
INTRODUCTION
Drug development studies consist of processes, that take a long time, and cost, furthermore, are not always successful in vivo trials [1,2]. The process of drug development timeline takes about 7–15 years for drug molecules to pass through various stages and become usable as drugs [3–5]. Moreover, drug development processes are complex stages that can be achieved as a result of cooperative studies in sciences such as chemistry, biology, and pharmacology [6]. To solve the problems that may be encountered, mathematical models have been developed that include in vivo and in vitro approaches and can evaluate physiological and pharmacological information together [7]. By using the developed in silico models, it is possible to reduce the time of drug development studies and costs [8–12].
In the pharmacy field, in silico tools have emerged as vital resources. One of the primary benefits of employing in silico approaches is their ability to predict drug properties according to the molecular structure [13]. In addition, they can predict absorption, distribution, metabolism, and excretion (ADME) properties [13,14], thereby reducing the need for extensive in vivo studies and leading to significant time and cost savings, ultimately accelerating drug production [15,16] by identifying and predicting the impact of drugs on biological systems, clinical use can be improved, side effects can be avoided, and treatments can be better selected and developed [17]. Several official authorities have recommended and even provided in silico tools for assessing chemicals in terms of hazard identification, risk assessment, and human health safety evaluation. Workflows have been established to guide the application of these in silico tools for chemicals risk assessment and computational toxicology [10].
When it comes to disadvantages, accurately predicting oral absorption and bioavailability using in silico methods can still pose challenges [18,19]. Some in silico software lack transparency in disclosing the underlying algorithms used for predictions. Despite recent advancements, there is still a gap in correlating in vivo, in vitro, and in silico ADME parameters [14].
In silico methods are applications based on calculating the properties of drugs (such as solubility and partition coefficients) and other chemical substances and their effects on the body with computer models [20–22]. These are applied using computer software at different stages in drug discovery and development. In silico methods include quantitative structure activity relationship (QSAR) methods which assess to evaluate the data arising from pharmacology laboratories. Molecular docking methods depend on drug and macromolecule interaction to provide atomic-level data. Quantum medicinal chemistry methods facilitate the assessment of the electronic structure and provide valuable insights into the chemical and biochemical processes related to drugs. Molecular dynamic methods are particularly helpful in modeling, exactly in fit effect of drug-macromolecule complexes. In addition, virtual screening efforts aid as a complementary technique to preclinical screening implementing on lead compounds. In the field of drug discovery, artificial intelligence methods are making significant progress and providing a significant number of innovative tools. Furthermore, the application of pharmacoinformatic techniques is highly demanded at each stage of the drug discovery and development process, including target identification, validation, 3-D structure prediction, medicinal and product chemistry, pharmacology (both in vitro and in vivo), pharmaceutics, formulation, drug delivery and disposition (pharmacodynamic and pharmacokinetic), preclinical and clinical trials, and postrelease study of drug-patient response. In the last years, these methods have gained increasing significance in certain fields, for instance, pharmacology, toxicology, and biotechnology [23]. This review aims to highlight the most common in silico method used in drug discovery and development.
IN SILICO CONCEPT
The term in silico, meaning “performed on computer or via computer simulation,” is derived from the concepts of in vivo and in vitro. The United States Environmental Protection Agency (EPA) defines the term in silico as the “integration of modern computing and information technology with molecular biology to improve agency prioritization of data requirements and risk assessment of chemicals” [24,25]. The European Union defines it as data models obtained without testing and uses them in the risk assessment of chemical substances [26]. Computer-aided methods have many advantages such as predicting the biological activity of the drug based on its structural features, determining its efficacy and side effects before clinical studies, limiting the use of animal experiments, helping the rational design of safe drug candidates, repositioning marketed drugs, and facilitating the drug development process [27–30].
Identification and analysis of a new drug’s efficacy, safety, toxicity, and drug specifications are very important in the drug discovery or formulation development stage. For this purpose, in vitro and in vivo experiments have been implemented for a long time. Due to the high use of experimental animals in in vivo studies, the high cost of these studies, and the long-time consumption. Scientists have sought alternative methods, leading to the widespread adoption of in vitro experiments utilizing invertebrates, cell cultures, and tissue-organ baths. Nevertheless, the demand for more efficient and ethical approaches continues to grow, prompting the development of in silico methods [31].
DATA SECURITY AND VALIDATION IN IN SILICO APPROACHES
External validation data sets and the diversity of the size and structure of the training data sets partially impact the estimated performance of in silico models [32,33]. In silico models and simulations are based on data obtained as a result of in vitro and in vivo experimental studies. Compliance and accuracy of these studies with scientific methodology are significant for the reliability of in silico methods [15]. According to the European Chemicals Agency (ECHA), the necessary steps for the reliability and acceptance of the data to be obtained by in silico methods should be carried out systematically and regularly. The procedures have been classified for in silico methods according to the Regulation of Registration, Evaluation, Authorization, and Restriction of Chemical Substances [34]. Details of this classification process are shown in Table 1. Applicability is an important factor in the validation of in silico methods. In other words, accurate predictions about physicochemical and structural properties and activity should be made by using a model or simulation. While determining this, an accuracy level is determined for the in silico method beforehand and when the method is applied to a chemical substance, it is analyzed mathematically and statistically whether it gives results at this accuracy level. In addition, parameters such as sensitivity and selectivity required for validation are also determined. While evaluating validation parameters in in silico methods, limiting factors such as data reliability, limited chemical substance groups, and applicability areas, in addition, the possibility that pharmacological and toxicological results may vary by different mechanisms should be considered in data analysis and evaluation studies [32].
IN SILICO METHODS
This section focuses on the most common in silico methods.
Structure activity relationship (SAR)
In 1868, Crum-Brown and Fraser suspected the quaternary ammonium character of curare [35]. Curare is a poison that causes muscle paralysis and blocks the action of the excitatory neurotransmitter acetylcholine on the muscle receptor. Analysis of its neuromuscular blocking effects in animals concluded that this physiological effect was the function of tubocurarine [36]. A little later, Richardson studied the increased hypnotic activity of aliphatic alcohols in relation to their molecular weight (MW). These studies formed the basis for the SAR model [37]. Currently, in silico modeling is employed for SAR analysis of pharmacological and toxicological activities. This modeling application involves a qualitative analysis of the chemical properties, as well as the biological and pharmacological effects of molecules. Functional groups, stereochemical structure, size and shape, chemical reactivity, resonance, and inductive effects are taken into account [38,39].
Quantitative SAR
In 1893, Richet noted the effect of physicochemical properties on pharmacological activity [40,41]. In the 1960s, Corwin Hansch showed the importance of the change in physicochemical properties that could lead to variation in biological activity (the structure-activity pattern) by examining certain structure modifications of the compounds [41,42].
 | Table 1. Process steps determined by ECHA for the reliability of in silico method (according to [34]). [Click here to view] |
The basis of the QSAR method is based on the tendency of structurally similar molecules to show similar biological activity. These models mathematically describe how the activity response of a target molecule that binds a ligand varies according to the structural properties of the ligand. QSAR is obtained by calculating the correlation between experimentally determined biological activity and various properties of ligand binders and is used to predict the activity of new drug molecule analogs. The success of a QSAR model depends on the molecular descriptors chosen and their ability to predict biological activity. The steps that take place in the QSAR model are as follows: active molecules that bind to the desired target molecule and their activities; database search or high throughput scanning result is defined. The number of bonds, atoms, functional groups, and surface area, that affect biological activity. After defining the structural or physicochemical molecular properties, a QSAR model is created between the biological activity and the defined properties of the drug molecules, and this model is used to optimize known active compounds to increase biological activity. Then, new optimized drug molecule activities are experimentally tested [43].
Machine learning approaches such as neural networks and support vector machine methods are used to construct QSAR models. Table 2 presents some of the machine learning algorithms used in some QSAR models [44].
