
INTRODUCTION
The second most common cancer among men worldwide, according to fatality rates, is prostate cancer (PC). The World Health Organization estimates that 1.4 million new instances of PC will be diagnosed globally in 2022, making up roughly 7.3% of all new cases of cancer in males. In addition, PC was responsible for nearly 375,000 deaths, or 3.8% of all fatalities in men from cancer. Numerous genetic and molecular variables affect PC [1]. The androgen receptor, Androgen Receptor (AR), a nuclear hormone receptor that binds to androgens [such as testosterone and dihydrotestosterone (DHT)] and controls gene expression in prostate cells, is one of the class of enzymes involved in molecular pathways that contribute to the onset and progression of the cancer [2]. Although it also plays a significant role in PC, AR signaling is necessary for healthy prostate growth and function [3]. The AR pathway can become hyperactivated in PC cells, causing unchecked growth and survival of cancer cells. The main therapeutic approach used for advanced PC is androgen deprivation therapy [4,5]. DHT, a more potent androgen, is created when testosterone is converted by the 5-alpha reductase enzyme. DHT has a stronger affinity for the androgen receptor and is essential for the growth and proliferation of both healthy prostate cells and PC cells. Finasteride and dutasteride are 5-alpha reductase inhibitors that are used to treat benign prostatic hyperplasia and have also been investigated for the prevention of PC [6–8].
Matrix metalloproteinases (MMPs), on the other hand, were a family of enzymes involved in the disintegration of extracellular matrix constituents. Some MMPs, including MMP-2 and MMP-9, have been discovered to be elevated in PC. Increased MMP production encourages the breakdown of the extracellular matrix, which can aid tumor invasion, metastasis, and angiogenesis. Poly (ADP-ribose) polymerase (PARP) enzymes are also said to be involved in DNA repair [9,10]. PARP inhibitors have demonstrated potential as a targeted treatment for patients with particular genetic abnormalities, such as BRCA1/2 mutations, in PC. These inhibitors stop the repair of DNA damage, which results in the buildup of DNA mistakes and the death of cancer cells. In addition, digital rectal examinations and blood tests for prostate-specific antigen are frequently used for PC screening. PC patients frequently have increased PSA levels, and the enzyme is necessary for the liquefaction of semen. Although its efficacy in screening is debatable because raised PSA levels can sometimes occur owing to noncancerous diseases, monitoring PSA levels can aid in the diagnosis, staging, and development of PC [11–14].
The aldo-keto reductase family member AKR1C3 catalyzes the conversion of less potent hormones to their more potent counterparts by driving nuclear receptor activation [15]. It is also known as 17-hydroxysteroid dehydrogenase type 5. Dehydroepiandrosterone, a weak androgen, is converted by the enzyme AKR1C3 into the more potent androgens testosterone and DHT, which can promote the proliferation of PC cells. It has been discovered that castration-resistant PC patients have elevated AKR1C3 expression, making them prospective targets. Non-steroidal anti-inflammatory drugs (NSAIDs), benzodiazepines, jasmonates, cinnamic acids, and compounds containing flavonoids are among the few AKR1C3 inhibitors that have been documented [16]. However, the demand for AKR1C3 inhibitors with nanomolar/sub-nano molar range of efficacy remains. To target the PC enzyme AKR1C3, we tried to create an effective, stable, and reliable model with strong forecasting of upcoming potent compounds.
Aldo-keto reductase 1C2 contributes to androgen metabolism in addition to AKR1C3. It has been discovered to be elevated in PC, notably in Castrate resistant prostate cancer (CRPC), and it is involved in the conversion of androstenedione to testosterone. Increased AKR1C2 expression may support androgen receptor activation and intratumoral androgen production, which favors PC cell proliferation and survival. Aldo-keto reductase 1B10, another isozyme, is similarly overexpressed in a number of malignancies, including PC. It contributes to the metabolism of carbonyl compounds and retinoids. Increased AKR1B10 expression has been linked to PC tumor invasion, progression, and a poor prognosis. It could lead to changes in cellular metabolism, resistance to apoptosis, and the multiplication of cancer cells. Although the precise processes through which these Ald-Keto Reductases (AKRs) contribute to the onset and progression of PC are unknown, they are still being researched.
Derivatives of isoquinolines have been investigated as potential anti-PC treatments in this regard. On PC cells, these substances are shown to have cytotoxic effects that stop cell development and trigger apoptosis. Inhibiting the androgen receptor signaling pathway, which is essential for the development and spread of PC, has also shown promise. Isoquinolines and their derivatives are found in a variety of natural products and are thought to be pharmacologically active due to their potential for expressing a wide range of biological activities, such as anti-glaucoma, anti-HIV, anti-tumor, anti-fungal, anti-tubercular, and anti-Parkinson’s disease [17–19]. A number of TetrahydroIsoquinoline (TIQ) compounds, including 1-methyl-TIQ and (R)-1,2-dimethyl-5,6-dihydroxy-TIQ [(R)-Nmethyl-salsolinol] among others, have been used to treat behavioral problems [20]. After being created from 2-methyl-1,4-naphthoquinone, benzo[g]isoquinoline-5,10-diones were tested against virulent strains, and better anti-tubercular potency was seen for derivatives with position-3 substitution. The acute cytotoxic doses of these substances are >128 M, while their minimum inhibitory concentrations range from 1.05 to 28.92 M, respectively [21].
The anti-neoplastic properties of different substituted isoquinoline1-carboxyaldhyde thiosemicarbazones (Compound 3) were examined by Liu et al. [2,23] Potential candidates include 4-amino and 4-(methylamino). Using the compound 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Compound 4), which was found to be the most effective anti-cancer agent, isoquinoline and -methylene-butyrolactone derivatives were examined [24]. Acetoxysubstituted 5,6-dihydropyrrolo[2,1-a]isoquinoline (Compound 5) plays a significant function in inhibiting estrogen receptors [25]. In addition, isoquinoline derivatives, such as Lamellarin D, function as a strong topoisomerase-I inhibitor and cause death in cancer cell lines via the mitochondria-mediated mechanism [26]. When compared to the drug tamoxifen, recently developed pyrrolo[2,1-a] isoquinoline derivatives and 1,2-diaryl-5,6-dihydropyrolo[2,1a]-isoquinoline derivatives (Compound 6) have demonstrated improved breast cancer suppression [27]. Fasudil (Compound 7), an Isoquinoline (ISOQ) sulphonamides-based mild Rho-kinase inhibitor, has been successfully tested for clinical investigations [28,29].
