
INTRODUCTION
Dementia is a progressive neurological illness that causes mental and behavioral decline, including, but not limited to, issues with communication, memory, reasoning ability, and other cognitive skills that impede day-to-day functioning. Such impairment in cognitive functions is commonly accompanied by deterioration in emotional control and social behavior. Vascular dementia, Alzheimer’s disease (AD), and Lewy body dementia are all types of dementia [1,2].
About 50,000,000 people are thought to be affected by it at the moment, with that figure rising to an estimated 152,000,000 by 2050 [3]. As the aging rates universally increase, the rate of dementia incidence continues to grow, starting to make dementia a major issue for public health in low-income regions. The cost to society is high, and it is not only the quality of life diminished by dementia. In nations with a low per capita income, the charge is about $ 900, whereas in countries with a lower to moderate income, such as Egypt, the cost may reach $3,200 [4]. When it comes to diagnosing dementia, neuro-psychometric testing has been shown to be the most effective in low-income regions. Studies have shown that the biomarkers indicating dementia are Aβ40, Aβ42, total tau, and phosphorylated tau (P-Tau). These biomarkers are considered the most sensitive markers for diagnosing AD and cognitive impairment [4].
Neuroinflammation is known to be implicated in the pathogenesis of dementia. It can be induced by systemic injection of lipopolysaccharides (LPSs), also known as endotoxins, which are complex molecules found in the outer membrane of Gram-negative bacteria, leading to the activation of pro-inflammatory signaling pathways via toll-like receptor 4 (TLR4), tau hyperphosphorylation, deposition of β-amyloid (Aβ) plaques, and ultimately cell apoptosis. LPS-induced neuroinflammation in the hippocampus is also associated with cognitive dysfunction, oxidative stress (OS), enhanced acetylcholinesterase activity, and activation of microglia and astrocytes [5]. Because of this, LPS has been used extensively in animal models to study the molecular underpinnings of cognitive impairment caused by neuroinflammation and to create a targeted therapeutic for alleviating neurological symptoms [6].
Up till now, there has been no curative therapy for dementia, and the most common currently used medications are acetylcholine esterase inhibitors that only delay the symptomatic progression and improve the cognitive and daily activities of patients with mild-to-moderate dementia. These drugs can be prescribed with memantine or used separately for moderate and severe cases [3]. Clinical studies of other therapeutic medicines targeting Aβ or P-tau as target pharmaceuticals have proven unsuccessful. As a result, novel therapeutic medication development is required [2].
Trimetazidine (TMZ), 1-(2,3,4-tri methoxy-benzyl) piperaciline hydrochloride, is an anti-ischemic agent [7]. Some studies proved that TMZ has anti-oxidative and anti-inflammatory properties [8]. This evidence illustrated that TMZ might be one of the appropriate candidates for counteracting neuroinflammation and prevention of cognitive decline following LPS injection. Therefore, this study was considered to elucidate the efficacy of TMZ in counteracting the pathological manifestations of dementia induced by LPSs through attenuating neuroinflammation and evaluating whether TMZ could differentially regulate the main biochemical and molecular markers in the pathological process of dementia.
MATERIAL AND METHODS
Materials
Chemicals and drugs
Pharmaceutical LPSs were purchased from Sigma-Aldrich Co., St Louis, MO; TMZ was obtained from Servier company, Egypt, whereas memantine was procured from COPAD Pharma Company, Cairo, Egypt. Any other chemicals used were locally purchased (Egypt) and were of high analytical grade.
Animals
Fifty male Sprague Dawley rats, adults weighing between 185 and 205 g, were maintained in the usual cages “under pathogen-free conditions and maintained under controlled room temperature and normal dark-light cycles. Animals were provided with standard chow and water ad libitum. Rats were adapted to these conditions for 2 weeks before the experimental protocol [9].”