QSAR models summarize the relationship between chemical structure and biological activity and predict the activities of new chemical molecules. Quantitative structure-property relationship (QSPR), in which a chemical property is defined as a variable, is a reliable statistical model for estimating the properties of new chemicals and analytical systems. Quantitative structure reactivity relationship, quantitative structure chromatography relationship, quantitative structure toxicity relationship, quantitative structure electrochemistry relationship, and quantitative structure bioavailability relationship are other approaches that can be given as examples [45–47].
 | Table 2. Machine learning algorithms are used in some QSAR models (according to [44]). [Click here to view] |
Previous studies have reported that QSAR models are divided into six categories of QSAR dimensions based on their molecular descriptor [48–51]. Table 3 provides a brief overview of these categories. Among these dimensions, the 3D-QSAR approach, a ligand-based drug design method, has proven to be instrumental in designing novel compounds. Chavda and Bhatt [52] conducted a study using four different 3D-QSAR techniques, including comparative molecular field analysis (CoMFA), comparative molecular similarity indices analysis (CoMSIA), molecular hologram QSAR (HQSAR), and topomer CoMFA, to design new B-Raf inhibitors using 28 synthetic B-Raf inhibitors. CoMFA correlated biological activity with steric and electrostatic parameters, while CoMSIA associated biological activity with hydrophobic, hydrogen bond donor, hydrogen bond acceptor, steric field, and electrostatic parameters. HQSAR correlated biological activity with the structural part of each group and atom of the molecules, providing essential insights into the impact of atoms, stereochemistry, and fragments on biological activity. The topomer CoMFA, aimed at overcoming CoMFA limitations, divided molecules into fragments, generating a model directly correlated with the molecule’s fragments. N-fold statistical validation yielded q2, r2, and r2pred values of 0.638, 0.969, and 0.848 in CoMFA, 0.796, 0.978, and 0.891 in CoMSIA, and 0.761, 0.973, and 0.852 in CoMSIA. For HQSAR analysis, statistical values were q2 = 0.984, r2 = 0.999, and r2pred = 0.634, with a best hologram length of 97. Topomer CoMFA showed a q2 value of 0.663 and an r2 value of 0.967. Contour map analysis of these 3D-QSAR techniques helped identify crucial features of purinylpyridine, facilitating the design of novel molecules as B-Raf inhibitors for melanoma cancer treatment [52].
Molecular dynamics (MD) simulation
MD simulation is a computational technique that calculates the forces between molecules and computes their motion through numerical integration. Starting with the positions of atoms from an identified crystal structure and randomly generated velocities, Newton’s equations are used to calculate the positions and velocities of the atoms at small time intervals. Through iterative steps, the forces are recalculated, and the simulation progresses. After an equilibration period (thousands of steps), during which the system (install) reaches the desired temperature and pressure, a production period begins, storing the molecular history for later analysis [53,54].
MD simulations have three essential applications in biomolecular dynamics. First, they bring biomolecular structures to life, providing insights into their natural dynamics in solution over different timescales. Second, MD simulations yield thermal averages of molecular properties, allowing the calculation of bulk properties of fluids and free energy differences for chemical processes, such as ligand binding, using time-averaged molecular properties that approach experimentally measurable ensemble averages, based on the ergodic hypothesis. Third, MD simulations explore the thermally accessible conformations of a molecule or complex [55]. MD simulations are commonly combined with various experimental structural biology methods, such as X-ray crystallography, cryoelectron microscopy, nuclear magnetic resonance, electron paramagnetic resonance, and Forster resonance energy transfer [54].
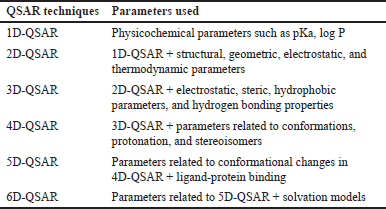 | Table 3. Groups of QSAR models according to their analysis capabilities (according to [48–51]). [Click here to view] |
Molecular docking
Molecular docking is a powerful technique that investigates how small molecules behave within the binding area of a target protein. As more protein structures are determined through X-ray crystallography or nuclear magnetic resonance, molecular docking has gained prominence as a valuable tool in drug discovery. It is now possible to perform docking against homology-modeled targets for proteins with unknown structures. Through docking approaches, the druggability of compounds as well as their specification against definite targets can be computed, aiding in lead optimization processes. Molecular docking programs use a search algorithm to iteratively evaluate the ligand’s conformation until it converges to the lowest energy state. Subsequently, an affinity scoring function (ΔG [U total in kcal/mol]) is applied to order the candidate poses by summing the electrostatic along with van der Waals energies. In addition, the driving forces behind these interactions in biological systems strive for complementarity between both the shape and electrostatics of the binding area surfaces and the ligand or substrate [56]. This comprehensive approach facilitates the identification of potential drug candidates and their interactions with the target protein, thus supporting the drug discovery process. Until 2016, no molecular modeling study has been conducted on ionone-based chalcones for anti-prostate cancer activity. Popular QSAR methods such as CoMFA and CoMSIA use 3D information to identify sites on molecules that can be modified to create more specific ligands, while HQSAR uses fingerprints to highlight sub-structural features significant for biological activity. In addition, molecular docking analysis provides insights into ligand-receptor interactions. By combining 3D-QSAR and docking, a more comprehensive understanding of the structural features at the protein’s binding area and protein-ligand interactions can be obtained to aid in the design of new potential molecules. The generated models in this study exhibited statistical precision with higher q2 and r2 values. The presence of bulky, negatively charged substituents with H-bond acceptors at specific positions increased the activity. Moreover, the hydrophobic property of the phenyl ring played a crucial role in the anti-cancer activities of ionone-based chalcones. These findings led to the design of twelve new anti-prostate cancer compounds (predicted high activity) [57]. In another investigation, Shahzadi et al. [58] synthesized MgO-doped cellulose nanocrystal grafted poly acrylic acid (CNC -g-PAA) hydrogel for antibacterial and anti-cancer activities. The hydrogel demonstrated improved bactericidal tendencies against both Gram-negative and Gram-positive bacteria, and molecular docking analyses were performed to evaluate the interactions between the nanocomposite hydrogel and biomolecules. The hydrogel also exhibited reactive oxygen species production by photocatalysis and showed promising potential for controlled drug delivery, with successful loading of the model anticancer drug Doxorubicin. In vitro cytotoxicity analysis further confirmed the enhanced antitumor efficiency of the nanocomposite hydrogels, suggesting their potential as carriers for innovative biomedical applications [58]. Furthermore, Shahzadi et al. [59] investigated the antibacterial and anti-arthritic effects of CNC-g-PAA and CNC-g-PAA doped with CaO. Molecular docking analysis was also conducted to evaluate the binding interaction between the targeted proteins and the synthesized nano-biomaterials. The results demonstrated improved antitumor effectiveness of CNC-g-PAA and CNC-g-PAA/CaO, suggesting their potential as delivery vehicles for multifunctional biomedical applications. These findings highlight the promising prospects of hydrogels in the field of biomedical research [59].
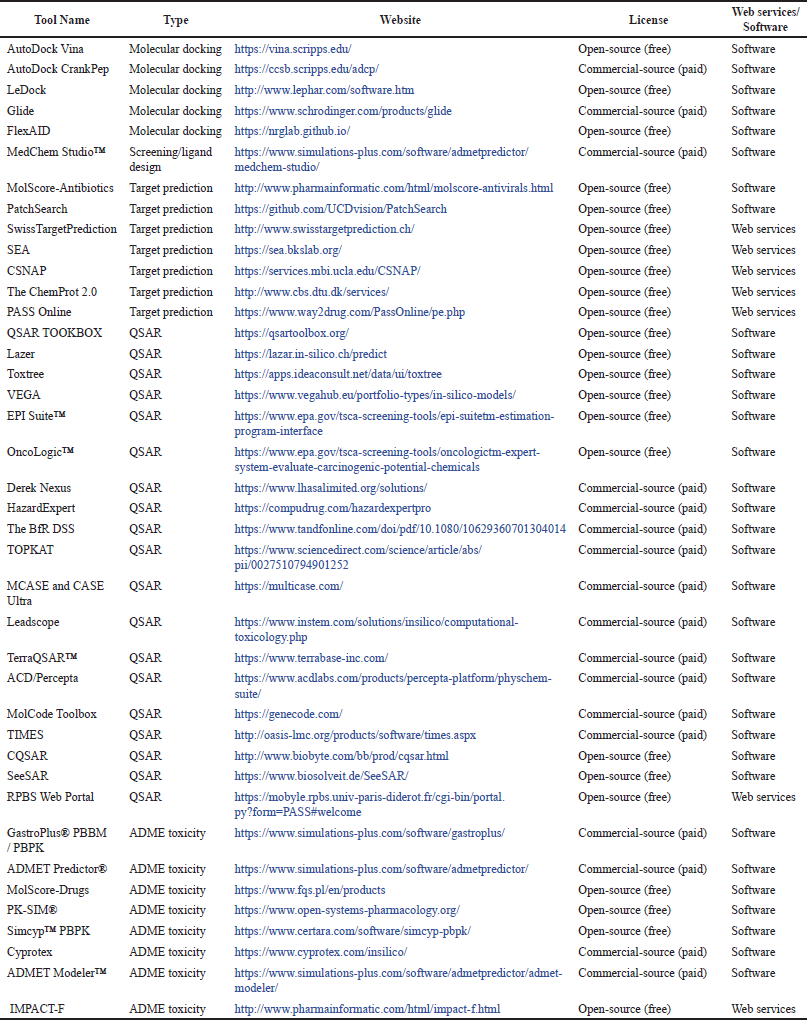 | Table 4. In silico tools are used for drug discovery and development. [Click here to view] |
EXPERT SYSTEMS
Most in silico methods are based on the knowledge of pharmacology and toxicology specialists. Information about the molecular structures of substances is often incomplete, or complex. For this reason, expert systems have been developed based on the explanation of different expert knowledge with various data processing methods and algorithms, as seen in Table 4. One such software is SAR which was created by combining QSAR and data banks and mathematically expresses the rules for a chemical molecule. The most important advantage of the QSAR method is that it can be evaluated with a specific mechanism when needed [39].