Structure-bioactivity relationships between isoquinoline derivatives and the AKR1C3 enzyme are revealed by molecular modeling-based QSAR studies using molecular representation of structure-property relationships (MoRSE) descriptors. These particular molecular descriptors are employed to store the structural data of molecules [30]. It provides a numerical representation of a molecule’s topology, connectivity, and atomic characteristics and is based on the idea of graph theory. In this regard, the development of an effective, stable, and robust model with strong forecasting of future effective ligands to target the PC enzyme AKR1C3 has been worked on.
EXPERIMENT AND METHODS
The software validates the multiple linear regression models created with QSARINS using the chemometric approaches [30]. The literature by Jamieson et al. [31] and Zheng et al. [32] was used to compile the dataset series of 3-(3, 4-dihydroisoquinolin-2(1H)-yl sulfonyl) benzoic acid derivatives with AKR1C3 inhibitory values. The dataset compounds’ IC50 values were transformed to their corresponding logarithmic values; pIC50 and numbering were given in accordance with references in the literature [31,32]. Compounds from the dataset are included in Table 1, along with their IC50 and pIC50 values.
Chemical structure preparation and 3-D optimization
Avogadro V1.2.0’s geometry optimization tool was used to optimize compound structures after structures were sketched using ACD Laboratories’ chemsketch 2017.2.1 [33] using the steepest descent technique and MMFF94 [34]. Where appropriate, several molecule representations were retrieved from Open Babel V2.4.1 [35]. Using the “energy” score function, the best conformer for each compound in the dataset was found using the Avogadro genetic algorithm program.
Data setup
To determine their respective molecular descriptor values, the aforementioned compounds were submitted to PaDEL software and other descriptor computation tools on chemdes—chemopy servers [36]. By eliminating all-zero value, missing value, and semi-constant or near-constant value (>50%) descriptors, the variables were pre-filtered and arranged. 678 descriptors with a value >0.85 were filtered out using pairwise correlation. 39 variables in total with cut-off correlation values greater than 0.35 were chosen for the investigation. A cut-off value of 6.00 pIC50 was used to select the compounds, and 64 compounds were used in the investigation. After excluding compounds S16, S36, S48, and S65, 64 molecules included in the study were divided into training and test sets in a 5:1 ratio based on response order. The best model follow-up method was covered in this article out of numerous models that were obtained from various trials.
Variable selection and model calculation
The QSARINS program offers a variety of options that examine several possible descriptor combinations [37,38]. Friedman’s “lack-of-fit” (LOF) function was used to assess the model’s fitness, and the genetic algorithm was applied to determine the link between structure and bioactivity. To investigate more alternatives, an LOF smoothness level of 1.0, population size of 200, mutation probability of 0.1, and maximum generations of 5,000 were chosen.
Model validation
The applicability domain and internal and external validation criteria were applied to QSARINS models. Every time Q2LMO was internally evaluated, 30% of the training set’s objects were randomly removed. To avoid chance correlation, the Y-scrambling technique was set to 5,000 iterations, which involves randomizing the response data. This requires that R2 and Q2loo values be logically higher than the scrambled ones and that Relative mean square error (RMSE) under prediction be lower than the scrambled ones. The concordance correlation coefficient was examined as an external validation metric for model repeatability. Descriptors specify the range of applications for a model that has undergone a leverage analysis. hi = xi (XT X)−1 xTi (i = 1, 2,..., m), where xi is the query compound’s descriptor row-value and m is the number of query compounds, was used to compute the leverage or crucial value (hat). For the training set, X was a n * p matrix, where n represents the number of samples and p is the number of model descriptors. The leverage cut-off value was 3 (p + 1)/n, which is the limit of the model domain. A leverage larger than h* for the training set indicates that the compound was very significant in the model’s determination, and the prediction in the test set (X outlier) will be made using extrapolation from the model. Y outlier was defined as a compound with a standardized residual greater than 2.5 (2.5 SD units).
RESULTS AND DISCUSSION
Model information
By applying MMFF94 and optimizing for geometry, the molecules in the chosen dataset were processed. Using the PaDEL program and the Chemopy-chemdes (RDKit and Bluedesc) server, around 3,000 descriptors were calculated. Based on the diversity of the dataset’s chemical and biological components, training and test groups were created. QSARINS was used to create a number of QSAR model equations. Some models had higher R2 and Q2LOO values, but their external validation was poor, and they had an excessive number of outliers. In general, a QSAR model should undergo cross-validation to assess both its resilience on the inside and predictability on the outside [39]. The statistical values that the model 1 developed using the aforementioned options,
Model 1
pIC50 = 6.7294 + 2.5913 (MoRSEP27) + 0.1929 (MoRSEE5) + 0.7471 (nF10HeteroRing)
ntr = 54, npred = 10, R2 = 0.6440, R2adj = 0.6226, R2-R2adj = 0.0214, LOF = 0.1803, RMSEtr = 0.3775, MAEtr = 0.3049, RSStr = 7.6945, CCCtr = 0.7834, s = 0.3923, F = 30.1461, Q2LOO = 0.5756, Q2LMO = 0.5606, R2Yscr = 0.0570, Q2Yscr = −0.1016, RMSEcv = 0.4122, MAEcv = 0.3311, PRESScv = 9.1731, CCCcv = 0.7423, R2ext = 0.9179, MAEext = 0.2294, PRESSext = 0.7599, RMSEext = 0.2757, CCCext = 0.8947, Q2F1 = 0.7802, Q2F2 = 0.7775, and Q2F3 = 0.8101.
This model showed up hat value of h* = 0.222 with one outlier (compound S10) in William’s plot at 2.5 SD unit level, with high “s” value and low Q2LOO and Q2LMO values. On removing the above compound S10, model 2 was generated and validated.
Model 2
pIC50 = 6.7216 + 2.4190 (MoRSEP27) + 0.2214 (MoRSEE5) + 0.8102 (nF10HeteroRing)
ntr = 53, npred = 10, R2 = 0.6905, R2adj = 0.6716, R2-R2adj = 0.0189, LOF = 0.1597, RMSEtr = 0.3544, MAEtr = 0.2920, RSStr = 6.6580, CCCtr = 0.8169, s=0.3686, F=36.4442, Q2LOO = 0.6285, Q2LMO = 0.6109, R2Yscr = 0.0569, Q2Yscr = −0.1052, RMSEcv = 0.3883, MAEcv = 0.3179, PRESScv = 7.9916, CCCcv = 0.7807, R2ext = 0.9130, MAEext = 0.2612, PRESSext = 0.9286, RMSEext = 0.3047, CCCext = 0.8805, Q2F1 = 0.7320, Q2F2 = 0.7280, and Q2F3 = 0.7712.
