
INTRODUCTION
Helicobacter pyroli is a well-known encyclopedic subject because it has been linked to a variety of gastrointestinal diseases and gastric cancers [1]. Proton pump inhibitors (PPIs)-established triple-drug therapy, which comprises PPIs, CLA, and metronidazole or amoxicillin (AMO) for decades, is widely used to treat H. pyroli infection. Nevertheless, due to CLA resistance, eradication rates have fallen below 80% [2]. Potassium competitive acid blockers are more efficient than PPIs, have a quicker onset of action, and maintain their ability to reduce acid secretion for longer periods of time. In addition, they limit the production of stomach acid. Furthermore, potassium competitive acid blockers can be administered whenever is convenient because they do not need stomach acid to activate them. The maintenance of a certain gastric pH level is crucial in facilitating antibiotic activity in the treatment of Helicobacter pylori infection. Potassium competitive acid blockers are known to hinder digestive secretions of the stomach mediated by hydrogen potassium ATPase. They possess stability to withstand acidic environments and are less affected by the CYP2C19 system compared to PPIs [1]. According to a recent study, vonoprazan (VON), a potassium competitive blocker, has demonstrated superiority over standard inhibitors of proton pump activity-based therapy in both resistance to CLA and non-resistance to strains of H. pylori. This study provided support for the approval of VON dual and triple therapies by the Food and Drug Administration in May 2022 [2]. In May 2022, the FDA granted approval for the usage of VON in the management of H. pylori infections. This medication had previously been authorized for the intervention of infection by H. pylori and other gastric-related diseases in various nations. The intervention results in a reduction of intragastric pH and sustains it to a greater extent compared to PPIs. This has been linked to increased rates of eradication [2].
CLA is a macrolide antibiotic, semisynthetic, and 14 membered containing two deoxy sugar moieties. It is a 6-o-methyl ether of erythromycin A. (Fig. 1) AMO is a penicillin derivative characterized by the presence of a 2-amino-2-(4-hydroxyphenyl) acetamido group at position 6 of the penam ring (Fig. 2) is the most commonly employed-lactam antibiotic for treating bacterial infections. VON is a pyrrole derivate and is chemically known as 1-[5-(2-fluorophenyl)-1-pyridin-3-ylsulfonylpyrrol-3-yl]-N-methylmethanamine (Fig. 3) is a new potassium competitive acid blocker [3]. In an aqueous solution, VON is soluble and stable over a wide pH range [4].
After conducting a thorough review of relevant literature, it was revealed that several studies have employed RP-HPLC to estimate the levels of CLA, AMO, and esomeprazole in rat plasma [5], as well as CLA, tinidazole, and lansoprazole by RP-HPLC [6]. In addition, reverse phase liquid chromatography was utilized to analyze ten related substances in vonprazan fumurate [7]. Furthermore, CLA and AMO were estimated alone and in combination with other drugs using different techniques [8–22]. A novel UPLC-based analytical approach is the subject of our current investigation. As lean laboratories become the norm, there is a pressing need to create efficient, cost-effective procedures. There are a number of HPLC procedures described in the literature, and they all need 30 minutes runtime to complete. However, the UPLC technique for simultaneously estimating CLA, AMO, and VON in a mixture has not been studied in the literature. The principal purpose of our research is to create a methodology that is both rapid and accurate, while also being cost-effective and reproducible. The technique was validated in accordance with the Q2 (R1) guidelines of the International Conference on Harmonization (ICH) protocol.
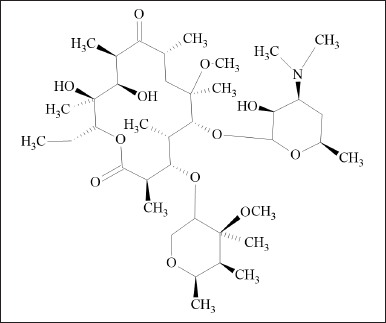 | Figure 1. Clarithromycin structure. [Click here to view] |
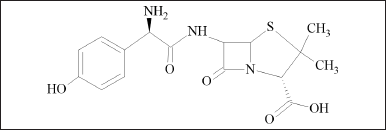 | Figure 2. Amoxicillin structure. [Click here to view] |
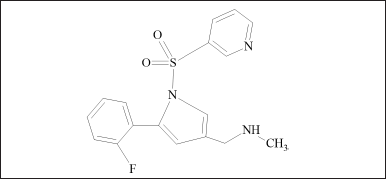 | Figure 3. Vonoprazan structure. [Click here to view] |
MATERIALS AND METHODS
Materials
Spectrum Pharma Research Solutions, located in Kukatpally, Hyderabad, provided CLA (99% purity), AMO (99% purity), VON (99% purity) active pharmaceutical ingredients and formulation voquezna triple pak (VON-20 mg tablets, CLA-500 mg tablets, and AMO capsules-500 mg capsules). Merck was the supplier for the HPLC grade monobasic potassium phosphate, acetonitrile, and orthophosphoric acid, as well as the methanol. Apart from the above-mentioned chemicals, the remaining reagents utilized for the experiment were of analytical grade. Through the MLpore Pvt. Ltd., Bangalore, ML-Q water purifying system, purified water was obtained for UPLC.