Sample applications of in silico methods
Computer-aided tools have proven to be greatly effective within the healthcare industry. They have been used in the development of distinctive molecules that have successfully demonstrated their therapeutic potential in clinical trials for various disorders. Some remarkable examples of the uses of computer-aided tools in the development of approved drugs include an angiotensin-converting enzyme (ACE, captopril) inhibitor used in cardiovascular diseases treatment and prevention, which was approved in 1981 as well as carbonic anhydrase inhibitor (dorzolamide) used for treating glaucoma and approved in 1995. In addition, saquinavir (approved in 1995), ritonavir, and indinavir (both approved in 1996) were approved as medications for the treatment of human immunodeficiency virus (HIV) in accordance with safety regulations [60]. Other examples can be found in Table 5.
AutoDock Vina
AutoDock Vina is a freely available software used for conducting molecular docking. The program was initially developed and implemented by Dr. Oleg Trott at The Scripps Research Institute’s Molecular Graphics Lab, which is now known as CCSB [61].
AutoDock CrankPep or ADCP
ADCP (AutoDock for peptides) is a specialized docking engine based on AutoDock, specifically designed for docking peptides. It combines techniques from the protein folding area with an effective representation of a rigid receptor using affinity grids. The process involves folding the peptide within the energy landscape obtained by the receptor, optimizing the interaction between the peptide and the receptor through a Monte–Carlo search, as a result, docked peptides are obtained. The program can handle peptides (3-D structures) within Protein Data Bank files or in the form of a sequence string [62].
LeDock
LeDock is a specialized software designed for fast, precise, and flexible docking of molecules into a protein. It has been shown to achieve a pose-prediction precision of over 90% on the Astex diversity group. For drug-like molecules, it typically takes about 3 seconds per run, making it a time-efficient tool. LeDock has been successfully utilized in high-throughput virtual screening campaigns, leading to the discovery of novel kinase inhibitors and bromodomain antagonists. One of its key features is its ability to directly use the SYBYL Mol2 format as input for small molecules [63].
FlexAID
FlexAID is an advanced docking algorithm capable of handling both small-molecules and peptides as ligands, with proteins/nucleic acids serving as targets. Its notable features include accommodating full ligand and target side-chain flexibility, adding versatility to the docking simulations. The scoring function employed by FlexAID is unique in its soft nature, reducing reliance on specific geometric criteria and instead focusing on surface complementarity. To fine-tune the scoring function’s energy parameters, a substantial dataset containing native and near-native conformations (less than 2Å root mean square deviation) of almost 1,500 complexes from the PDBbind database was used as true positive examples. Remarkably, it has demonstrated superior predictive capabilities compared to well-established software such as AutoDock Vina and FlexX when predicting binding poses. This superiority is especially evident in cases where target flexibility is essential, as often encountered when applying homology models [64].
MedChem Studio™
MedChem Studio™ represents a comprehensive cheminformatics software bundle, encompassing a wide range of tools for essential drug discovery and development tasks, including high throughput screening analysis, prioritization, lead identification, de novo design, lead optimization, and scaffold hopping. An attractive feature is the “VIEWER” mode, which does not require a license and facilitates collaboration among scientists with different expertise. In addition, the software offers MedChem Designer™, a valuable molecular drawing tool, freely accessible from MedChem Studio. It grants users the ability to input or modify structures, visualize metabolites, define structure queries, and offer other valuable functionalities to enhance the software’s versatility [65].
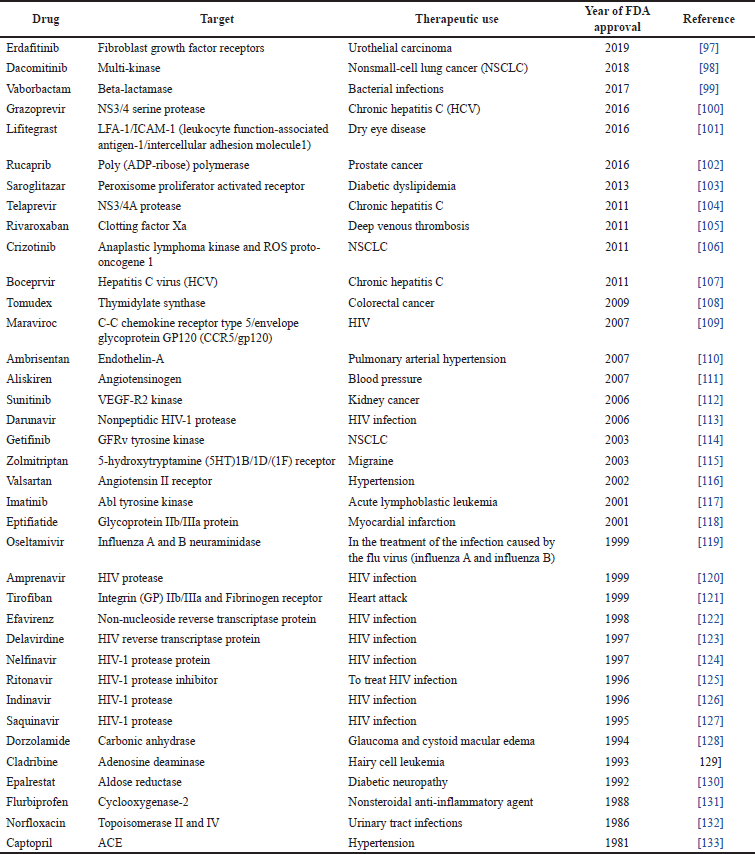 | Table 5. Drugs developed by computer-aided approaches (according to [96]). [Click here to view] |
MolScore-Antibiotics
MolScore-Antibiotics serves as a valuable tool for distinguishing between antibiotics and nonantibiotics. This scoring system assigns a probability value between 0 and 1 to a compound, indicating the likelihood of possessing antibiotic activity. With its capability to assess compounds’ potential antibiotic properties, MolScore-Antibiotics proves beneficial in guiding the process of selecting compounds for focused biological screening, particularly in prioritizing compounds from extensive collections. Our expert system’s analysis demonstrated that many compound databases from external suppliers have a limited number of compounds with antibiotic activity. As a result, MolScore-Antibiotics enables efficient cherry-picking of interesting antibiotic compounds, as exemplified in the selection of antibiotics from a database consisting of 195.064 compounds [66].
PatchSearch
PatchSearch is an innovative tool designed to facilitate the identification of potential off-target proteins by searching for structurally conserved binding sites across the entire surface of a protein. This powerful method employs a quasi-clique approach, allowing for a flexible consideration of binding area atoms without imposing overly strict distance conservation constraints. In essence, PatchSearch identifies dense subgraphs, or quasi-cliques, on the protein surface [67].
SwissTargetPrediction
SwissTargetPrediction offers a range of unique capabilities. First, it allows users to integrate 2-D and 3-D similarity values with known ligands. Second, the tool delivers results for five distinct species, enabling researchers to explore drug-target interactions across different organisms. Finally, SwissTargetPrediction permits users to map predictions based on target homology, facilitating the transfer of target predictions within and between organisms. These exceptional features make SwissTargetPrediction a valuable asset in drug discovery and target identification research [68].
Similarity ensemble approach (SEA)
The SEA employs ligand-based chemical similarity to establish relationships among proteins. This method enables quick searching of extensive compound databases and the creation of cross-target similarity maps [69].
Chemical similarity network analysis pull-down (CSNAP) web
CSNAP is a computational technique used to identify compound targets through network similarity graphs. By placing query and reference compounds on the network connectivity map, a graph-based neighbor counting method ranks the consensus targets within the neighborhood of every query ligand. CSNAP proves valuable in high-throughput target drug discovery as well as off-target prediction for compound sets obtained from either phenotype-based or cell-based chemical screens [70].
ChemProt-2.0
ChemProt-2.0 is a publicly accessible compilation of several chemical-protein annotation resources, enriched with diseases and clinical result information. This updated database now includes over 1.15 million compounds plus 5.32 million bioactivity measurements of all these for 15,290 proteins. Each protein is associated with quality-scored human protein–protein interaction information, comprising more than half a million interactions, which facilitates the study of diseases and biological outcomes through protein complexes. Notably, ChemProt-2.0 integrates therapeutic effects and adverse drug reactions, offering insights into proteins linked to clinical results. To enhance its functionality, the database employs new chemical structure fingerprints computed using the SEA [71].
QSAR toolbox
The toolbox is a user-friendly and free software application designed to facilitate reproducible plus transparent chemical hazard evaluation. It provides various functionalities, including the retrieval of experimental data, simulation of metabolism, and profiling of chemical properties. This valuable information and tools enable users to identify structurally and mechanistically known analogs and chemical classifications, which can be utilized for read-across and trend analysis, effectively filling data gaps in hazard evaluation [72].