The model 2 showed up hat value of h* = 0.226 with one outlier (compound S3) in William’s plot at a 2.5 SD unit level, with betterment in statistical values. However, on removing the above compound S3, model 3 was generated and validated.
 | Table 1. Dataset compounds with akr1c3 inhibitory activity. [Click here to view] |
Model 3
pIC50 = 6.6108 + 2.5415 (MoRSEP27) + 0.1927 (MoRSEE5) + 0.8701 (nF10HeteroRing)
ntr = 52, npred = 10, R2 = 0.7279, R2adj = 0.7109, R2-R2adj = 0.0170, LOF = 0.1427, RMSEtr = 0.3341, MAEtr = 0.2794, RSStr = 5.8054, CCCtr = 0.8425, s=0.3478, F=42.8065, Q2LOO = 0.6736, Q2LMO = 0.6641, R2Yscr = 0.0590, Q2Yscr = −0.1064, RMSEcv = 0.3660, MAEcv = 0.3043, PRESScv = 6.9641, CCCcv = 0.8116, R2ext = 0.9085, MAEext = 0.2770, PRESSext = 1.0756, RMSEext = 0.3280, CCCext = 0.8673, Q2F1 = 0.6907, Q2F2 = 0.6850, and Q2F3 = 0.7379.
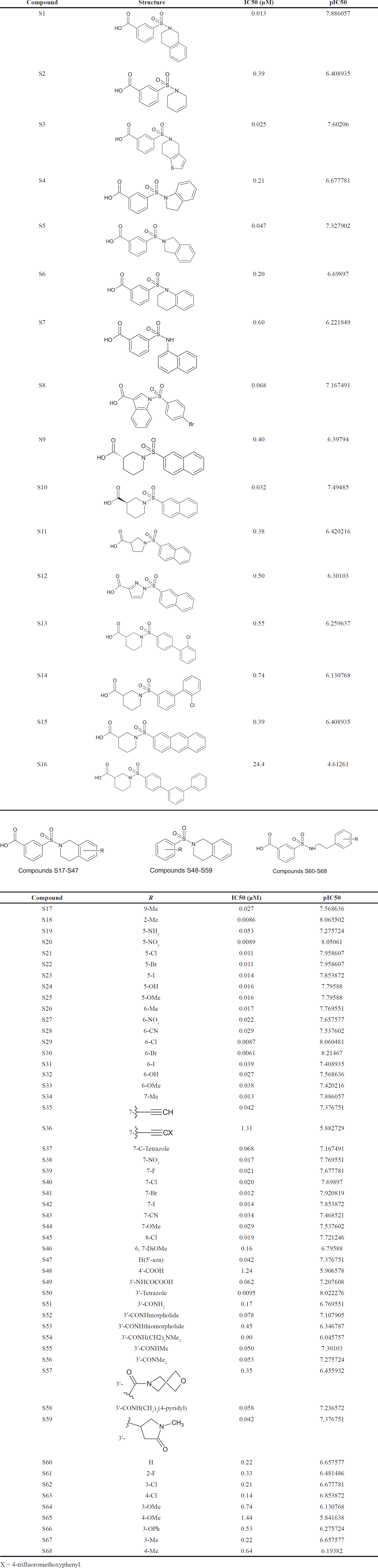
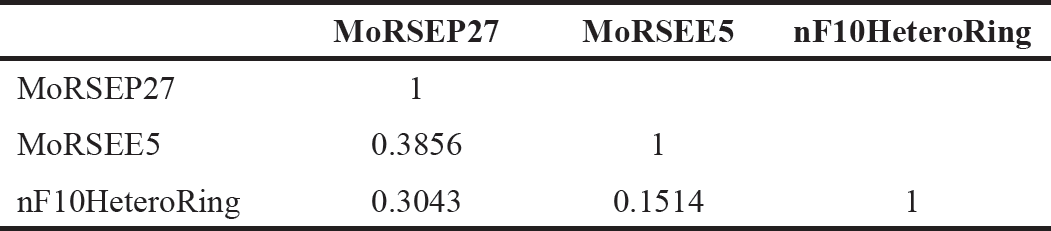
Model 3 demonstrated good validation values and good criteria fitting. Model 3 outperformed earlier models in terms of internal validation parameters and had no outliers in William’s plot. Table 2 shows the model 3 descriptor correlation matrix. The scatter plot shows the “AKR1C3 inhibitory activities of 3-(3, 4-dihydroisoquinolin-2(1H)-yl sulfonyl) benzoic acid derivatives” in comparison to calculated results, and Figure 1 shows predicted values that are consistent with corresponding experimental results. Figure 2 shows the Kxy, the inter-correlation between descriptors, and response versus Q2LMO of model 3, along with the Leave many out (LMO) parameter values surrounding the model parameters, indicating the robustness and stability of the model. The correlation values of model 3 are substantially higher than those following endpoint scrambling, and a broken relationship between structure and responses can be shown. Figure 3 shows the Y-scramble plot of Kxy versus R2Yscr and Q2Yscr.
Williams plot in Figure 4 of the standardized residuals versus leverage values illustrates the applicability domain of the model, allowing one to determine whether or not the molecules are situated within it. Leverage values were discovered to be lower from the plot than the warning h* of 0.231. The threshold values of 0.70 and concordance correlation coefficient parameter values of more than 0.80 are being approached by Q2F1 and Q2F2 values, respectively. According to these findings, there is no random association and a real connection between the structural features of 3-(3, 4-dihydroisoquinolin-2(1H)-yl sulfonyl) benzoic acid derivatives and their AKR1C3 inhibitory action.