Equipment
Acquity UPLC System from Waters Corporation, equipped with a TUV detector, was used to develop a novel method. To process signals, the Empower 2 software was utilized. Hibar bis phosphonate C18 Columns with dimensions of 100 × 2.1 mm × 2 μm were utilized during the process of method development. In addition to that, we made use of a weighing balance (PI-214), a pH meter Thermo Fischer), a vortex mixer (Remi CM-101), and a centrifuge (CM 01).
METHOD
Operating conditions
A Hibar C18 Bis phosphonate column (100 × 2.1 mm × 2 μ) regulated to 30°C was used for separation, with a mobile phase proportion of 60:40 of 0.1 N monobasic potassium phosphate buffer (pH 3.8) and acetonitrile. The analysis was performed for 4 minutes at 210 nm with an injection load of 1 μL and a flow velocity of 0.2 ml/min.
Preparation of solutions
0.1N mono basic Potassium buffer (pH 3.8)
Accurately weighing 1.36 g of mono basic potassium phosphate in a standard flask of 1,000 ml capacity, adding approximately 900 ml of ml-Q water, sonicating it 10 minutes, and then adding 1ml triethyamine and bringing the pH to 3.8 with dilute phosphoric acid.
Mobile phase composition
It is a combination of mono basic phosphate buffer and acetonitrile in the proportion of 60:40 (v/v).
Diluent preparation
The mobile phase is utilized as a diluent.
CLA standard stock solution (1,000 μg/ml)
In a 50 ml standard flask (cleaned and dried), precisely weigh 50 mg of clarithromycin standard, add on 10 ml of mobile phase, homogenize for 10 minutes, bring the volume up to the final volume using diluent, and mix well.
AMO standard stock solution (1,000 μg/ml)
In a 50 ml standard flask (cleaned and dried), precisely weigh 50 mg of AMO standard, add 10 ml of mobile phase, homogenize for 10 minutes, bring the volume up to the final capacity using diluent, and mix well.
Vonoprozan standard stock solution (40 μg/ml)
In 50 ml standard flask (cleaned and dried), precisely weigh 2 mg of VON standard, add 10 ml of mobile phase, homogenize for 10 minutes, and with the diluent, bring up to the final volume and mix well.
Standard working solution (100 μg/ml of CLA, 100 μg/ml of AMO, and 4 μg/ml of VON)
From the aforementioned two standard stock solutions (CLA and AMO stock solutions), 1 and 0.1 ml of VON standard stock solution were taken and diluted to 10 ml in a volumetric flask with diluent and mixed well.
Sample stock solution
The average weight of 10 VON and CLA tablets and 10 AMO capsules was determined by weighing the formulation. The tablets and powder were then finely ground. In a 100 ml standard flask, we weighed out equivalent to one tablet of CLA (500 mg), oone tablet of VON (20 mg), and one capsule of AMO (500 mg). Then, we added 5 ml of acetonitrile and sonicated the mixture. Diluent was added to make up the volume to 100 ml, mixed well, and then filtered using a 0.22 m finer porosity PVDF membrane filter. The solutions of 5,000 μg/ml CLA, 5,000 μg/ml AMO, and 200 μg/ml VON were obtained.
Sample working solution
A volume of 0.2 ml of the stock solution of the sample (CLA, 5,000 μg/ml AMO, and 200 μg/ml VON) was placed into a 10 ml standard flask and subsequently diluted using mobile phase, mixed well. The final concentrations obtained were CLA and AMO 100 μg/ml, and VON 4 μg/ml.