Lazar
Lazar is a valuable tool utilized for predicting the toxic properties of chemical structures. In addition, it employs the QSAR statistical approach to generate predictions for a query structure by utilizing a database of experimentally determined toxicity data. The Lazar software model has demonstrated impressive performance in external validation datasets, achieving an accuracy (86%) along with a sensitivity (78%) in the carcinogenicity test, while attaining a remarkable accuracy (95%) for the mutagenicity test [73].
Toxtree
Toxtree is a valuable and freely available QSAR tool designed to assess the Cramer class of a chemical compound and evaluate its relative toxic hazard. Toxtree is a collaborative effort between Ideaconsult Ltd. and the Joint Research Centre of the European Commission [74].
VEGA
VEGA places a strong emphasis on generating transparent, reasonable, reproducible, and verifiable data in its models. To achieve this, they have optimized a series of tools that establish connections between the outcomes obtained for the target chemical and those obtained for structurally related compounds. These tools facilitate a reproducible read-across procedure, which involves extracting required values for the target compound depending on identified values for similar substances. This read-across strategy is made possible through the implementation of independent algorithms that go beyond QSAR models. These algorithms take advantage of identifying similar compounds as well as analyze the importance of descriptors and fragments for the chemical of interest plus the associated compounds [75].
EPI Suite™
The QSPR models available in EPI Suite™ have found extensive application in predicting physicochemical characteristics and half-lives of chemicals, particularly for screening-level hazard evaluation. These models were developed based on property data obtained from training sets, primarily comprising anthropogenic chemicals, including persistent organic pollutants, organochlorine pesticides, personal care products, modern pesticides, and industrial chemicals [76].
OncoLogic™
OncoLogic™, developed in collaboration with the EPA’s structure-activity team (SAT), is a unique knowledge-based software. The SAT consists of globally recognized experts responsible for assessing the carcinogenic potential of newly developed chemicals within the United States or those imported for marketing purposes. The objectives behind creating OncoLogic™ encompass several key aspects: Offering industry-specific guidance on crucial elements for developing safer chemicals. Providing a comprehensive source of information for all stakeholders, explaining the rationale behind identifying potential cancer hazards associated with chemicals. Promoting research initiatives to bridge existing knowledge gaps in this field [77].
HazardExpert
CompuDrug’s HazardExpert stands as a crucial software tool, enabling the initial estimation of toxic symptoms caused by organic compounds in both humans and animals. Notably, HazardExpert incorporates a robust model that considers the bioavailability of the compounds. Its predictive capabilities surpass human experts, delivering toxic effect estimations with remarkable precision. HazardExpert offers toxicity prediction for organic compounds on the basis of toxic fragments, with results provided for seven distinct toxicity classes, including oncogenicity, mutagenicity, teratogenicity, membrane irritation, sensitivity, immunotoxicity, and neurotoxicity. In addition, the software calculates bioavailability built on pKa and logP, as well as bioaccumulation. Users can further predict toxicity for metabolites [78].
The BfR decision support system (DSS)
The DSS developed by the German Federal Institute for Risk Assessment (BfR) aims to evaluate specific hazardous properties of pure chemical substances, which include skin and eye irritation and/or corrosion. Serving as a rule-based system, the BfR-DSS has significant applicability in the regulatory framework classification of chemical substances within the European Union [79].
TOPKAT
TOPKAT aims to predict chemical carcinogens, focused on its capability to foresee the carcinogenicity of chemicals examined by the National Toxicology Program. However, TOPKAT’s performance proved to be inadequate when attempting to distinguish potential rodent carcinogens and noncarcinogens within the studied dataset. The TOPKAT database consists of identified carcinogens and noncarcinogens, and the software attempts to identify chemicals that are most “similar” to unidentified compounds. Nonetheless, when observing six examples, the chemicals deemed “similar” by the software exhibited no apparent connection to the chemical of interest concerning metabolism or mechanism of carcinogenicity [80].
MCASE and CASE Ultra
CASE Ultra is a computer-based toxicology software designed to detect structural alerts associated with toxicity through (QSAR) analysis. The (QSAR) models in CASE Ultra undergo validation following Organization for Economic Co-Operation and Development guidelines and are accompanied by QSAR model reporting format reports. The software provides models for various toxicological endpoints, including bacterial mutagenicity/ICH M7, genotoxicity, carcinogenicity, skin sensitization, acute toxicity, endocrine disruption, reproductive toxicity, developmental toxicity, cardiotoxicity, hepatotoxicity, renal toxicity, ADME, and ecotoxicity [81].
LeadScope
LeadScope is an innovative computer software that seamlessly connects chemical and biological data, providing medicinal chemists with a powerful platform to visualize and interactively investigate extensive collections of chemicals, their properties, and biological activities. Within the software, chemical structures are intelligently categorized into a vast taxonomy of recognizable structural features, encompassing functional groups, aromatic rings, and heterocycles. All of these structural elements are further combined with general substituents, representing the fundamental construction blocks of medicinal chemistry [82].
TerraQSAR™
TerraQSAR™ computer programs have been meticulously crafted to offer rapid and dependable assessments of both the biological effects and physicochemical properties of organic compounds. The program provides valuable output data, including computed effect or property values represented in pT (log1/C) and mg/l (for rat and mouse intravenous: mg/kg b.w.), as well as the MW of the compounds [83]. For those seeking accurate and efficient estimations, TerraQSAR™ proves to be a valuable tool in the field.
ACD/Percepta
ACD/PhysChem Suite comprises multiple prediction modules that deliver accurate assessments of physicochemical properties based on molecular structure. It enables the estimation of essential properties, such as aqueous solubility, logD, logP, pKa, boiling point, Sigma, and other molecular specifications, specifically for organic compounds. Users can examine the calculated outcomes using sorting and plotting tools, ensuring the reliability of predicted physicochemical values. Moreover, the suite facilitates investigations into QSPR, structural modifications, and lead optimization to achieve desired target profiles. To enhance the applicability to novel chemical space, predictors can be trained with experimental data. ACD/PhysChem Suite also accommodates custom models and in-house prediction algorithms, offering flexibility for diverse scientific applications [84].
MolCode toolbox
The Molcode toolbox is an exceptional computational expert system developed for rapid and reliable prediction of crucial biomedical and environmental properties of chemicals and materials. It relies on proprietary techniques that map compound properties onto extensive sets of molecular descriptors, which include thousands of descriptors derived from quantum chemical theory, meticulously considering the intricate spatial and electronic structures of molecules. Ahead of this computation, molecular mechanics is employed to perform a comprehensive conformational search of extensive compounds. Using the Molcode toolbox, users have the flexibility to load their own compound structures, make adjustments to encoded compounds, or even create and optimize entirely new ones [85].
CQSAR
David Elkins initially developed the first program for data searching in 1970, but its usage was cumbersome due to the encoding of structures in the Wisswesser line notation and the requirement to use IBM cards, resulting in slow and inconvenient operations. Consequently, the current C-QSAR program is the result of over 35 years of continuous research and development. The program was expertly designed and authored by David Hoekman, incorporating the widely adopted simplified molecular input line entry system notation (originated by David Weininger) for entering chemical structures. In addition, it effectively employs the Merlin searching program [86].
Ressource Parisienne en Bioinformatique Structurale (RPBS) web portal
RPBS is a collaborative effort involving multiple teams, with the goal of providing exclusive services in the domain of structural bioinformatics through a single-entry point. The expertise offered spans from sequence and structure analysis to modeling of protein as well as the design of drugs. However, not all aspects are currently addressed on the RPBS server. The server itself encompasses a wide range of tools, meticulously designed to holistically cover diverse areas of structural bioinformatics. As of now, the P-server section is only partially functional. In addition, RPBS offers access to proprietary software developed by their teams. Among other RPBS tools, some are specialized in handling 3-D structures, namely SA-Search, employed for discovering structural similarities and relying on a structural alphabet plus Scit, used for comparison side-chain conformations. Furthermore, RPBS maintains different compilations of commercially achievable organic compounds that prove useful for conducting structure-based in silico testing experiments [87].
GastroPlus®
GastroPlus® is an advanced software designed for modeling and conducting simulations of various properties of drugs or chemicals. These encompass release rate, absorption, bioavailability, pharmacodynamics, and pharmacokinetics. The software is equipped to predict drug-to-drug interactions, effects on animals, and virtual patient populations. It also facilitates researchers in modifying pharmacodynamic models based on observed data and employing the fitted models to forecast pharmacodynamic changes resulting from alterations in drug or chemical dosage, dosage form, and dosing regimen. Moreover, GastroPlus® enables the creation of in vitro–in vivo correlations and predictions of absorption and systemic distribution/elimination for large molecules [88].
ADMET Predictor®
ADMET Predictor® is advanced computer software designed specifically for QSAR modeling of ADMET properties. It provides estimations for more than 140 ADMET properties, offering a comprehensive analysis of drug properties. The software allows users to build QSAR and QSPR models by applying both in-house and publicly available data sources through a proprietary software program. Its user-friendly interface facilitates easy manipulation and visualization of data [89].