In vivo study
The animals were arbitrarily categorized into five groups (10 rats/group): (1) The negative control group, which was intraperitoneally injected with a vehicle [0.5 ml of phosphate buffer saline (PBS)] four times/week for 2 weeks. (2) The untreated dementia-induced group was intraperitoneally injected with LPS (250 μg/kg ) dissolved in PBS four times/week for 2 weeks [10]. (3) TMZ 20 group denoted the dementia-triggered rats, which were administered per oral 20 mg/kg TMZ (dissolved in PBS) daily for 4 weeks, following the induction with LPS for 2 weeks [11]. (4) TMZ 40 group constituted the dementia-triggered rats treated orally with 40 mg/kg TMZ daily for 28 days [12]. (5) Memantine group comprised the dementia-triggered rats, which were administered orally with 10 mg/kg memantine (dissolved in PBS) as a reference drug daily for 4 weeks [13].
Methods
After animal treatment was over, the relevant behavioral assessments were performed, including the object recognition test (ORT), Y-maze, and Morris water maze (MWM).
Object recognition test
“Three days before testing, animals were subjected to 3 consecutive days of training in the dark open box, and each rat was also allowed to explore the apparatus without objects for 2 minute/day. On the testing day, a session of 2 minutes of two trials (T1 and T2) was performed. In the “sample” trial (T1), two identical objects were positioned in two opposite corners of the apparatus. The rat was located in the middle of the apparatus and was left to discover these two identical objects. After T1, the rat was placed back in its home cage, and an inter-trial interval of 1 hour was given. Subsequently, the “choice” trial (T2) was performed, where a new object replaced one of the objects that were presented in T1. Rats were exposed again to two different objects: the familiar (F) and the new one (N). To avoid the presence of a smell effect, the apparatus and the objects were carefully cleaned with alcohol solution after each trial. The objects’ discovery was as follows; directing the nose toward the object at a distance of no more than 2 cm or touching the object with the nose. The times spent by rats exploring each object in T1 and T2 were documented manually using a stopwatch. Many variables were calculated as follows: a) The total time spent exploring the two identical objects in T1, b) the whole time spent exploring the two different objects in T2, c) the discrimination between F and N in T2 was measured by comparing the time spent discovering F with exploring N, and d) discrimination index (DI) represents the difference in exploration time expressed as a proportion of the total time spent exploring the 2 objects in T2. DI was calculated as follows: DI = N−F/N+F” [14,15].
Y-maze task
“The spatial memory function was evaluated by the Y maze task using the rewarded alternation. Each trial in this test comprised two runs. The first one was the sample run: during which one of the goal arms was closed to force the rat to enter a particular arm, where it was rewarded with food. Before the consequent choice run, the second arm was opened, and the rat chose to enter either arm. If it entered the alternate arm, it received a food reward. Thus, correct performance depends mainly on memorizing which arm was entered in the sample run to guide proper response in choosing one. Each rat was subjected to five trials, with a 10-minute interval between them. The percentage of rewarded alternations was finally calculated” [16–18].
MWM test
MWM was carried out in a container (pool-like) about 180 cm in width, with a depth of 70 cm deep and an output faucet. The container was loaded with water to a depth of 35 cm with a white, non-poisonous, water-soluble stain. The temperature of the pool was retained at 26°C ± 1°C by the addition of warm water. A small, movable platform (about 9 cm in diameter) was placed in the middle of one of the pool’s four imaginary quadrants. Before the acquisition phase or probing test, the rats were given 1 minute of free swimming.
“In the acquisition phase, rats were exposed to three training sessions daily, each of 60 seconds, for 4 successive days. During each trial, animals were left free to find the platform in the target quadrant. Once the rat positioned the platform, it was permitted to remain on it for 10 seconds. However, if an animal failed to reach the platform within 60 seconds, it was gently placed on the platform for 20 seconds. The escape latency was calculated as the average of the total time taken in all trials of each day of the acquisition phases to locate the platform. It was used as a measure of spatial learning. On the 5th day, a probe test (retrieval trial) was executed where the platform was removed, and each rat was placed in the water facing the pool wall starting from the quadrant opposite the platform quadrant and was allowed to explore the pool for 60 seconds. The time spent swimming in the target quadrant and the time spent in the non-target quadrant searching for the removed platform were considered the index of retrieval” [19].