 | Table 2. Correlation matrix of model 3. [Click here to view] |
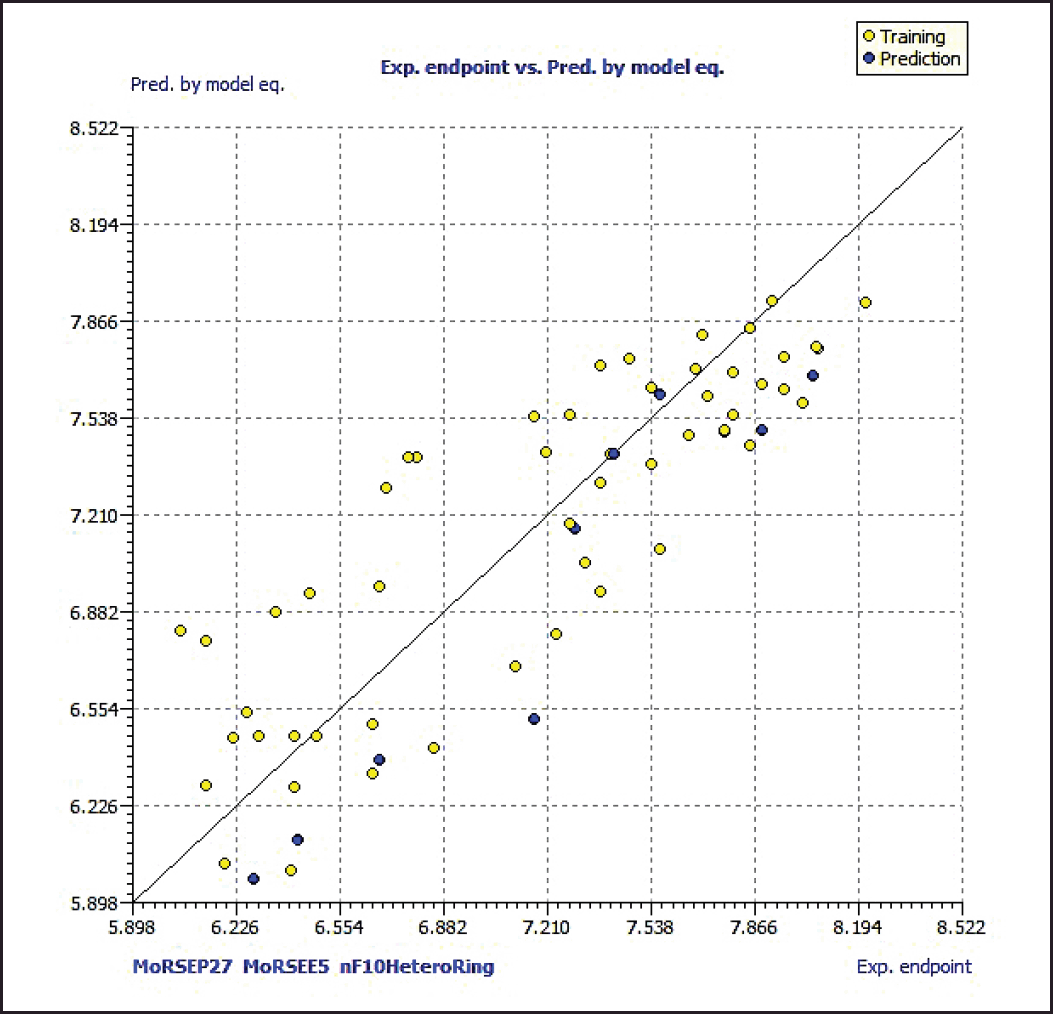 | Figure 1. Scatter plot of dataset compounds. [Click here to view] |
The model 3 with molecular descriptors contributed structural data pertaining to predicted bioactivity, and the data gained is compared to investigations of the dataset’s structure-activity relationships. The residuals data for chemical bioactivity when compared to the best experimental and QSAR models, are shown in Table 3.
Molecular descriptors information
3-D molecular representations of structure based on electron diffraction descriptors, also called 3-D-MoRSE [40] descriptors provide molecule structure information derived from euclidean interatomic distances, scattering parameter (0–31 integer values), and weighting by atomic mass (MoRSEM15, where 15 is scattering parameter), atomic properties such as atomic