MolScore-Drugs
Amidst the diverse array of structures found in marketed drugs, molecules exhibiting biological activity share frequent characteristics. Through a thorough analysis of these intricate drug patterns, they have developed an expert system capable of distinguishing between drugs and nondrugs. For instance, MolScore-Drugs near 0 signifies the lowest predicted probability, while MolScore-Drugs near 1 indicates the highest predicted probability with an interesting ADME-profile. This expert system is founded on a collection of robust models. Leveraging SARs, we can estimate the drug-like chemical space effectively. In addition, structure-property relationships derived from their in-house ADME/Tox-database enable the prediction of ADMET properties and identification of potential risks, ultimately reducing clinical failures [90].
PK-Sim®
PK-Sim® is an extensive software tool designed for all body physiologically based pharmacokinetic (PBPK) modeling. It offers quick access to all pertinent anatomical and physiological parameters for humans and animal models (the most common preclinical), including mouse, rat, minipig, dog, and monkey, from its integrated database. The software also provides access to various PBPK calculation methods, streamlining model building and parameterization processes. PK-Sim® automatically considers relevant generic passive processes. For example, distribution through blood flow and specific active processes, like metabolization by specific enzymes. While PK-Sim® is user-friendly and suitable for nonmodeling experts, it allows slight structural model adjustments. Unlike many other PBPK modeling tools, PK-Sim® provides varied model structures to cater to critical distinctions between small and large drug molecules. Most notably, PK-Sim® seamlessly integrates with the expert modeling software tool MoBi®, granting full access to all model details, extensive modifications, and extensions. This capability facilitates the creation of custom systems pharmacology models tailored to the challenges of innovative drug research and development [91].
Simcyp™ PBPK
The Simcyp Simulator stands as the pharmaceutical industry’s most complicated PBPK platform. Its capabilities encompass diverse applications, such as determining initial dosing for human trials, optimizing clinical study designs, assessing novel drug formulations, setting dosages for unstudied populations, and conducting simulated bioequivalence analyses besides foreseeing drug–drug interactions. Simcyp’s versatility extends across small molecules, biological compounds, ADCs, generic drugs, and emerging modality drugs. By linking in vitro to in vivo (ADME), as well as pharmacokinetic plus pharmacodynamic outcomes, Simcyp empowers the exploration of clinical scenarios and informed decision-making throughout drug development. Hence, Simcyp PBPK models offer comprehensive descriptions of drug behavior in tissues and organs. Every single organ can be represented by one or multiple physiological compartments. The drug concentration in each compartment is calculated through the integration of systems information, drug information, and trial design information [92].
Cyprotex
Cyprotex specializes in in vitro–in silico ADME-Tox services, covering a wide range of offerings. This encompasses in vitro ADME screening that aids discovery projects, as well as regulatory in vitro ADME and drug-drug interaction studies in the course of preclinical and clinical study and development. The company also provides specialized mechanistic in vitro human and animal toxicity models, such as 3-D models and MEA electrophysiology, along with PBPK and QSAR modeling expertise. Their comprehensive in vitro ADME and DMPK services contain metabolism studies, permeability and transporter assessments, solubility and physicochemical attribute evaluations, protein binding analysis, and pharmacokinetic and bioanalysis services. Cyprotex’s data has been highly validated and trusted by over 1,700 clients across pharmaceutical, biotechnology, cosmetics, healthcare companies, academic, and government associations [93].
ADMET modeler
ADMET modeler serves as a valuable QSAR/QSPR model building within ADMET Predictor®. This module efficiently automates the challenging and time-consuming task of constructing high-quality predictions of QSAR and QSPR models using experimental data. Seamlessly integrating with ADMET Predictor, it takes advantage of the platform’s descriptors as data and incorporates the chosen final model back into ADMET Predictor as an extra predicted property [94].
IMPACT-F
The assessment of oral bioavailability relies on robust computational models derived from the extensive PACT-F knowledge base, the largest repository of bioavailability data worldwide. Predicting human oral bioavailability early has numerous advantages, such as aiding in the selection of bioavailable drug candidates, and significantly reducing the risk of clinical failures compared to animal trials. The results are promptly available, ensuring confidentiality and reliability as no data or information leaves the company. Moreover, this approach enhances the potential of novel drugs by enabling a more precise determination of the optimal oral drug dose for first-in-human clinical trials. IMPACT-F, the novel expert system, is widely adopted by pharmaceutical companies across various therapeutic areas, including diabetes, inflammation, antivirals, autoimmune diseases, and cancer. It serves as a valuable tool for selecting and prioritizing drug candidates, optimizing prodrugs, and evaluating oral bioavailability before proceeding to clinical trials in humans. IMPACT-F stands out for its user-friendly interface, eliminating the need for chemical synthesis or animal experiments, and its superior reliability compared to animal trials, yielding immediate and crucial insights for future drug discovery and development. Ultimately, it plays a vital role in enhancing the efficiency and safety of human clinical trials [95].
DISCUSSION AND CONCLUSION
Drug discovery, development, and analysis studies involve a long and laborious process that requires time and high cost. The discovery of new drug molecules in the past; was done by examining the effects of molecules on known diseases through clinical observations, screening tests, and metabolism studies. Although this method was long and inefficient, it led to the discovery of many molecules until the 1980s. Studies for the development of new methods in order to increase the efficiency of the drug discovery, development, and analysis process and to achieve success in a short time with lower costs have yielded results. One of the alternative methods developed is in silico testing approaches based on computer simulations and mathematical algorithms.
In silico testing approaches, are approaches that limit the use of experimental animals used in in vivo experiments and reduce the time and cost required for the drug molecule to be marketed. Today, studies such as the discovery of the precursor compound and the optimization of the precursor compound are carried out through computer-assisted drug discovery and design. As a result of the rapid development of computational chemistry and biological sciences, computer-aided drug design methods continue to be successfully applied to accelerate the research and development process of drug molecules. With the development of artificial intelligence technology and machine learning, which are powerful data mining (DM) tools, the use of in silico methods such as QSAR, DM, molecular docking, molecular placement, and PBPK has increased. Due to in silico methods, which can be used in a wide variety of fields such as pharmacology, toxicology, cosmetology, and physiology, the discovery, preclinical analysis, and clinical studies of a drug molecule can be done easily. With these methods, the 3-D structures of drug molecules are examined, and their activities are estimated. The binding states of the ligand and the receptor are analyzed. Gastric and intestinal simulations are created with physiology-based pharmacokinetic models, and it is possible to examine the solubility, bioavailability, ADME, and toxicity properties of the drug. In silico testing approaches, there are disadvantages such as not always paying attention to pharmacokinetic properties and the possibility of obtaining erroneous results, but these methods have an important place in drug discovery, development, and analysis studies and are used progressively.
From the aforementioned introduction, it is easy to see that using in silico methods can be recommended for the prediction of a drug’s in vivo performance through drug discovery or preformulation study, however, the used algorithms and data sets should be considered. Recently, it was observed that the in silico research studies have increased, which contributed to the development of in silico modeling.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Lennernäs H, Lindahl A, Van Peer A, Ollier C, Flanagan T, Lionberger R, et al. In vivo predictive dissolution (IPD) and biopharmaceutical modeling and simulation: future use of modern approaches and methodologies in a regulatory context. Mol Pharm. 2017;14(4):1307–14.
2. Morgan S, Grootendorst P, Lexchin J, Cunningham C, Greyson D. The cost of drug development: a systematic review. Health Policy (New York) [Internet]. 2011;100(1):4–17. CrossRef
3. Schmid EF, Smith DA. Pharmaceutical R&D in the spotlight: why is there still unmet medical need? Drug Discov Today. 2007;12(23–24):998–1006.
4. Kaitin KI. Deconstructing the drug development process: the new face of innovation. Clin Pharmacol Ther. 2010;87(3):356–61.
5. DiMasi JA. The value of improving the productivity of the drug development process: faster times and better decisions. Pharmacoeconomics. 2002;20(SUPPL. 3):1–10.
6. Drews J. Drug discovery: a historical perspective. Science. 2000;287(5460):1960–4.
7. Williams DP, Shipley R, Ellis MJ, Webb S, Ward J, Gardner I, et al. Novel in vitro and mathematical models for the prediction of chemical toxicity. Toxicol Res (Camb). 2013;2(1):40–59.
8. Yu LX, Ellison CD, Hussain AS. Predicting human oral bioavailability using in silico models. In: Krishna R, editor. Applications of pharmacokinetic principles in drug development. Boston, MA: Springer; 2004. pp 53–74.
9. Hamada M. In silico approaches to RNA aptamer design. Biochimie [Internet]. 2018;145:8–14. CrossRef
10. Lorenz S, Amsel AK, Puhlmann N, Reich M, Olsson O, Kümmerer K. Toward application and implementation of in silico tools and workflows within benign by design approaches. ACS Sustain Chem Eng. 2021;9:37.