Blood and tissue sampling
After performing the behavioral tests, after an overnight fast, the Sprague Dawley rats’ blood was obtained, following receiving 10 mg/kg Midazolam (via IP route) for complete anesthesia, from the tail vein to separate the serum samples following spin at 400× g for 10 minutes at 5°C for biochemical analyses. After blood collection, the rats were sacrificed under complete anesthesia, and the brains were quickly dissected, thoroughly rinsed with ice-cold PBS , and blotted dry. The whole brain of a certain number of animals in each group was fixed in 10% neutral formalin buffer for 24 hours for histological and immunohistochemical examinations. While the whole brains of the rest of the animals in each group were dissected, hippocampi were extracted, immediately snap-frozen in cold nitrogen in the liquid state, and for the study of gene expression, kept at −80°C.
Biochemical determinations
Aβ1-42 residues (Aβ1-42), P-Tau, tau protein, and neurogranin serum levels were evaluated using ELISA kits bought from Wuhan Fine Biotech Co. LTD, Wuhan, China, according to the associated manufacturer’s manuals. While APL25, APL27, APL28, and 3-methoxy4-hydroxyphenyl glycol (MHPG) were analyzed in the serum using ELISA kits supplied by SinoGeneClon Biotech Co., Hangzhou, China.
Molecular analyses
Total RNA was extracted from the hippocampi tissues of rats in each group using an RNeasy mini kit for total RNA purification from animal cells (Cat#74104, Qiagen, Hilden, Germany) according to the provided pamphlet. The integrity of the isolated RNA was assessed by a NanoDrop 2000 (Thermo “Fisher Scientific, Rockford, IL) using a 260/280 nm ratio. After that, RNA was reverse transcribed into cDNA using Revert Aid first strand cDNA synthesis kit (Thermo Fisher Scientific, Vilnius, Lithuania) according to the manufacturer’s instructions” as per Mahmoudet al., [20]. Then, the mRNA levels of TLR4, cyclooxygenase-2 (COX-2), tumor necrosis factor-α (TNF-α), Caspase-3, BCL-2, nuclear factor kappa B (NF-κB), and inducible nitric oxide synthase (iNOS) genes were assessed using QuantiTect SYBR Green polymerase chain reaction (PCR) Kit (Qiagen, Hilden, Germany), according to the manufacturer’s protocol. DNA-Technology Real-Time PCR device (DTlite 4, Moscow, Russia) was employed to perform the quantitative PCR cycles. “The thermal conditions were adjusted as follows; initial denaturation cycle at 94°C for 15 minutes, followed by 40 cycles of denaturation at 94°C for 15 seconds, annealing at 60°C for 30 seconds, and extension at 72°C for 30 seconds” as per Mahmoud et al., [20]. Relative target mRNA quantification versus control was calculated using the comparative threshold cycle (Ct) method (2-ΔΔCt ). All values were normalized to the GAPDH gene. The primer pairs of the target genes are listed in Table 1.
Histological examination of brain tissue
After fixation of rat brain tissue samples in 10% formalin for 1 day, the specimen was treated sequentially with dilutions of diethyl alcohol and evaporated in xylene. A paraplast tissue embedding medium was used to infiltrate the cells, and subsequently, they were embedded. Five microns thick sagittal brain slices taken by the rotatory microtome were placed on glass slides to show off the many hippocampus structures in the specimens. Hematoxylin and eosin staining of tissue slices was performed as a standard procedure before microscopic analysis [29].
Immunohistochemical investigation
Immunohistochemical staining
In accordance with the manufacturer’s guidelines, immunohistochemical staining for ionized calcium-binding adapter molecule 1 (IbA-1) was conducted. The secondary antibody Horseradish peroxidase (HRP) secondary antibodies Envision kit (DAKO, Hamburg, Germany) was incubated with the anti-IbA-1 antibody (ab108539—Abcam, Cambridge, USA—1:100) for 20 minutes after blocking the antigen-retrieved brain tissue slices with 3% hydrogen peroxide for 15 minutes. After that, “the tissue sections were PBS-washed and incubated with diaminobenzidine for 10 minutes. Counterstaining with hematoxylin was performed, followed by dehydration and clearing in xylene. Tissue sections were then cover-slipped for microscopic examination,” as per Abbas et al. [30].
