taylor R. Three-dimensional pharmacophore methods in drug discovery. J Med Chem. 2010;53(2):539-58. CrossRef
12. Chang C, Ekins S, Bahadduri P, Swaan PW. Pharmacophore-based discovery of ligands for drug transporters. Adv Drug Deliv Rev. 2006;58(12-13):1431-50. CrossRef
13. Krishnamurthy N, Grimshaw AA, Axson SA, Choe SH, Miller JE. Drug repurposing: a systematic review on root causes, barriers and facilitators. BMC Health Serv Res. 2022;22(1):970. CrossRef
14. Knox C, Wilson M, Klinger CM, Franklin M, Oler E, Wilson A, et al. DrugBank 6.0: the DrugBank Knowledgebase for 2024. Nucleic Acids Res. 2024;52(D1):D1265-75. CrossRef
15. Su HX, Yao S, Zhao WF, Li MJ, Liu J, Shang WJ, et al. Anti-SARS-CoV-2 activities in vitro of shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol Sin. 2020;41(9):1167-77. CrossRef
16. Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA. PHASE: a new engine for pharmacophore perception, 3D QSAR Model Development, and 3D Database Screening: 1. Methodology and preliminary results. J Comput Aided Mol Des. 2006;20(10-11):647-71. CrossRef
17. Mafethe O, Ntseane T, Dongola TH, Shonhai A, Gumede NJ, Mokoena F. Pharmacophore model-based virtual screening workflow for discovery of inhibitors targeting Plasmodium falciparum Hsp90. ACS Omega. 2023;8(41):38220-32. CrossRef
18. Yang Y, Yao K, Repasky MP, Leswing K, Abel R, Shoichet BK, et al. Efficient exploration of chemical space with docking and deep learning. J Chem Theory Comput. 2021;17(11):7106-19. CrossRef
19. Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739-49. CrossRef
20. Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, et al. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55(2):351-67. CrossRef
21. Muralidar S, Ambi SV, Sekaran S, Krishnan UM. The emergence of COVID-19 as a alobal pandemic: understanding the epidemiology, immune response and potential therapeutic targets of SARS-CoV-2. Biochimie. 2020;179:85-100. CrossRef
22. Moghadas SM, Shoukat A, Fitzpatrick MC, Wells CR, Sah P, Pandey A, et al. Projecting hospital utilization during the COVID-19 outbreaks in the United States. Proc Natl Acad Sci. 2020;117(16):9122-6. CrossRef
23. Brooks SK, Webster RK, Smith LE, Woodland L, Wessely S, Greenberg N, et al. The psychological impact of quarantine and how to reduce it: rapid review of the evidence. Lancet. 2020;395(10227):912-20. CrossRef
24. Thomson EC, Rosen LE, Shepherd JG, Spreafico R, da Silva Filipe A, Wojcechowskyj JA, et al. Circulating SARS-CoV-2 Spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell. 2021;184(5):1171-87. CrossRef
25. Chan JF-W, Yuan S, Chu H, Sridhar S, Yuen K-Y. COVID-19 drug discovery and treatment options. Nat Rev Microbiol. 2024;22(4):391–407. CrossRef
26. Turatto F, Sassano M, Goletti M, Severoni S, Grossi A, Parente P. Ensuring equitable access to the COVID-19 vaccine: the experience of a local health unit in Rome, Italy. Healthcare. 2022;10(11):2246. CrossRef
27. Elfiky AA. SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp) Targeting: an in silico perspective. J Biomol Struct Dyn. 2021;39(9):3204-12. CrossRef
28. Almaraz-Giron MA, Calderon-Jaimes E, Carrillo AS, Diaz-Cervantes E, Alonso EC, Islas-Jacome A, et al. Search for non-protein protease inhibitors constituted with an indole and acetylene core. Molecules. 2021;26(13):3817. CrossRef
29. Rodrigues L, Bento Cunha R, Vassilevskaia T, Viveiros M, Cunha C. Drug repurposing for COVID-19: a review and a novel strategy to identify new targets and potential drug candidates. Molecules. 2022;27(9):2723. CrossRef
30. Stalinskaya AL, Martynenko NV, Shulgau ZT, Shustov AV, Keyer VV, Kulakov IV. Synthesis and antiviral properties against SARS-CoV-2 of Epoxybenzooxocino[4,3-b]Pyridine Derivatives. Molecules. 2022;27(12):3701. CrossRef
31. Chapman RL, Andurkar SV. A review of natural products, their effects on SARS-CoV-2 and their utility as lead compounds in the discovery of drugs for the treatment of COVID-19. Med Chem Res. 2022;31(1):40-51. CrossRef
32. Srivastava V, Yadav A, Sarkar P. Molecular docking and ADMET study of bioactive compounds of Glycyrrhiza glabra against main protease of SARS-CoV-2. Mater Today Proc. 2022;49:2999-3007. CrossRef
33. Ray AK, Sen Gupta PS, Panda SK, Biswal S, Bhattacharya U, Rana MK. Repurposing of FDA-approved drugs as potential inhibitors of the SARS-CoV-2 main protease: molecular insights into improved therapeutic discovery. Comput Biol Med. 2022;142:105183. CrossRef
34. Pantsar T, Poso A. Binding affinity via docking: fact and fiction. Molecules. 2018;23(8):1899. CrossRef
35. Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10(5):449-61. CrossRef
36. Elsaman T, Ahmad I, Eltayib EM, Suliman Mohamed M, Yusuf O, Saeed M, et al. Flavonostilbenes natural hybrids from Rhamnoneuron balansae as potential antitumors targeting ALDH1A1: molecular docking, ADMET, MM-GBSA calculations and molecular dynamics studies. J Biomol Struct Dyn. 2024;42(6):3249-66. CrossRef
37. Challapa-Mamani MR, Tomas-Alvarado E, Espinoza-Baigorria A, Leon-Figueroa DA, Sah R, Rodriguez-Morales AJ, et al. Molecular docking and molecular dynamics simulations in related to Leishmania donovani: an update and literature review. Trop Med Infect Dis. 2023;8(10):457. CrossRef
38. Ghahremanian S, Rashidi MM, Raeisi K, Toghraie D. Molecular dynamics simulation approach for discovering potential inhibitors against SARS-CoV-2: a structural review. J Mol Liq. 2022;354:118901. CrossRef
39. Kandeel M, Al-Nazawi M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020;251:117627. CrossRef
40. Chatterjee S, Maity A, Chowdhury S, Islam MA, Muttinini RK, Sen D. in silico analysis and identification of promising hits against 2019 novel coronavirus 3C-like main protease enzyme. J Biomol Struct Dyn. 2021;39(14):5290-303. CrossRef
41. Chan WW, Grover M. The COVID-19 pandemic and post-infection irritable bowel syndrome: what lies ahead for gastroenterologists. Clin Gastroenterol Hepatol. 2022;20(10):2195-7. CrossRef
42. Brenner DM, Sayuk GS, Gutman CR, Jo E, Elmes SJR, Liu LWC, et al. Efficacy and safety of eluxadoline in patients with irritable bowel syndrome with diarrhea who report inadequate symptom control with loperamide: RELIEF phase 4 study. Am J Gastroenterol. 2019;114(9):1502-11. CrossRef