11. Gomes D, Silvestre S, Duarte AP, Venuti A, Soares CP, Passarinha L, et al. In silico approaches: a way to unveil novel therapeutic drugs for cervical cancer management. Pharmaceuticals. 2021;14(8):741.
12. Barh D, Tiwari S, Jain N, Ali A, Santos AR, Misra AN, et al. In silico subtractive genomics for target identification in human bacterial pathogens. Drug Dev Res. 2011;72(2):162–77.
13. Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: applications to targets and beyond. Br J Pharmacol. 2007;152(1):21–37.
14. Pelkonen O, Turpeinen M, Raunio H. In vivo-in vitro-in silico pharmacokinetic modelling in drug development. Clin Pharmacokinet. 2011;50(8):483–91.
15. Valerio LG. Application of advanced in silico methods for predictive modeling and information integration. Expert Opin Drug Metab Toxicol. 2012;8(4):395–8.
16. Riyaphan J, Pham DC, Leong MK, Weng CF. In silico approaches to identify polyphenol compounds as α-glucosidase and α-amylase inhibitors against type-II diabetes. Biomolecules. 2021;11(12):1877.
17. Hodos RA, Kidd BA, Shameer K, Readhead BP, Dudley JT. In silico methods for drug repurposing and pharmacology. WIREs Syst Biol Med [Internet]. 2016 May 1;8(3):186–210. CrossRef
18. Könczöl Á, Dargó G. Brief overview of solubility methods: recent trends in equilibrium solubility measurement and predictive models. Drug Discov Today Technol [Internet]. 2018;27:3–10. Available from: https://www.sciencedirect.com/science/article/pii/S1740674918300015
19. Montanari F, Ecker GF. Prediction of drug–ABC-transporter interaction—recent advances and future challenges. Adv Drug Deliv Rev [Internet]. 2015;86:17–26. Available from: https://www.sciencedirect.com/science/article/pii/S0169409X15000204
20. Richards WG. Computer-aided drug design. Pure Appl Chem [Internet]. 1994;66(8):1589–96. CrossRef
21. Niederquell A, Wyttenbach N, Kuentz M. New prediction methods for solubility parameters based on molecular sigma profiles using pharmaceutical materials. Int J Pharm [Internet]. 2018;546(1–2):137–44. CrossRef
22. Watanabe-Matsumoto S, Yoshida K, Meiseki Y, Ishida S, Hirose A, Yamada T. A physiologically based kinetic modeling of ethyl tert-butyl ether in humans—an illustrative application of quantitative structure-property relationship and Monte Carlo simulation. J Toxicol Sci. 2022;47(2):77–87.
23. Bharatam PV. Computer-aided drug design BT—drug discovery and development: from targets and molecules to medicines. In: Poduri R, editor. Singapore: Springer Singapore; 2021. pp 137–210. CrossRef
24. Raunio H. In silico toxicology—non-testing methods. Front Pharmacol. 2011;2:33.
25. Kavlock RJ, Ankley GT, Blancato JN, Collette TW, Fowle III J, Francis EZ, et al. A framework a computational toxicology research program [Internet]. Washington, DC: EPA 600/R-03/065 (NTIS PB2005-105438); 2003. Available from: https://nepis.epa.gov/Exe/ZyPDF.cgi/100046MA.PDF?Dockey=100046MA.PDF
26. Rotter S, Beronius A, Boobis AR, Hanberg A, van Klaveren J, Luijten M, et al. Overview on legislation and scientific approaches for risk assessment of combined exposure to multiple chemicals: the potential EuroMix contribution. Crit Rev Toxicol [Internet]. 2018;48(9):796–814. CrossRef
27. Huang HJ, Yu HW, Chen CY, Hsu CH, Chen HY, Lee KJ, et al. Current developments of computer-aided drug design. J Taiwan Inst Chem Eng [Internet]. 2010;41(6):623–35. CrossRef
28. Kapetanovic IM. Computer-aided drug discovery and development (CADDD): in silico-chemico-biological approach. Chem Biol Interact. 2008;171(2):165–76.
29. Bajorath J. Computer-aided drug discovery. F1000Research. 2015;4 F1000 Faculty Rev-630.
30. Yu W, MacKerell AD Jr. Computer-aided drug design methods. Methods Mol Biol.2017;1520:85–106.
31. Titz B, Schneider T, Elamin A, Martin F, Dijon S, Ivanov N V, et al. Analysis of proteomic data for toxicological applications BT—computational systems toxicology. In: Hoeng J, Peitsch MC, editors. New York, NY: Springer New York; 2015. pp 257–84. CrossRef
32. Contrera JF. Validation of Toxtree and SciQSAR in silico predictive software using a publicly available benchmark mutagenicity database and their applicability for the qualification of impurities in pharmaceuticals. Regul Toxicol Pharmacol [Internet]. 2013;67(2):285–93. CrossRef
33. Tomalia DA, Nixon LS, Hedstrand DM. Engineering critical nanoscale design parameters (CNDPs): a strategy for developing effective nanomedicine therapies and assessing quantitative nanoscale structure-activity relationships (QNSARs) [Internet]. Pharmaceutical applications of dendrimers. Amsterdam, Netherlands: Elsevier; 2019. 3–47 pp. CrossRef
34. ECHA. Guidance on information requirements and chemical safety assessment. Chapter R. 6: QSARs and grouping of chemicals. Helsinki, Finland: European Chemicals Agency; 2008.
35. Karczmar AG, Lindstrom J, Christopoulos A. History of research on nicotinic and muscarinic cholinergic receptors BT—exploring the vertebrate central cholinergic nervous system. In: Karczmar AG, editor. Boston, MA: Springer US; 2007. pp 151–62. CrossRef
36. Bowman WC. Neuromuscular block. Br J Pharmacol. 2006;147(SUPPL. 1):277–86.
37. Nelson BK, Brightwell WS, Mackenzie DR, Khan A, Burg JR, Weigel WW, et al. Teratological assessment of methanol and ethanol at high inhalation levels in rats. Toxicol Sci. 1985;5(4):727–36.
38. Mohapatra A. Software tools for toxicology and risk assessment [Internet]. Information resources in toxicology. London, San Diego, Cambridge, Oxford: Academic Press; 2020. 791–812 pp. CrossRef
39. Hartung T, Hoffmann S. Food for thought... on in silico methods in toxicology. ALTEX. 2009;26(3):155–66.
40. Kulkarni AS, Kasabe AJ, Bhatia MS, Bhatia NM, Gaikwad VL. Quantitative structure–property relationship approach in formulation development: an overview. AAPS PharmSciTech. 2019;20(7):1–10.
41. Waring MJ. Lipophilicity in drug discovery. Expert Opin Drug Discov. 2010;5(3):235–48.
42. Fujita T. In memoriam professor Corwin Hansch: birth pangs of QSAR before 1961. J Comput Aided Mol Des. 2011;25(6):509–17.
43. Leelananda SP, Lindert S. Computational methods in drug discovery. Beilstein J Org Chem. 2016;12:2694–718.
44. Lavecchia A. Machine-learning approaches in drug discovery: methods and applications. Drug Discov Today [Internet]. 2015;20(3):318–31. CrossRef
45. Hemmateenejad B, Yazdani M. QSPR models for half-wave reduction potential of steroids: a comparative study between feature selection and feature extraction from subsets of or entire set of descriptors. Anal Chim Acta. 2009;634(1):27–35.
46. Yousefinejad S, Hemmateenejad B. Chemometrics tools in QSAR/QSPR studies: a historical perspective. Chemom Intell Lab Syst. 2015;149:177–204.
47. Roy K, Das SK, Narayan R. Chapter 1 QSAR / QSPR modeling?: introduction. A primer on qsar/qspr modeling: fundamental concepts. New York, NY: Springer Berlin Heidelberg; 2015. pp 1–7.
48. Lill MA. Multi-dimensional QSAR in drug discovery. Drug Discov Today. 2007;12(23–24):1013–7.
49. Vedani A, Briem H, Dobler M, Dollinger H, McMasters DR. Multiple-conformation and protonation-state representation in 4D-QSAR: the neurokinin-1 receptor system. J Med Chem. 2000;43(23):4416–27.
50. Perkins R, Fang H, Tong W, Welsh WJ. Quantitative structure-activity relationship methods: perspectives on drug discovery and toxicology. Environ Toxicol Chem. 2003;22(8):1666–79.
51. Wang T, Yuan XS, Wu MB, Lin JP, Yang LR. The advancement of multidimensional QSAR for novel drug discovery—where are we headed? Expert Opin Drug Discov [Internet]. 2017;12(8):769–84. CrossRef
52. Chavda J, Bhatt H. 3D-QSAR (CoMFA, CoMSIA, HQSAR and topomer CoMFA), MD simulations and molecular docking studies on purinylpyridine derivatives as B-Raf inhibitors for the treatment of melanoma cancer. Struct Chem. 2019;30:2093–107.