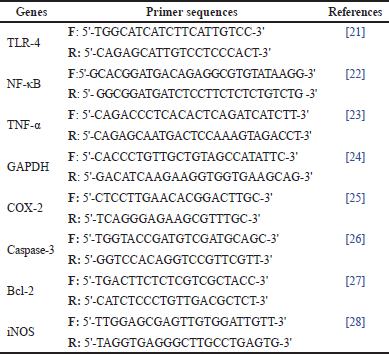 | Table 1. List of primer sequences of neuroinflammation-related genes used in qRT-PCR. [Click here to view] |
Quantitative immunohistochemical evaluation
Six far unplanned areas of the hippocampal CA3 subregions were scanned and analyzed to determine the mean positive counts of IbA-1/++ microglial cells in each immunostained tissue section with anti-IbA-1. “All morphological examinations, photographs as well as quantitative analyses were recorded using Leica Application system modules for histological analysis (Leica Microsystems GmbH, Wetzlar, Germany)” as per Ibrahim and Abdel Rasheed [29].
Statistical analysis
The current data are expressed as the mean ± SD. “Data were processed by one-way ANOVA followed by the Tukey Post hoc test except for the immunohistochemical scoring; statistical analysis was carried out by nonparametric Kruskal–Wallis H-test, followed by Dunn’s test. p < 0.05 was considered to be statistically significant. GraphPad Prism software (version 9; GraphPad Software, Inc., San Diego, CA) was employed to perform the statistical analysis and to establish the graphical representation” [9,31].
RESULTS
Behavioral tests
Object recognition test
As depicted in Figure 1, dementia, TMZ (20 or 40 mg/kg), and memantine groups exhibited shorter T1 time (ORT) than the negative control group by 63%, 70%, 69%, and 82%, respectively. Dementia and memantine groups depicted significantly shorter T2 time (ORT) than the negative control group by almost 40% and 44%, respectively. Conversely, the treatment with TMZ (20 or 40 mg/kg) elongated the T2 time (ORT) compared to the untreated dementia group by 1.7 and 2-fold, respectively, with statistically comparable time to the negative control group. The ORT novel object time was reduced in dementia and memantine-treated rats by 82% and 51% versus the negative control group, respectively. This diminution was reversed by the treatment with TMZ (20 or 40 mg/kg) by six and seven-fold compared to the dementia group, respectively. The results were statistically comparable to the negative control group.
Y-maze task
Figure 2 represents the Y-maze test performed by all the experimental groups. The dementia-induced rats, as well as dementia rats treated with TMZ (20 mg/kg), experienced a decline in the percentage of rewarded alternations of rats in the Y-maze apparatus by 75% versus the healthy rats. On the other side, administration with TMZ (40 mg/kg) or memantine depicted a prominent elevation in rats’ rewarded alternations by about 3.6 and 2.9-fold, respectively, versus the untreated dementia group, reaching the normal values.
Morris water maze
On the fourth and fifth training days, the dementia-induced rats portrayed significantly higher training time latency than the negative control group by approximately 1.7 and 2-fold, respectively. In the probe trial, treatment with either TMZ (20 or 40 mg/kg) or memantine decreased the exploration time in the target quadrant by 27%, 42% and 27% compared to the dementia group, respectively (Fig. 2). Noteworthy, the exploration time of rats treated with TMZ (40 mg/kg) reached the normal values.
Biochemical determinations
Figure 3 constitutes the influence of treatment with TMZ (20 or 40mg/kg) and memantine on the serum levels of dementia-related biomarkers in dementia-triggered rats. The positive dementia rats discovered a substantial rise in the serum tau and P-Tau levels along with a substantial decline in the serum levels of Aβ (Aβ1-42), neurogranin, APL1b25, APL1b27, APL1b28, and MHPG against the normal control rats. Whereas, administration of either TMZ (20 or 40 mg/kg) or memantine in the dementia-afflicted rats significantly decreased the serum levels of tau and P-Tau with a concurrent significant amelioration of the serum levels of Aβ (Aβ1-42), neurogranin, APL1b25, APL1b27, APL1b28, and MHPG (p > 0.05) [except for TMZ 20 group that showed a slight improvement in the serum APL1b28 level] in comparison with the untreated dementia rats. Worth mentioning, treatment with TMZ (40 mg/kg) significantly modulates the serum levels of Aβ (Aβ1-42), APL1b25, APL1b27, APL1b28, and MHPG when compared with TMZ 20 group. Moreover, the TMZ 40 group experienced a significant refinement in serum levels of APL1b28 and MHPG versus the memantine-treated group. Furthermore, the dementia rats treated with memantine significantly attenuated the serum levels of Aβ (Aβ1-42) and APL1b25 versus those treated with TMZ (20 mg/kg).