Optimization of the method
Trials were run using different stationary phases, mobile phase, and buffer pH chromatographic settings. The method was thoroughly optimized using the observations for peak symmetry, theoretical plates, and low retention time.
Validation
The method optimized was validated regarding linearity, specificity, accuracy, precision, detection and quantification limit, and system suitability parameters as per ICH guidelines [23].
System suitability
It guarantees that the developed method is fit for its intended use. The chromatograms taken under ideal conditions were subjected to the system appropriateness test to evaluate a number of factors, including column efficiency (plates >2,000), resolution (Rs) (>1.5), capacity factor, and peak tailing. Six duplicates of a standard solution containing CLA, AMO, and VON, 100, 100, and 4 μg/ml, respectively, were introduced into the system, and the system suitability variables were assessed.
Specificity
The UPLC method’s specificity was assessed by introducing a blank sample, a placebo and sample solution, and a degraded sample obtained from the degradation investigation in line with the requirements of the ICH.
Linearity
Linearity of standard drugs was obtained at a range of concentration of 25–150, 25–150, and 1–6 μg/ml for CLA, AMO, and VON, respectively. These solutions were introduced into the UPLC apparatus, and a correlation coefficient was calculated by plotting the concentration against the peak area.
Precision
Multiple samples are analyzed to determine an analytical procedure’s precision. Measurements of homogeneous samples should be conducted under the same conditions each time. A method’s precision is expressed by its variance and standard deviation (SD). Six replicates of CLA, AMO, and VON solution concentrations of 100, 100, and 4 μg/ml, respectively, were injected to test the method’s precision. Six replicates of each sample solution of concentration of 100, 100, and 4 μg/ml of CLA, AMO, and VON were injected into the chromatographic apparatus on two different days over the course of a week to establish intermediate precision.
Accuracy
The method’s accuracy defines the degree of correlation between a measurement’s result and its actual value. In accordance with ICH standards, a recovery study was conducted to confirm the accuracy. Samples and standards of concentrations of 100, 100, and 4 μg/ml of CLA, AMO, and VON were produced, subjected to sonication, and subsequently filtered. The Standard solution was then spiked at levels of 50%, 100%, and 150% of the sample stock solution by adding 0.5, 1, and 1.5 ml, respectively. The samples were injected in triplicates. The percentage recovery was evaluated by using the following equation (Eq. 1):
% Recovery = Concentration Recovered/Concentration Injected × 100. (1)
Robustness
The robustness study involved making minor adjustments to the parameters of the developed method, such as mobile phase ratio, flow velocity, and temperatures such as mobile phase composition (±10%) (Buffer and acetonitrile) 66:44 and 54:36 flow rate (±10%) 0.22 and 0.18 ml/min, and temperature (±10%) 38.5oC and 31.5oC. Sample preparations were injected five times with the above adjustments, and RSD values of peak areas were computed.
Limit of detection and limit of quantification
Mathematical equations 2 and 3 were used to calculate the detection limit (LOD) and quantification limit (LOQ) of CLA, AMO, and VON. The calibration curve’s SD was used to estimate LOD and LOQ as follows:
LOD = SD.3.3/slope (2)
LOQ = SD.10/slope (3)
where SD is the SD.
Assay
The analysis of the marketed formulation of CLA, AMO, and VON was conducted to ascertain the drug’s amount and percentage purity using the following equation (Eqs. 4 and 5):
Amount of drug present in dosage form = a/b ×c/d × Average weight (4)
where a is sample area, b is standard area, c is standard dilution, and d is sample dilution.
Percentage purity = Amount of drug present/Label claim ×100. (5)
Forced degradation studies and stability studies
The investigation focused on assessing the stability of CLA, AMO, and VON sample working solutions for two distinct purposes. The primary objective was to assess the stability of CLA, AMO, and VON as well as to demonstrate the selective analysis under different high-stress situations. The second objective aimed to ascertain the stability of sample solutions during a 24-hours period at ambient temperature. In accordance with ICH guidelines [24], stress conditions were applied to sample stock solutions CLA, AMO, and VON. Subsequently, the degraded sample solution was introduced and separated using the developed method as described below.
Forced degradation in acid conditions
We added 0.2 ml of stock solution of the sample containing CLA, AMO, and VON to 1 ml of 2N HCl. Refluxing the mixture at 60°C for 30 minutes. Diluting the solution creates solutions with concentrations of 100, 100, and 4 μg/ml. A 10 μl was then introduced to UPLC, and the resulting chromatograms were documented.