53. Gini G. QSAR methods. In: Benfenati E, editor. In silico methods for predicting drug toxicity. Methods in molecular biology. New York, NY: Humana Press; 2016. pp 1–20. CrossRef
54. Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron [Internet]. 2018;99(6):1129–43. CrossRef
55. Hansson T, Oostenbrink C, Van Gunsteren WF. Molecular dynamics simulations. Curr Opin Struct Biol. 2002;12(2):190–6.
56. Pagadala NS, Syed K, Tuszynski J. Software for molecular docking: a review. Biophys Rev [Internet]. 2017;9(2):91–102. CrossRef
57. Sharma R, Dhingra N, Patil S. CoMFA, CoMSIA, HQSAR and molecular docking analysis of ionone-based chalcone derivatives as antiprostate cancer activity. Indian J Pharm Sci. 2016;78(1):54–64.
58. Shahzadi I, Islam M, Saeed H, Haider A, Shahzadi A, Haider J, et al. Formation of biocompatible MgO/cellulose grafted hydrogel for efficient bactericidal and controlled release of doxorubicin. Int J Biol Macromol [Internet]. 2022;220:1277–86. CrossRef
59. Shahzadi I, Islam M, Saeed H, Shahzadi A, Haider J, Haider A, et al. Facile synthesis of copolymerized cellulose grafted hydrogel doped calcium oxide nanocomposites with improved antioxidant activity for anti-arthritic and controlled release of doxorubicin for anti-cancer evaluation. Int J Biol Macromol [Internet]. 2023;235:123874. CrossRef
60. Prasad S, Srivastava A, Singh N, Singh H, Saluja R, Kumar A, et al. Chapter 21—present and future challenges in therapeutic designing using computational approaches. In: Parihar A, Khan R, Kumar A, Kaushik AK, Gohel H, editors. Computational approaches for novel therapeutic and diagnostic designing to mitigate SARS-CoV-2 infection. Revolutionary strategies to combat pandemics. London, San Diego, Cambridge, Oxford: Academic Press; 2022. pp 489–505. Available from: https://www.sciencedirect.com/science/article/pii/B9780323911726000200
61. Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: new docking methods, expanded force field, and python bindings. J Chem Inf Model [Internet]. 2021 Aug 23;61(8):3891–8. CrossRef
62. Center for Computational Strucural Biology. ADCP [Internet]. Sacramento, CA: The Funded Research Laboratories; 2012–2018 [cited 2023 Jul 16]. Available from: https://ccsb.scripps.edu/adcp/
63. Apolo Scientific Computing Center’s Documentation. LeDock [Internet]. Medellin, Colombia: The Scientific Computing Center at Universidad EAFIT; [cited 2023 Jul 16]. Available from: https://apolo-docs.readthedocs.io/en/latest/software/applications/leDock/index.html
64. Najmanovich Research Group. Resources [Internet]. Montreal, Canada: Université de Montreal; [cited 2023 Jul 16]. Available from: http://biophys.umontreal.ca/nrg/resources.html
65. Simulations Plus. MedChem Studio.: 1–24. Available from: https://www.simulations-plus.com/wp-content/uploads/MedChem4.0LinearEuro.pdf
66. Pharmainformatic. MolScore-Antibiotics. Emden, Germany: PharmaInformatic Boomgaarden; 2004–2021 [cited 2023 Jul 16].
67. Rey J, Rasolohery I, Tufféry P, Guyon F, Moroy G. PatchSearch: a web server for off-target protein identification. Nucleic Acids Res [Internet]. 2019 Jul 2;47(W1):W365–72. CrossRef
68. Gfeller D, Grosdidier A, Wirth M, Daina A, Michielin O, Zoete V. SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014;42(W1):32–8.
69. Keiser MJ, Roth BL, Armbruster BN, Ernsberger P, Irwin JJ, Shoichet BK. Relating protein pharmacology by ligand chemistry. Nat Biotechnol [Internet]. 2007;25(2):197–206. CrossRef
70. Lo YC, Senese S, Li CM, Hu Q, Huang Y, Damoiseaux R, et al. Large-scale chemical similarity networks for target profiling of compounds ?dentified in cell-based chemical screens. PLoS Comput Biol. 2015;11(3):1–23.
71. Kim Kjærulff S, Wich L, Kringelum J, Jacobsen UP, Kouskoumvekaki I, Audouze K, et al. ChemProt-2.0: visual navigation in a disease chemical biology database. Nucleic Acids Res [Internet]. 2013 Jan 1;41(D1):D464–9. CrossRef
72. Qsartoolbox. QSAR toolbox [Internet]. Paris, France: Organisation for Economic Co-operation and Development; 2023 [cited 2023 Jul 16]. Available from: https://qsartoolbox.org/
73. Merdekawati F. In silico study of pyrazolylaminoquinazoline toxicity by lazar, protox, and admet predictor. J Appl Pharm Sci. 2018;8(9):119–29.
74. ChemSafetyPro. Toxicology and health risk assessment; 2015–2021 [Internet]. Shrewsbury, UK: ChemSafetyPro; 2018 [cited 2023 Jul 16]. Available from: https://www.chemsafetypro.com/Topics/CRA/How_to_Use_Toxtree_to_Determine_Cramer_Class_and_Estimate_Toxic_Hazard.html
75. VEGA HUB. About VEGA HUB [Internet]. Milan, Italy: Istituto di Ricerche Farmacologiche Mario Negri IRCCS; [cited 2023 Jul 16]. Available from: https://www.vegahub.eu/about-vegahub/
76. Rodríguez-Leal I, MacLeod M. The applicability domain of EPI SuiteTM for screening phytotoxins for potential to contaminate source water for drinking. Environ Sci Eur [Internet]. 2022;34(1):96. CrossRef
77. EPA. OncoLogicTM user guide [Internet]. Washington, DC: Environmental Protection Agency; 2009–2010 [cited 2023 Jul 16]. Available from: https://www.epa.gov/sites/default/files/2015-09/documents/usermanual.pdf
78. CompuDrug. HazardExpert Pro [Internet]. Bal Harbour, FL: CompuDrug Ltd.; 2000–2013 [cited 2023 Jul 16]. Available from: http://www.compudrug.com/hazardexpertpro
79. Saliner AG, Tsakovska I, Pavan M, Patlewicz G, Worth AP. Evaluation of SARs for the prediction of skin irritation/corrosion potential-structural inclusion rules in the BfR decision support system. SAR QSAR Environ Res. 2007;18(3–4):331–42.
80. Prival MJ. Evaluation of the TOPKAT system for predicting the carcinogenicity of chemicals. Environ Mol Mutagen [Internet]. 2001 Jan 1;37(1):55–69. CrossRef
81. MultiCase. CASE Ultra [Internet]. Mayfield Heights, OH: MultiCase; 2023 [cited 2023 Jul 16]. Available from: http://www.multicase.com/case-ultra-models#skin_eye_tox_bundle
82. Roberts G, Myatt GJ, Johnson WP, Cross KP, Blower PE. LeadScope: software for exploring large sets of screening data. J Chem Inf Comput Sci [Internet]. 2000 Nov 1;40(6):1302–14. CrossRef
83. TerraBase Inc. Overv?ew [Internet]. Hamilton, Canada: TerraBase Inc; 1996–2021 [cited 2023 Jul 16]. Available from: https://www.terrabase-inc.com/
84. ACDLABS. PhysChem Suite [Internet]. Toronto, Canada: Advanced Chemistry Development; 2022 [cited 2023 Jul 16]. Available from: https://www.acdlabs.com/products/percepta-platform/physchem-suite/
85. Molcode. Molcode [Internet]. Available from: http://itekem.fr/DocumentsPDF/Molcode Toolbox 08.12.2009.pdf
86. BioByte. A general approach to the organization of quantitative structure-activity relationships in chemistry and biology. Claremont, CA: BioByte Corp.; 2000. Available from: http://www.biobyte.com/bb/prod/qsarman2k.pdf
87. Alland C, Moreews F, Boens D, Carpentier M, Chiusa S, Lonquety M, et al. RPBS: a web resource for structural bioinformatics. Nucleic Acids Res. 2005;33(suppl_2):W44–9.