charges (MoRSEC9), atomic number (MoRSEN26), Sanderson electronegativity (MoRSEE12), atomic van der Waals volume (MoRSEV23), atomic polarizability (MoRSEP4), and unweighted (MoRSEU13).
A simplified equation to determine the MoRSE function:
where rij is the Euclidean distance b/w ith and jth atoms, s is the scattering parameter, Ai and Aj are different atomic properties used as weights, and N is the total number of atoms. Each functional term depends on distance and acts as a radial basis function.
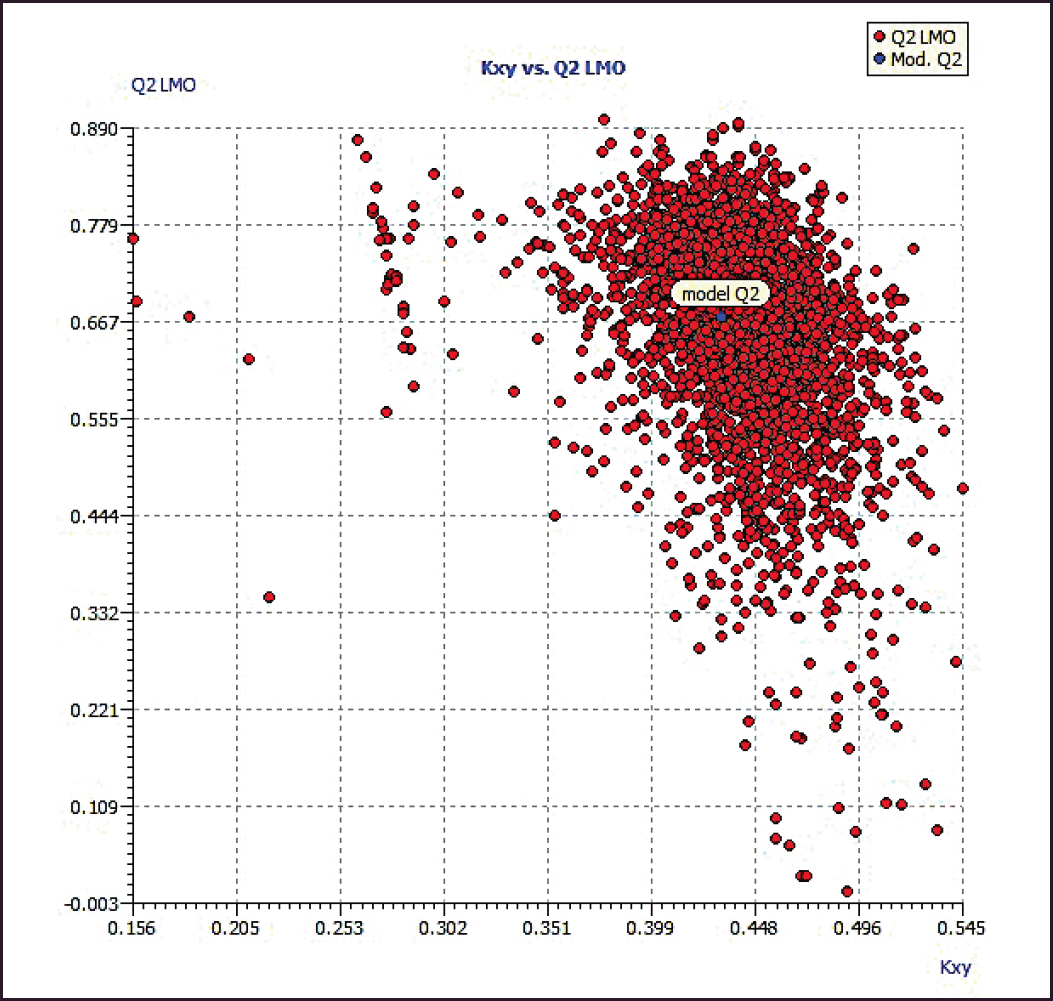 | Figure 2. The LMO scatter plot (plot of Kxy vs. Q2LMO). [Click here to view] |
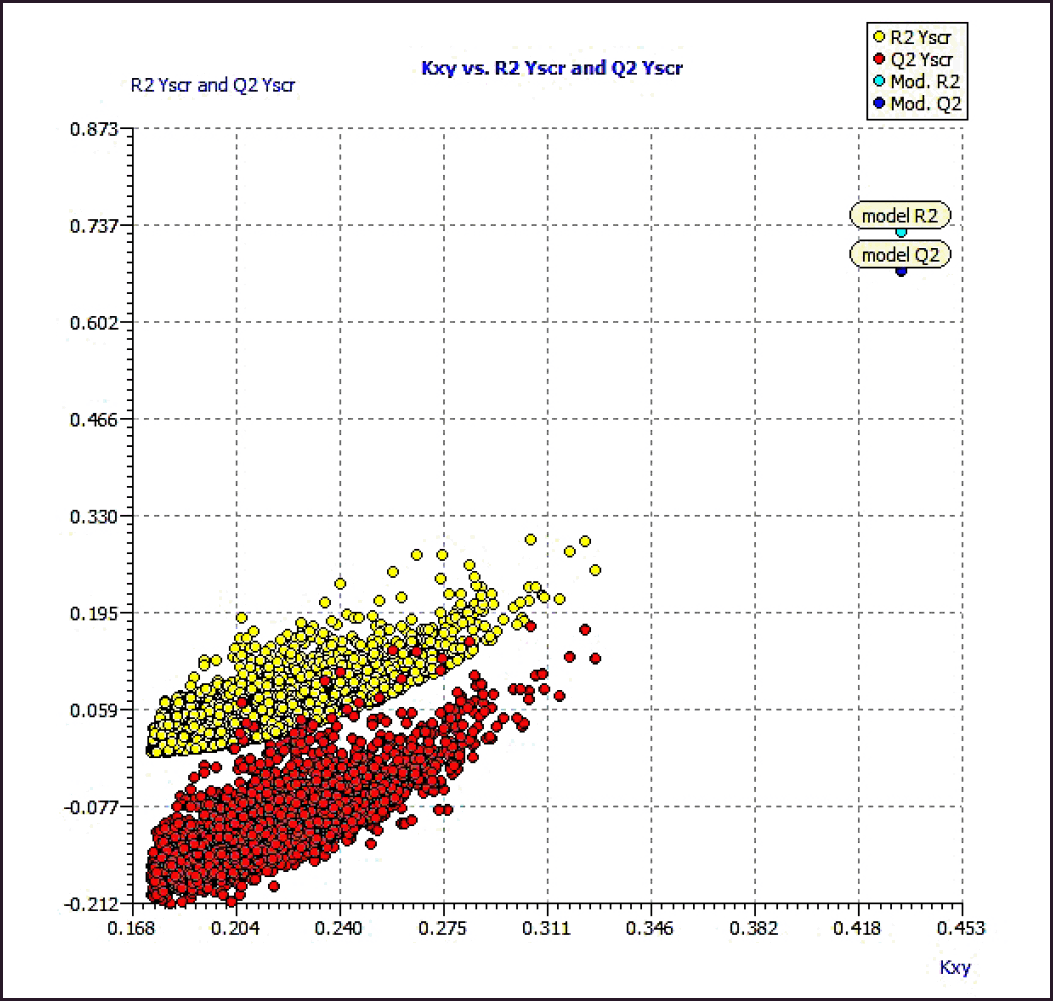 | Figure 3. Y-Scramble plot (plot of Kxy vs. R2 and Q2LOO from Y-Scrambling procedure). [Click here to view] |
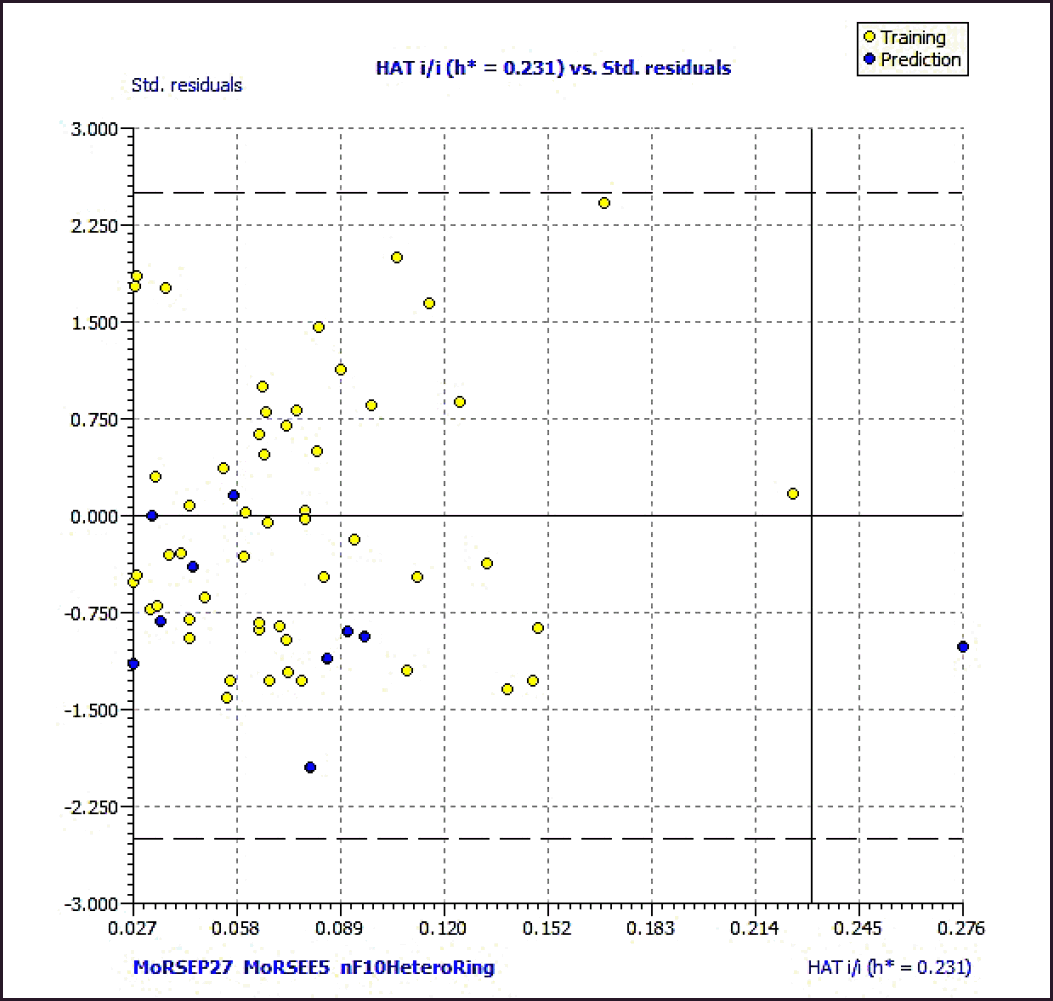 | Figure 4. Williams plot of the best model. The dashed lines are the cut-off 2.5σ and the warning value of hat (h*, 0.231). [Click here to view] |