Forced degradation in alkaline conditions
We added 0.2 ml of stock solution of the sample containing CLA, AMO, and VON to 1 ml of 2N NaOH. Refluxing the mixture at 60°C for 30 minutes. Diluting the solution creates solutions with concentrations of 100, 100, and 4 μg/ml. A volume of 10 μl was then introduced into UPLC, and documenting the resulting chromatograms.
Forced degradation by oxidation
We added 0.2 ml of stock solution of the sample containing CLA, AMO, and VON to 1 ml of 20% hydrogen peroxide. Refluxing the mixture at 60°C for 30 minutes. Diluting the solution creates solutions with concentrations of 100, 100, and 4 μg/ml. A volume of 10 μl was introduced into UPLC, and the resulting chromatograms were documented.
Forced degradation by exposure to heat
The sample stock solution was subjected to a temperature of 105oC for 6 hours in order to investigate the process of dry heat degradation. Diluting the solution creates solutions with concentrations of 100, 100, and 4 μg/ml. Then, 10 μl of diluted solutions was introduced into UPLC, and documenting the resulting chromatograms.
Forced degradation by exposure to UV light
The drug’s photochemical stability was further investigated by subjecting sample stock solutions to UV light in a UV cabinet for 7 days. The resulting solution was diluted to yield 100, 100, and 4 μg/ml solution, and then 10 μl were injected into the system to record chromatograms and determine how well the sample held up over time using UPLC.
Forced degradation in neutral conditions
Refluxing the mixture of stock solution of the sample at 60°C for 30 minutes. The solution obtained was diluted to create solutions with concentrations of 100, 100, and 4 μg/ml. A volume of 10 μl was introduced into UPLC, and documenting the resulting chromatograms.
Solution stability
The standard solution and sample solutions of CLA, AMO, and VON stability were assessed by storing them in volumetric flasks that were tightly sealed with a secure cap at room temperature, maintaining a known concentration for a duration of 24 hours. At the conclusion of the study, the sample solutions were analyzed for percentage purity in comparison to freshly prepared standard solutions.
RESULTS AND DISCUSSION
Optimization of method
Our objective throughout the development of a method was to optimize the separation efficiency, minimize the runtime, and maximize the sensitivity. Based on their respective structures, it was determined that all three medications exhibited polar and ionic characteristics. Consequently, the reverse phase technique was chosen, necessitating the use of a non-polar Hibar C18 bis phosphonate column, deemed suitable for the process. In order to optimize and validate a UPLC method, it is customary to optimize the mobile phase composition subsequent to the selection of a suitable column. Acetonitrile was utilized as an organic solvent in combination with 0.1 N mono basic phosphate buffer (pH adjusted to 3.8 with dilute orthophosphoric acid), which has a positive effect on the ionization of the drugs. On the Hibar C18 bisphosphonate column, peak height is not related to drug concentration when using phosphate buffer (pH 6.0) and acetonitrile in a 50:50 mix, and the plate count was not enough. With phosphate buffer (pH 7.4) and acetonitrile in a ratio of 45:55, peaks were merging with insufficient Rs. By setting the pH to 3.8 and slightly raising the buffer-to-solvent mix ratio, the process was further refined. The mobile phase proportion of 60:40 (v/v) of 0.1 N monobasic potassium phosphate buffer (pH 3.8) and acetonitrile, elution of compounds took place within 3 minutes and was rapid, with excellent Rs and peak symmetry.
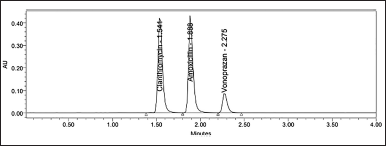
Acquity Hibar bis phosphonate C18 column form waters corporation of dimensions 100 × 2.1 mm, 2 μm, with a rate of flow of 0.2 ml/min, the temperature of column oven at 30oC, a 0.1N mono basic phosphate (pH 3.8) and acetonitrile in the proportion of 60:40 (v/v) as mobile phase and detection wavelength of 210 nm was used to obtain final optimized conditions (Fig. 4).
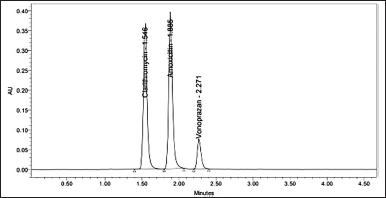 | Figure 4. Standard Chromatogram of Clarithromycin, Amoxicillin and Vonoprazan. [Click here to view] |
Validation
To demonstrate the method’s adequacy, the optimized method was validated according to the ICH guidelines, including system suitability test, specificity through degradation studies of the mixture, linearity, precision, accuracy, robustness, detection, and quantification limit.