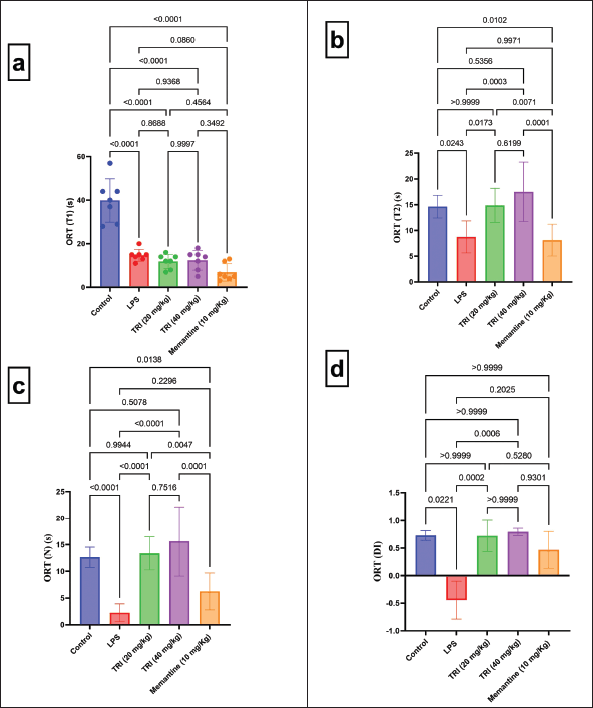 | Figure 1. Effect of TMZ and memantine administration on ORT in different experimental groups. (a) ORT T1 with F = 38.61 (DFn = 4, DFd = 30, and p < 0.0001); (b) ORT T2 with F = 9.932 (DFn = 4, DFd = 30, and p < 0.0001); (c) ORT N with F = 17.63 (DFn = 4, DFd = 30, and p < 0.0001); and (d) ORT DI with K = 23.36 (colums = 5, total number = 30, and p < 0.0001). Data are presented as mean ± SD except for ORT DI, which is presented as median ± IQR. [Click here to view] |
Molecular analyses
Figure 4 comprises the influence of TMZ and memantine administration on the transcriptional levels of neuroinflammation-related genes in the dementia-induced model. Dementia rats experienced a significant over-expression of TLR4, NF-κB, TNF-α, COX-2, iNOS, and caspase 3, accompanied by a significant down-expression of BCL-2 compared to the negative control group. On the contrary, dementia-induced rats treated with either TMZ (20 or 40 mg/kg) or memantine demonstrated a significant diminution in the gene activity patterns of TLR4, NF-κB, TNF-α, COX-2, iNOS, and caspase 3 accompanied by a significant upregulation of BCL-2 gene in comparison with the untreated dementia-induced rats. Notably, dementia rats treated with 40 mg/kg of TMZ revealed a significant down-expression of TLR4, iNOS, and COX-2, parallel with a significant elevation of the gene expression level of BCL-2 with respect to those treated with either 20 mg/kg TMZ or memantine.