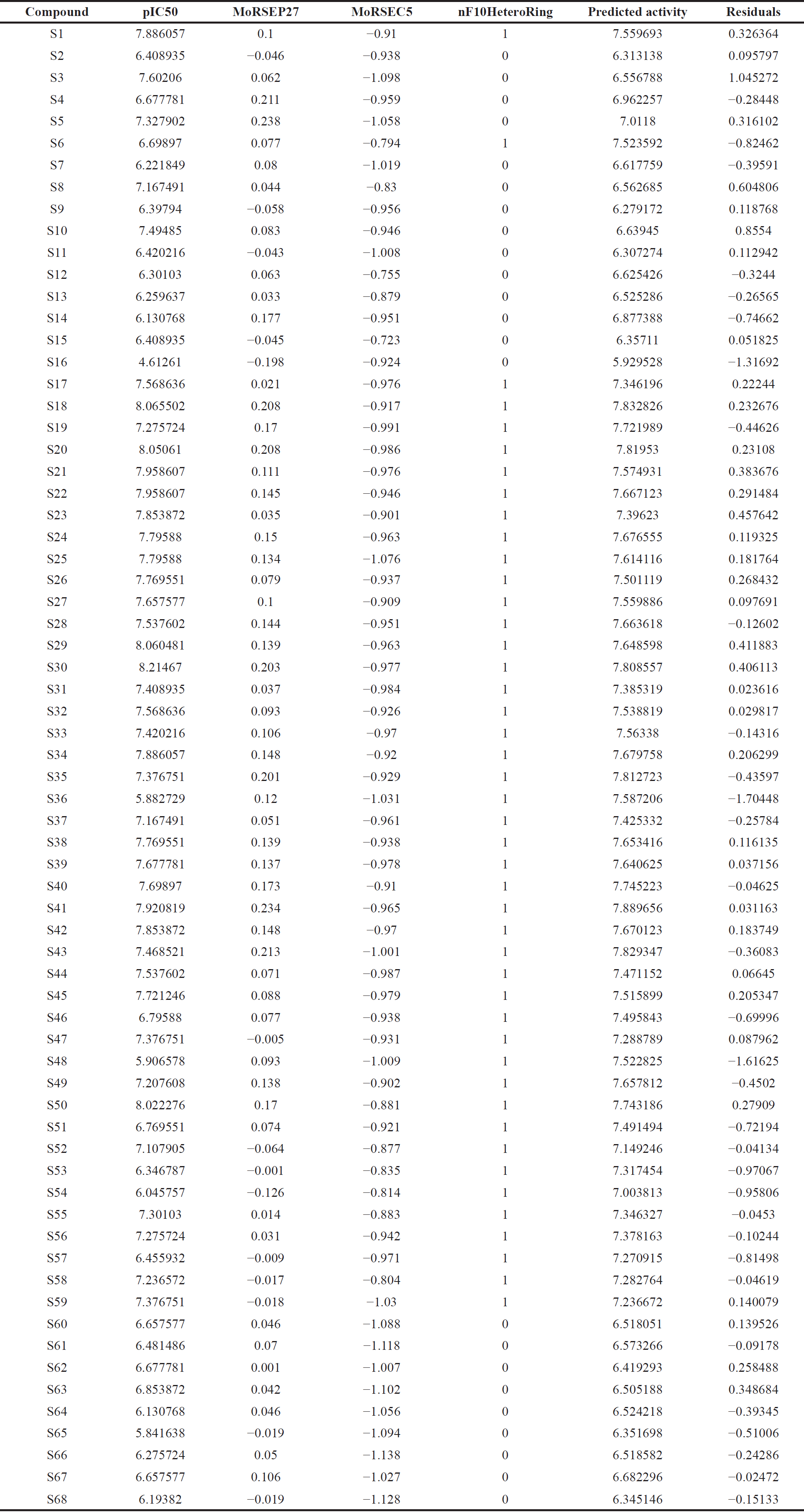 | Table 3. Residuals from model equation versus experimental values. [Click here to view] |
The compound information can be sensitized using specific molecule fragments by weighting descriptors. Weighting atomic partial charge, for instance, reflects the distance between atoms with an excessive or insufficient electron density. The 3-D-MoRSE descriptors weighted with schemes where the role of hydrogen is diminished should exhibit lower variation with increasing scattering parameters. With an increase in scattering, the atomic mass, van der Waals volume, atomic number, and polarizability weightings exhibit the least relative variation, according to published research. The dynamics of the cumulative sum of the 3-D-MoRSE terms can be used to determine the value of different interatomic distances.