System suitability
Six replicate injections of the standard solution were used to evaluate the system suitability parameters. CLA, AMO, and VON each had a retention time of 1.55, 1.89, and 2.28 minutes, respectively. The Rs was found to be between 3.68 and 3.98. The number of plates corresponded to 4,555, 6,723, and 8,915. It was observed that the tailing factor was 1.22, 1.28, and 1.31. The relative SD (RSD) of retention time, tailing factor, and plate count was well below 2% (Table 1).
Specificity
The specificity of the method illustrated that there were no interventions due to the presence of additives or eluent at 210 nm, proving that the optimized method was specific for CLA, AMO, and VON. No peaks were obtained at retention times of drugs due to excipients or eluent or degradation products if present (Figs. 5–8).
Linearity
The linearity of Standard drugs was obtained at a concentration range between 25–150, 25–150, and 1–6 μg/ml for CLA, AMO, and VON, respectively, indicating that the method was suitable at an overbearing range of concentration for the drugs. The linear regression equations and coefficient of determination were found to be y = 15,152x + 8,650.5, y = 14,182x + 4,669.5, y = 80,662x + 2,315.5, and R2 = 0.9991, R2 = 0.9998, R2 = 0.9998, respectively. Thus, it is possible to determine the concentration by graphs of calibrations (Figs. 9–11).
 | Table 1. System suitability parameter. [Click here to view] |
 | Figure 5. Chromatogram of blank solution. [Click here to view] |
 | Figure 6. Chromatogram of placebo solution. [Click here to view] |
 | Figure 7. Chromatograms of sample solution. [Click here to view] |
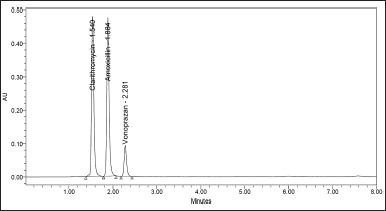 | Figure 8. Chromatograms of sample solution spiked with placebo. [Click here to view] |
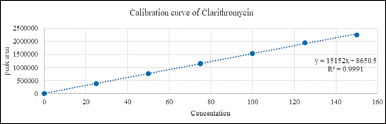 | Figure 9. Calibration curve of clarithromycin. [Click here to view] |
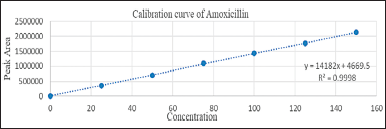 | Figure 10. Calibration curve of amoxicillin. [Click here to view] |
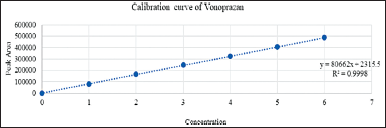 | Figure 11. Calibration curve of Vonoprazan. [Click here to view] |
Precision
RP UPLC assay methods precision was evaluated as part of the repeatability assessment. The relative SD (RSD) values for CLA, AMO, and VON were determined to be 0.8, 0.8, and 0.5, respectively. In the context of intermediate precision, the areas were observed, and the relative SD (%RSD) was computed. The resulting %RSD was determined to be less than 2% (Tables 2 and 3).
Accuracy
The accuracy of the CLA, AMO, and VON was assessed using the percentage recovery method. The mean recovery rates were found to be 99.74%, 99.07%, and 99.85% for CLA, AMO, and VON, respectively. The comprehensive results of the percentage recovery are displayed (Table 4).
Robustness
Consideration was given to variations in mobile phase proportions, flow velocity, and temperature of the column oven to establish the method’s robustness. The outcomes were compiled in (Table 5). The robustness test was passed because the variation between the initial results and the sample results of the robustness test was less than 2.0%.