Histological observations
Figure 5 illustrates the microscopic investigation of brain tissue samples (Hippocampal/CA3) of the different experimental groups. Brain tissue samples obtained from negative control rats showed normal organized histological features of hippocampal layers, including pyramidal neurons with intact nuclear details (black arrow) and intact intercellular brain matrix. Samples obtained in the dementia-induced group revealed significant neuronal injury and loss, with several instances of deteriorated or necrotic hypereosinophilic neurons losing their subcellular details with moderate intercellular edema (blue arrow) and minor records of apparent intact pyramidal neurons (black arrow). Moderate higher records of reactive glial cell infiltrates were also shown (arrowhead). On the contrary side, hippocampal sections of the TMZ 20 group demonstrated moderate neuroprotective efficacy with few records of degenerative neuronal changes (blue arrow) alternated with many figures of well-organized apparent intact neurons (black arrow); however; persistence of higher forms of glial cell infiltrates was observed (arrowhead). Interestingly, the microscopic observation of hippocampal sections taken from the TMZ 40 group indicated a remarkable protective efficacy with minimal scattered records of degenerative neuronal changes (blue arrow) and apparent intact neurons (black arrow) with milder glial cell infiltrates (arrowhead). The hippocampal samples of the memantine group displayed lower scattered records of degenerative neuronal changes (blue arrow) and intact neurons (black arrow) with milder glial cell infiltrates (arrowhead).
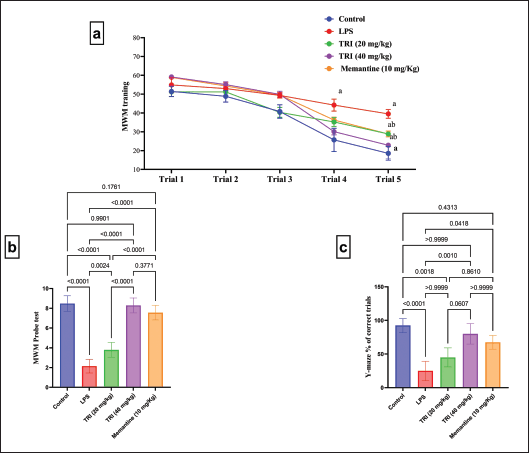 | Figure 2. Influence of TMZ and memantine administration on Y-maze and MWM tasks in different groups. (a) MWM training; (b) MWM probe test with F = 105.5 (DFn = 4, DFd = 30, and p < 0.0001); and (c) Y-maze % correct trial with K = 31.65 (colums = 5, total number = 30, and p < 0.0001). Data are presented as mean ± SD except for Y-maze, which are presented as median ± IQR. a Significance versus negative group and b Significance versus dementia group at p < 0.05. [Click here to view] |
Immunohistochemical findings
IbA-1 expression in the hippocampal tissue was determined by immunohistochemical technique to estimate the activation of microglia in response to inflammation triggered by LPS administration. Figure 6 illustrates the representative photomicrographs of IbA-1 immunostaining of hippocampal tissue of the dissimilar groups. Hippocampal sections attained from the negative control group showed a lack of IbA-1 immunostaining. In contrast, those obtained from the dementia group demonstrated significantly elevated expression of IbA-1 as compared to the negative control group, indicating the microglial activation following LPS supplementation. On the opposite side, the hippocampal sections of dementia-induced rats treated with 20 mg/kg TMZ revealed moderate IbA-1 immunoreactive cells. While those dementia rats received 40 mg/kg, TMZ displayed a substantial drop in the number of IbA-1 immunoreactive cells compared to dementia-induced rats. The hippocampal tissue sections of the dementia rats treated with memantine demonstrated low expression of IbA-1.