88. Simulations Plus. GastroPlus® [Internet]. Washington, DC: Simulations Plus; 2023 [cited 2023 Jul 16]. Available from: https://www.simulations-plus.com/software/gastroplus/
89. Simulations Plus. ADMET Predictor® [Internet]. Washington, DC: Simulations Plus; 2023 [cited 2023 Jul 16]. Available from: https://www.simulations-plus.com/software/admetpredictor/?gclid=CjwKCAjw5c6LBhBdEiwAP9ejG8ftGtTSZjxIEpbABdzW_2xoCHPQdEJBjumwXK2UvFx6zwyfSmAyyxoCOFUQAvD_BwE
90. Pharmainformatic. MolScore-Drugs [Internet]. Emden, Germany: PharmaInformatic Boomgaarden; 2004–2021 [cited 2023 Jul 16]. Available from: https://www.pharmainformatic.com/html/molscore-drugs.html
91. GitHub. PK-Sim® [Internet]. GitHub; 2023 [cited 2023 Jul 16]. Available from: https://github.com/Open-Systems-Pharmacology/PK-Sim
92. Certara. SimcypTM PBPK [Internet]. Princeton, NJ: Certara; 2023 [cited 2023 Jul 16]. Available from: https://www.certara.com/software/simcyp-pbpk/
93. Evotec. Cyprotex—an Evotec company [Internet]. Hamburg, Germany: Evotec; 2023 [cited 2023 Jul 16]. Available from: https://www.evotec.com/en/execute/cyprotex
94. Simulations Plus. ADMET ModelerTM [Internet]. Washington, DC: Simulations Plus; 2023 [cited 2023 Jul 16]. Available from: https://www.simulations-plus.com/software/admetpredictor/admet-modeler/
95. Pharmainformatic. IMPACT-F. Emden, Germany: PharmaInformatic Boomgaarden; 2004–2021 [cited 2023 Jul 16].
96. Vemula D, Jayasurya P, Sushmitha V, Kumar YN, Bhandari V. CADD, AI and ML in drug discovery: a comprehensive review. Eur J Pharm Sci [Internet]. 2023;181:106324. CrossRef
97. Murray CW, Newell DR, Angibaud P. A successful collaboration between academia, biotech and pharma led to discovery of erdafitinib, a selective FGFR inhibitor recently approved by the FDA. Medchemcomm. 2019;10(9):1509–11.
98. Reed JE, Smaill JB. The discovery of dacomitinib, a potent ?rreversible EGFR ?nhibitor. Comprehensive accounts of pharmaceutical research and development: from discovery to late-stage process development (ACS Symposium Series; vol. 1239) [Internet]. Washington, DC: American Chemical Society; 2016. vol. 1, pp 207–33. CrossRef
99. Hecker SJ, Reddy KR, Lomovskaya O, Griffith DC, Rubio-Aparicio D, Nelson K, et al. Discovery of cyclic boronic acid QPX7728, an ultrabroad-spectrum inhibitor of serine and metallo-β-lactamases. J Med Chem. 2020;63(14):7491–507.
100. Harper S, McCauley JA, Rudd MT, Ferrara M, DiFilippo M, Crescenzi B, et al. Discovery of MK-5172, a macrocyclic hepatitis C virus NS3/4a protease inhibitor. ACS Med Chem Lett. 2012;3(4):332–6.
101. Abidi A, Shukla P, Ahmad A. Lifitegrast: a novel drug for treatment of dry eye disease. J Pharmacol Pharmacother. 2016;7(4):194–8.
102. White AW, Almassy R, Calvert AH, Curtin NJ, Griffin RJ, Hostomsky Z, et al. Resistance-modifying agents. 9. Synthesis and biological properties of benzimidazole ?nhibitors of the DNA repair enzyme poly(ADP-ribose) polymerase. J Med Chem [Internet]. 2000 Nov 1;43(22):4084–97. CrossRef
103. Agrawal R. The first approved agent in the Glitazar’s class: saroglitazar. Curr Drug Targets. 2014;15(2):151–5.
104. Kwong AD, Kauffman RS, Hurter P, Mueller P. Discovery and development of telaprevir: an NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nat Biotechnol [Internet]. 2011;29(11):993–1003. CrossRef
105. Perzborn E, Roehrig S, Straub A, Kubitza D, Misselwitz F. The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor. Nat Rev Drug Discov. 2011;10(1):61–75.
106. Cui JJ, Tran-Dubé M, Shen H, Nambu M, Kung P-P, Pairish M, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal–epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med Chem. 2011;54(18):6342–63.
107. Njoroge FG, Chen KX, Shih N-Y, Piwinski JJ. Challenges in modern drug discovery: a case study of boceprevir, an HCV protease inhibitor for the treatment of hepatitis C virus infection. Acc Chem Res. 2008;41(1):50–9.
108. Rutenber EE, Stroud RM. Binding of the anticancer drug ZD1694 to E. coli thymidylate synthase: assessing specificity and affinity. Structure. 1996;4(11):1317–24.
109. Veljkovic N, Vucicevic J, Tassini S, Glisic S, Veljkovic V, Radi M. Preclinical discovery and development of maraviroc for the treatment of HIV. Expert Opin Drug Discov. 2015;10(6):671–84.
110. Rivera-Lebron BN, Risbano MG. Ambrisentan: a review of its use in pulmonary arterial hypertension. Ther Adv Respir Dis. 2017;11(6):233–44.
111. Cohen NC. Structure-based drug design and the discovery of aliskiren (Tekturna): perseverance and creativity to overcome a R&D pipeline challenge. Chem Biol Drug Des. 2007;70(6):557–65.
112. Sun CL, Christensen JG, McMahon G. Discovery and development of sunitinib (SU11248): a multitarget tyrosine kinase inhibitor of tumor growth, survival, and angiogenesis. Kinase inhibitor drugs. Hoboken, NJ: John Wiley & Sons; 2009. pp 1–39.
113. Ghosh AK, Dawson ZL, Mitsuya H. Darunavir, a conceptually new HIV-1 protease inhibitor for the treatment of drug-resistant HIV. Bioorg Med Chem. 2007;15(24):7576–80.
114. Barker A, Andrews D. Discovery and development of the anticancer agent gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase. Introduction to biological and small molecule drug research and development. Waltham, Oxford, Amsterdam: Elsevier; 2013. pp 255–81.
115. Fischer J, Ganellin CR. Analogue-based drug discovery. Chem Int IUPAC. 2010;32(4):12–5.
116. Aulakh GK, Sodhi RK, Singh M. An update on non-peptide angiotensin receptor antagonists and related RAAS modulators. Life Sci. 2007;81(8):615–39.
117. Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289(5486):1938–42.
118. Gao Q, Yang L, Zhu Y. Pharmacophore based drug design approach as a practical process in drug discovery. Curr Comput Aided Drug Des. 2010;6(1):37–49.
119. Lew W, Chen X, Kim CU. Discovery and development of GS 4104 (oseltamivir) an orally active influenza neuraminidase inhibitor. Curr Med Chem. 2000;7(6):663–72.
120. Lyle TA. Ribonucleic acid viruses: antivirals for human ?mmunodeficiency virus. Comprehensive medicinal chemistry II. Amsterdam, The Netherlands: Elsevier; 2007. pp 329–71.
121. Lynch JJ, Cook JJ, Sitko GR, Holahan MA, Ramjit DR, Mellott MJ, et al. Nonpeptide glycoprotein IIb/IIIa inhibitors. 5. Antithrombotic effects of MK-0383. J Pharmacol Exp Ther. 1995;272(1):20–32.
122. Best BM, Goicoechea M. Efavirenz—still first-line king? Expert Opin Drug Metab Toxicol [Internet]. 2008 Jul 1;4(7):965–72. CrossRef
123. Adams WJ, Aristoff PA, Jensen RK, Morozowich W, Romero DL, Schinzer WC, et al. Discovery and development of the BHAP nonnucleoside reverse transcriptase inhibitor delavirdine mesylate. Pharm Biotechnol. 1998;11:285–312.
124. Aruksakunwong O, Promsri S, Wittayanarakul K, Nimmanpipug P, Lee VS, Wijitkosoom A, et al. Current development on HIV-1 protease inhibitors. Curr Comput Aided Drug Des. 2007;3(3):201–13.
125. Kim EE, Baker CT, Dwyer MD, Murcko MA, Rao BG, Tung RD, et al. Crystal structure of HIV-1 protease in complex with VX-478, a potent and orally bioavailable inhibitor of the enzyme. J Am Chem Soc. 1995;117(3):1181–2.
126. Wlodawer A, Vondrasek J. Inhibitors of HIV-1 protease: a major success of structure-assisted drug design. Annu Rev Biophys Biomol Struct. 1998;27(1):249–84.
127. De Clercq E. The history of antiretrovirals: key discoveries over the past 25 years. Rev Med Virol. 2009;19(5):287–99.
128. Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2012;27(6):759–72.
129. Warnke C, Wiendl H, Hartung H-P, Stüve O, Kieseier BC. Identification of targets and new developments in the treatment of multiple sclerosis—focus on cladribine. Drug Des Devel Ther. 2010;4:117–26.
130. Zhu C. Aldose reductase inhibitors as potential therapeutic drugs of diabetic complications. London, UK: IntechOpen; 2013. vol. 2.
131. Ashraf Z, Alamgeer, Kanwal M, Hassan M, Abdullah S, Waheed M, et al. Flurbiprofen–antioxidant mutual prodrugs as safer nonsteroidal anti-inflammatory drugs: synthesis, pharmacological investigation, and computational molecular modeling. Drug Des Devel Ther. 2016;10:2401–19.
132. Takahashi H, Hayakawa I, Akimoto T. The history of the development and changes of quinolone antibacterial agents. Yakushigaku Zasshi. 2003;38(2):161–79.
133. Cushman DW, Ondetti MA. History of the design of captopril and related inhibitors of angiotensin converting enzyme. Hypertension. 1991;17(4):589–92.