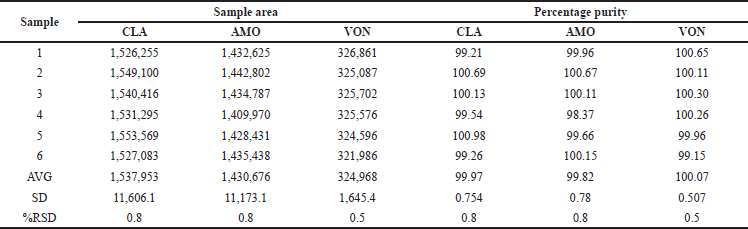 | Table 2. Results of method precision. [Click here to view] |
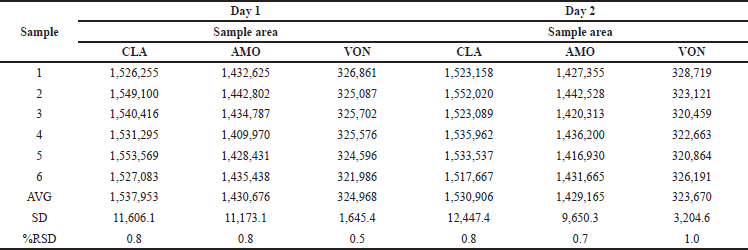 | Table 3. Results of intermediate precision. [Click here to view] |
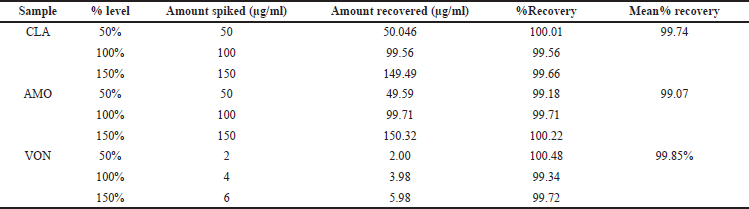 | Table 4. Results of % recovery accuracy. [Click here to view] |
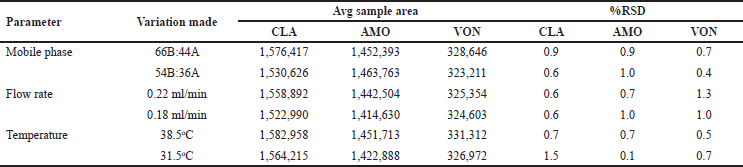 | Table 5. Results of robustness. [Click here to view] |
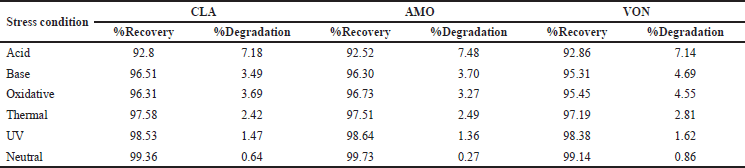 | Table 6. Results of forced degradation studies. [Click here to view] |
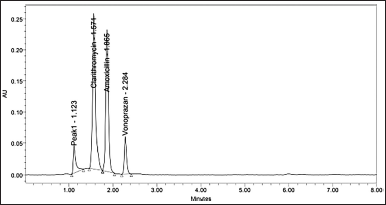 | Figure 12. Chromatogram of acid degradation. [Click here to view] |
LOD and LOQ
The LOD of CLA, AMO, and VON were found to be 0.39, 0.31, and 0.07 μg/ml, and the LOQ of CLA, AMO, and VON was found to be 1.19, 0.93 and 0.21 μg/ml, respectively, determined by regression analysis.
Assay of dosage form
Six sets of samples were prepared and evaluated. The observed results of the test were 99.97% with RSD 0.5% for CLA, 99.82% and RSD 0.8% for amoxicillin, and 100.07% and RSD 0.5% for VON.
Forced degradation studies
Degradation studies were conducted on the formulation, and the samples were injected after they had degraded. Acidic conditions resulted in CLA degradation of about 7.18%, amoxicillin degradation of about 7.48%, and VON degradation of about 7.14%, with about one significant degradation peak noted (Fig. 12). Alkali conditions resulted in CLA degradation of about 3.49%, amoxicillin degradation of about 3.79%, and VON degradation of about 4.69% with about no significant degradation peak. Oxidative stress resulted in CLA degradation of about 3.69%, amoxicillin degradation of about 3.27%, and VON degradation of about 4.55%, and thermal stress resulted in CLA degradation of about 2.42%, amoxicillin degradation of about 2.49%, and VON degradation about 2.81% with no significant degradation peak. The degradation was less than 2% for both drugs under the remaining conditions, i.e., photostability and neutral conditions with no degradation peaks. Table 6 summarizes the forces degradation results.
 | Table 7. Analyte solutions stability. [Click here to view] |
Analyte solution stability
Stored standard solution and test preparation were studied for their stability, revealing that both solutions were stable for up to 24 hours. After 24 hours, the assay values were statistically identical to the initial value, with no detectable loss (Table 7).