Ring count descriptor, nF10HeteroRing, meaning a number of 10-membered fused rings containing hetero atoms (N, O, P, S, or halogens), reflected molecular topological complexity.
SAR studies of the dataset
On analyzing the bioactivity results from the original dataset [4,5], as shown in Table 1, compounds 1 to 16 having quinoline rings have shown activity compared to others. Similarly, compounds 48 to 59 with substitutions on the R position of the quinoline ring:
1. R substituted with the nitro group at the fifth position of the quinoline ring (compound 20) has shown an increase in bioactivity compared to the sixth position (compound 27) and seventh position (compound 38).
2. R substituted with methoxy, hydroxyl, and cyano groups at the fifth position of the quinoline ring (compounds 25, 24, and 28, respectively) have shown an increase in bioactivity compared to other positions.
3. R substituted with electron-withdrawing groups (Cl and Br) group at the sixth position of the quinoline ring (compounds 29 and 30 for Cl and Br, respectively) have shown an increase in bioactivity compared to the fifth position (compounds 21 and 22), seventh position (compounds 40 and 41), and eighth position (compound 45). At the same time, the Iodine group at the sixth position has shown no increment in activity (compound 31).
4. R substituted with electron donating group (Methyl) group at the second position of the quinoline ring (compound 18) has shown an increase in bioactivity compared to other positions.
Compounds 48 to 59 with substitutions on the R position of the quinoline ring:
1. Substituted carbamides at the third position with thiomorpholide (compound 53) have shown decrement in activity compared with its morpholide (compound 52) derivative.
Compounds 60 to 68 with substitutions on the R position of the quinoline ring:
1. Compared with the substitution of the electron-withdrawing group (chlorine) at the third and fourth position, the electron-donating group (methyl) has shown decrement in activity.
2. The Methoxy group at the third position has shown better activity compared with the fourth position.
CONCLUSION
QSAR best-fit model developed using 3-D-MoRSE (weighted by polarizability and Sanderson’ electronegativity indices) and 2D-Ring count descriptors was validated using statistical parameters to establish a meaningful relationship between chemical structures of 3-(3, 4-dihydroisoquinolin-2(1H)-yl sulfonyl) benzoic acid derivatives and bioactivity of AKR1C3 inhibition. The obtained QSAR model provides insights for better lead generation and optimization, which were discussed in section 3.3, structure-activity relationship for AKR1C3 inhibitory activity. It is worth noting that the electron-withdrawing functional groups at the fifth and sixth position of the Isoquinoline ring provided better lead bioactive compounds (S20, S29, and S30). However, further conformational analysis of the compounds will provide planarity, isomerism, and energy information through which better compounds can be designed. Overall, these results would serve as a significant guideline for the discovery and design of novel AKR1C3 inhibitors.
ACKNOWLEDGMENTS
The authors thank Prof. Gramatica P, University of Insubria, Italy, for providing QSARINS software and Research Council, GITAM University, Hyderabad.
AUTHOR CONTRIBUTIONS
Concept and design: Gopinath P
Data acquisition: Gopinath P, Durgaprasad B, Bidhubhusan K
Data analysis/interpretation: Gopinath P, Ashishranjan D, RamReddy G
Drafting manuscript: Gopinath P, Bidhubhusan K, Ravisankara Reddy D
Critical revision of the manuscript: Gopinath P, Asishranjan D, Durgaprasad B, RamReddy G.
Statistical analysis
Gopinath P, Ashishranjan D, Raghuveer P
Supervision and Final approval
Gopinath P, Durgaprasad B, Bidhubhusan K, Ramalingeswara Rao K, Ravisankara Reddy D, Subramanyam S, Raghuveer P
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Sekhoacha M, Riet K, Motloung P, Gumenku L, Adegoke A, Mashele S. Prostate cancer review: genetics, diagnosis, treatment options, and alternative approaches. Molecules. 2022 Sep 5;27(17):5730. CrossRef
2. Bluemn EG, Nelson PS. The androgen/androgen receptor axis in prostate cancer. Curr Opin Oncol. 2012;24:251–7. CrossRef
3. Molina A, Belldegrun A. Novel therapeutic strategies for castration resistant prostate cancer: inhibition of persistent androgen production and androgen receptor mediated signaling. J Urol. 2011;185:787–94. CrossRef
4. Stein MN, Singer EA, Patel N, Bershadskiy A, Sokoloff A. Androgen synthesis inhibitors in the treatment of castration-resistant prostate cancer. Asian J Androl. 2014;16:387–400. CrossRef
5. Abidi A. Cabazitaxel: a novel taxane for metastatic castration-resistant prostate cancer-current implications and future prospects. J Pharmacol Pharmacother. 2013;4:230–7. CrossRef
6. Jacob A, Raj R, Allison DB, Myint ZW. Androgen rceptor signaling in prostate cancer and therapeutic strategies. Cancers (Basel). 2021 Oct 28;13(21):5417. CrossRef
7. Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18:41–58. CrossRef
8. Asangani I, Blair IA, Van Duyne G, Hilser VJ, Moiseenkova-Bell V, Plymate S, et al. Using biochemistry and biophysics to extinguish androgen receptor signaling in prostate cancer. J Biol Chem. 2021 Jan-Jun;296:100240. CrossRef
9. Risdon EN, Chau CH, Price DK, Sartor O, Figg WD. PARP inhibitors and prostate cancer: to infinity and beyond BRCA. Oncologist. 2021 Jan;26(1):e115–29. CrossRef
10. Teyssonneau D, Margot H, Cabart M, Anonnay M, Sargos P, Vuong NS, et al. Prostate cancer and PARP inhibitors: progress and challenges. J Hematol Oncol. 2021 Mar 29;14(1):51. CrossRef
11. He Y, Xu W, Xiao YT, Huang H, Gu D, Ren S. Targeting signaling pathways in prostate cancer: mechanisms and clinical trials. Signal Transduct Target Ther. 2022 Jun 24;7(1):198. CrossRef
12. Gasmi A, Roubaud G, Dariane C, Barret E, Beauval JB, Brureau L, et al. Overview of the development and use of Akt inhibitors in prostate cancer. J Clin Med. 2021 Dec 29;11(1):160. CrossRef
13. Beinhoff P, Sabharwal L, Udhane V, Maranto C, LaViolette PS, Jacobsohn KM, et al. Second-generation Jak2 inhibitors for advanced prostate cancer: are we ready for clinical development? Cancers (Basel). 2021 Oct 17;13(20):5204. CrossRef
14. Rana Z, Diermeier S, Hanif M, Rosengren RJ. Understanding failure and improving treatment using HDAC inhibitors for prostate cancer. Biomedicines. 2020 Jan 30;8(2):22. CrossRef
15. Penning TM. Aldo-Keto reductase (AKR) 1C3 inhibitors: a patent review. Expert Opin Ther Pat. 2017;27(12):1329–40. CrossRef
16. Pippione AC, Carnovale IM, Bonanni D, Sini M, Goyal P, Marini E, et al. Potent and selective aldo-keto reductase 1C3 (AKR1C3) inhibitors based on the benzoisoxazole moiety: application of a bioisosteric scaffold hopping approach to flufenamic acid. Eur J Med Chem. 2018;150:930–45. CrossRef
17. Kakhkia S, Shahosseinia S, Zarghi A. Pyrrolo[2,1-a] isoquinoline-based derivatives as new cytotoxic agents. Iran J Pharm Res. 2016;15(4):743–51.