CONCLUSION
A novel technique developed for simultaneous quantification of CLA, AMO, and VON in a mixture using the reverse phase ultra performance liquid chromatography (RP-UPLC) technique was rapid, accurate, precise, and reproducible, and was validated as per ICH guidelines, satisfactory results were obtained for all the characteristics tested. In comparison to the previously published RP-HPLC approach [25], the method proposed in this study is more sophisticated. The utilization of the UPLC methodology resulted in a reduction in both the overall analysis duration to less than 3 minutes and the amount of solvents consumed. The decreased retention time resulted in cost reduction for the estimation of CLA, AMO, and VON medication in both API (Active Pharmaceutical Ingredients) and medicinal formulations, while also enhancing sensitivity and speed. Therefore, rendering it appropriate for regular laboratory analysis. This approach also possesses the capability to distinguish and isolate all the degradation peaks from the peaks corresponding to CLA, AMO, and VON. Therefore, it can be utilized as a means of assessing stability and doing routine quality control examinations for CLA, AMO, and VON.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
FINANCIAL SUPPORT
There is no financial support to report.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Kim SY, Chung JW. Best Helicobacter pylori eradication strategy in the era of antibiotic resistance. Antibiotics (Basel). 2020;9(8):436. 10.3390/antibiotics9080436 CrossRef
2. Crist C. Vonoprazan-based therapy for resistant H. pylori superior to standard care-a look at the data behind the FDA approval. GI and Hepatol News. 2022 Aug 9 [cited 2022 Sep 21]. Available from: https://www.mdedge.com/gihepnews/article/256930/upper-gi-tract/vonoprazan-based-therapy-resistant-h-pylori-superior
3. PubChem [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2004-.PubChem Compound Summary for CID 15981397, Vonoprazan; [cited 2023 Oct. 13]. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/Vonoprazan
4. Otake K, Sakurai Y, Nishida H, Fukui H, Tagawa Y, Yamasaki H, et al. Characteristics of the novel potassium-competitive acid blocker vonoprazan fumarate (TAK-438). Adv The. 2016;33(7):1140–57. 10.1007/s12325-016-0345-2 CrossRef
5. Mustafa A, Ali H, Bibi R, Hassan A, Khan S, Khan SA. Simultaneous determination of amoxicillin, clarithromycin and esomeprazole in mice plasma after oral administration by reverse phase HPLC method. J Chromatogr Sep Tech. 2021;12(1):1–7. Available from https://www.longdom.org/open-access/simultaneous-determination-of-amoxicillin-clarithromycin-and-esomeprazolein-mice-plasma-after-oral-administration-by-reverse-phase-61739.html
6. Lal B, Jaimini M, Kapoor DU. Development and validation of a RP HPLC method for the simultaneous estimation of clarithromycin, tinidazole and lansprazole in fixed dose combination. Trop J Pharm Life Sci. 2021;8(4):1–10. Available from https://informativejournals.com/journal/index.php/tjpls/article/view/69
7. Zhiqiang L, Aoxue L, Yang L, Guopeng W, Xinjing C, Hao Q, et al. Development of a stability-indicating HPLC method for simultaneous determination of ten related substances in vonoprazan fumarate drug substance. J Pharm Anal. 2018;149:133–142. 10.1016/j.jpba.2017.11.011 CrossRef
8. Smerikerova M, Bozhanov S, Maslarska V. Development and validation of a RP HPLC method to quantify amoxicillin, tinidazole, esomeprazole and lansaprazole in a mixture. Indian J Pharm Sci. 2019;81(6):1122–1127. 10.36468/pharmaceutical-sciences.611 CrossRef
9. Hakan Aktas A, Mine Sandag A. Liquid chromatographic—chemometric techniques for the simultaneous HPLC determination of lansoprazole, amoxicillin and clarithromycin. J Chromatogr Sci. 2017;55(8):798–804. 10.1093/chromsci/bmx039 CrossRef
10. Jain V, Jain NK. Analytical method development and validation for simulataneous determination of amoxicillin, omeprazole and rifabutin in bulk and in a synthetic mixture by UV spectroscopy. J Drug Deliv Ther. 2019;9(4):788–94. 10.22270/jddt.v9i4.4211
11. Gangishetty S, Surajpal V. RP-HPLC method development and validation for simultaneous estimation of clarithromycin and paracetamol. ISRN Anal Chem. 2013;2013:1–5. 10.1155/2013/948547 CrossRef
12. Alam, M, Hossain S, Bhadra S, Kumar U, Rouf A.S. Development and validation of RP-HPLC method for quantitation of clarithromycin in matrix tablet dosage form. Dhaka Univ J Pharm Sci. 2017;16(1):69–75. 10.3329/dujps.v16i1.33384. CrossRef
13. Tzouganaki Z, Koupparis M, Development and validation of an HPLC method for the determination of the macrolide antibiotic clarithromycin using evaporative light scattering detector in raw materials and pharmaceutical formulations. Mediterr. J. Chem. 2017;6(4):133–141. 10.13171/mjc64/01706211420-tzouganaki CrossRef
14. Osman A, Sumaiya F, Anas R. Evaluation and validation of a UPLC method for estimation of amoxyclav in oral dosage form. World J Pharm Life Sci. 2020;6(9):107–113. Available from https://www.researchgate.net/publication/344218584_EVALUATION_AND_VALIDATION_OF_A_UPLC_METHOD_FOR_ESTIMATION_OF_AMOXYCLAV_IN_ORAL_DOSAGE_FORM
15. Tavakoli N, Varshosaz J, Dorkoosh F, Zargarzadeh MR. Development and validation of a simple HPLC method for simultaneous in vitro determination of amoxicillin and metronidazole at single wavelength. J Pharm Biomed Anal. 2007;43(1):325–329. 10.1016/j.jpba.2006.06.002 CrossRef
16. Marcel S, Tito U, Ines I, Pierre NJ. Validation of HPLC-UV method for determination of amoxicillin Trihydrate in capsule. Annals Adv Chem. 2018;2:56–72. 10.29328/journal.aac.1001014
17. Beg S, Kohli Swain S, Hasnain MS. Development and validation of RP-HPLC method for quantitation of amoxicillin trihydrate in bulk and pharmaceutical formulations using box-Behnken Experimental design. J Liq Chromatogr Related Technol. 2012;35(3):393–406. 10.1080/10826076.2011.601493 CrossRef
18. Tippa DMR, Singh N. Development and validation of stability indicating HPLC method for simultaneous estimation of amoxicillin and clavulanic acid in injection. Am J Anal Chem. 2010;1:95–101. 10.4236/ajac.2010.13013 CrossRef
19. Sahoo NK, Sahu M, Algarsamy V, Srividya B, Sahoo CH. Validation of assay indicating method development of amoxicillin in bulk and one of its marketed dosage form by RP-HPLC. Annals Chromatogr Sep. 2016;2(1):1014. CrossRef
20. Gebretsadik H, Kahsay G, Eticha T, Gebretsadikan T. A validated new RP-HPLC method for simultaneous determination of amoxicillin, ampicillin and cloxacillin in pharmaceutical formulations. Acta Chromatogr. 2022;1–10. 10.1556/1326.2022.01043 CrossRef
21. Deshmukh N, Patel R, Das PK. Development of quantitative method for analysis of meropenem and amoxycillin by RP HPLC method. Pharm Biol Eval. 2016;3(4):431–436. Available from https://www.academia.edu/27695300/Development_of_quantitative_method_for_analysis_of_Meropenem_and_Amoxycillin_by_RP_HPLC_method
22. De Marco BA, Hisano Natori JS, Fanelli S, Tótoli EG, Nunes Salgado HR. Characteristics, properties and analytical methods of amoxicillin: a review with green approach. Crit Rev Anal Chem. 2017;47(3):267–277. 10.1080/10408347.2017.1281097 CrossRef
23. International Conference of Harmonization (ICH) of technical requirements for registration of pharmaceuticals for Human use an Use Q2:(R1), Harmonized tripartite guideline, Validation of Analytical procedures, 6th Nov 1996. Available from https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf
24. International Conference of Harmonization (ICH) of technical requirements for registration of pharmaceuticals for human use Q1A:(R2), harmonized tripartite guideline, stability testing of new drug substance and products, 6th Nov 1996. Available from: https://database.ich.org/sites/default/files/Q1A%28R2%29%20Guideline.pdf
25. Umeshbhai PY, Patel SR. Spectroscopic and chromatographic method development and validation for the estimation of antimicrobial agents and their combinations in synthetic mixture and assay method for the pharmaceutical dosage forms. Atmiya University; 2022. Available from http://hdl.handle.net/10603/449869