18. Heravi M, Nazari N. Bischler-napieralski reaction in total synthesis of isoquinoline-based natural products. An old reaction, a new application. Curr Org Chem. 2015;19(24):2358–408.
19. Pashev AS, Burdzhiev NT, Stanoeva ER. Synthetic approaches toward the benzo [a] quinolizidine system. A review. Org Prep Proced Int. 2016;48(6):425–67.
20. Ishiwata K, Koyanagi Y, Abe K, Kawamura K, Taguchi K, Saitoh T, et al. Evaluation of neurotoxicity of TIQ and MPTP and of parkinsonism-preventing effect of 1-MeTIQ by in vivo measurement of pre-synaptic dopamine transporters and post-synaptic dopamine D2 receptors in the mouse striatum. J Neurochem. 2001;79(4):868–76.
21. Smets RJ, Torfs E, Lemière F, Cos P, Cappoen D, Tehrani KA. Synthesis and antitubercular activity of 1-and 3-substituted benzo [g] isoquinoline-5, 10-diones. Org Biomol Chem. 2019;17(11):2923–39.
22. Liu MC, Lin TS, Penketh P, Sartorelli AC. Synthesis and antitumor activity of 4-and 5-substituted derivatives of isoquinoline-1-carboxaldehyde thiosemicarbazone. J Med Chem. 1995;38(21):4234–43.
23. Liu MC, Lin TS, Sartorelli AC. Chemical and biological properties of cytotoxic alpha-(N)-heterocyclic carboxaldehyde thiosemicarbazones. Prog Med Chem. 1995;32:1–35.
24. Yan Y, En D, Zhuang Z, Guo Y, Liao WW. Synthesis of densely functionalized α-methylene γ-butyrolactones via an organocatalytic one-pot allylic-alkylation–cyclization reaction. Tetrahedron Lett. 2014;55(2):479–82.
25. Kakhki S, Shahosseini S, Zarghi A. Design and synthesis of pyrrolo [2, 1-a] isoquinoline-based derivatives as new cytotoxic agents. Iran J Pharm Res. 2016;15(4):743.
26. Facompré M, Tardy C, Bal-Mahieu C, Colson P, Perez C, Manzanares I, et al. A novel potent inhibitor of topoisomerase I. Cancer Res. 2003;63(21):7392–9.
27. Kakhki S, Shahosseini S, Zarghi A. Design, synthesis and cytotoxicity evaluation of new 2-aryl-5, 6-dihydro pyrrolo[2, 1-a]isoquinoline derivatives as topoisomerase inhibitors. Iran J Pharm Res. 2014;13:71–7.
28. Feng Y, Lograsso PV, Defert O. Rho kinase (ROCK) inhibitors and their therapeutic potential. J Med Chem. 2016;59(2):269–300.
29. Oliveira RG, Guerra FS, Mermelstein CD, Fernandes PD, Bastos IT, Costa FN, et al. Synthesis and pharmacological evaluation of novel isoquinoline N-sulphonylhydrazones designed as ROCK inhibitors. J Enzyme Inhib Med Chem. 2018;33(1):1181–93.
30. Gopinath P, Kathiravan MK. QSAR and docking studies on triazole benzene sulfonamides with human carbonic anhydrase IX inhibitory activity. J Chemom. 2019;33(12):e3189. CrossRef
31. Jamieson SMF, Brooke DG, Heinrich D, Atwell GJ, Silva S, Denny WA, et al. 3-(3, 4-dihydroisoquinolin-2(1H)-yl sulfonyl) benzoic acids: highly potent and selective inhibitors of the type 5 17-β-hydroxysteroid dehydrogenase AKR1C3. J Med Chem. 2012;55(17):7746–58. CrossRef
32. Zheng X, Wu Y, Wu D, Wang X, Zhang C, Guo X, et al. 3D-QSAR studies of 3-(3,4-dihydroisoquinolin-2(1 H )-yl sulfonyl)benzoic acids as AKR1C3 inhibitors: highlight the importance of molecular docking in conformation generation. Bioorg Med Chem Lett. 2016;26(23):5631–8. CrossRef
33. Available from: https://www.acdlabs.com/resources/freeware/chemsketch/
34. Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: an advanced semantic chemical editor, visualization and analysis platform. J Cheminformatics. 2012;4(1):1–7. CrossRef
35. O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open babel: an open chemical toolbox. J Cheminformatics. 2011;3(1):1–4. CrossRef
36. Dong J, Cao DS, Miao HY, Liu S, Deng BC, Yun YH, et al. ChemDes: an integrated web-based platform for molecular descriptor and fingerprint computation. J Cheminformatics. 2015;7(1):1. CrossRef
37. Gramatica P, Chirico N, Papa E, Cassani S, Kovarich S. QSARINS: a new software for the development, analysis, and validation of QSAR MLR models. J Comput Chem. 2013;34:2121–32. CrossRef
38. Gramatica P, Chirico N, Cassani S. QSARINS-chem: insubria datasets and new QSAR/QSPR models for environmental pollutants in QSARINS. J Comput Chem. 2014;35:1036–44. CrossRef
39. Consonni V, Ballabio D, Todeschini R. Evaluation of model predictive ability by external validation techniques. J Chemometrics. 2010;24:194–201. CrossRef
40. Devinyak O, Havrylyuk D, Lesyk R. 3D-MoRSE descriptors explained. J Mol Graph Model. 2014;54:194–203. CrossRef