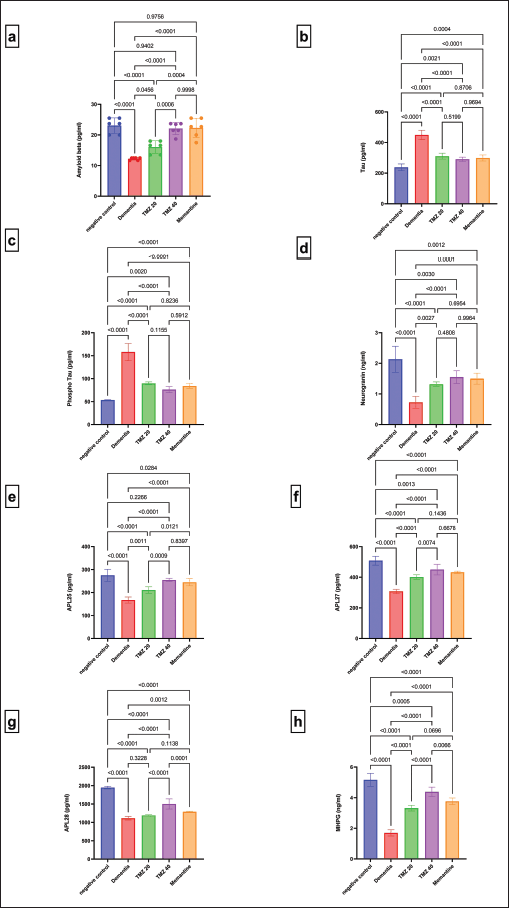 | Figure 3. Influence of TMZ and memantine administration on serum levels of dementia-related biomarkers in dementia rats. (a) Aβ with F = 28.16 (DFn = 4, DFd = 25, and p < 0.0001); (b) Tau with F = 79.38 (DFn = 4, DFd = 25, and p < 0.0001); (c) P-Tau with F = 108.5 (DFn = 4, DFd = 25, and p < 0.0001); (d) neurogranin with F = 25.53 (DFn = 4, DFd = 25, and p < 0.0001); (e) APL25 with F = 39.16 (DFn = 4, DFd = 25, and p < 0.0001); (f) APL27 with F = 64.01 (DFn = 4, DFd = 25, and p < 0.0001); (g) APL28 with F = 144.0 (DFn = 4, DFd = 25, and p < 0.0001); and (h) MHPG with F = 129.2 (DFn = 4, DFd = 25, and p < 0.0001). Data are presented as mean ± SD. [Click here to view] |
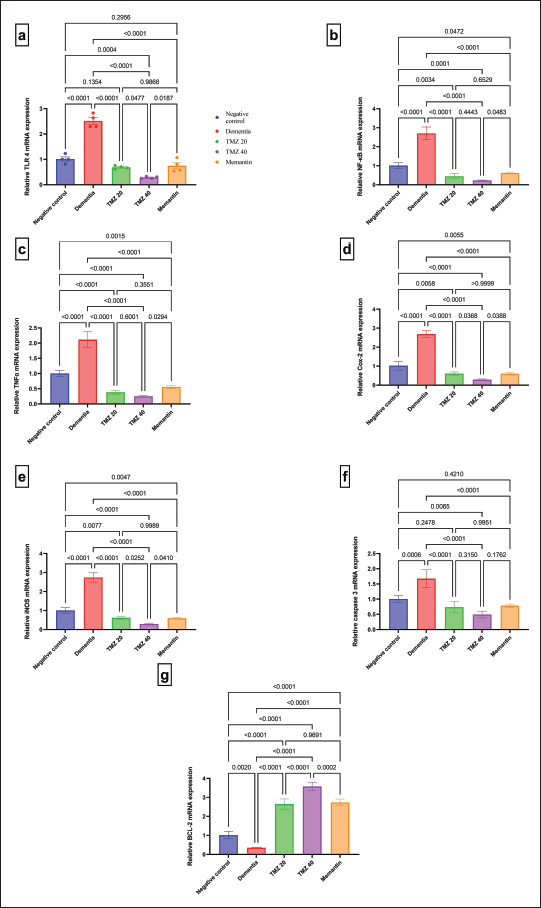 | Figure 4. Impact of TMZ (20 or 40 mg/kg) and memantine administration on gene expression patterns of (a) TLR4 with F = 89.35 (DFn = 4, DFd = 15, and p < 0.0001); (b) NF-κB with F = 124.6 (DFn = 4, DFd = 15, and p < 0.0001); (c) TNF-α with F = 139.4 (DFn = 4, DFd = 15, and p < 0.0001); (d) COX-2 with F = 189.7 (DFn = 4, DFd = 15, and p < 0.0001); (e) iNOS with F = 209.7 (DFn = 4, DFd = 15, and p < 0.0001); (f) Caspase 3 with F = 26.38 (DFn = 4, DFd = 15, and p < 0.0001); and (g) BCL-2 with F = 182.6 (DFn = 4, DFd = 15, and p < 0.0001) in different groups. Data are presented as mean ± SD. [Click here to view] |
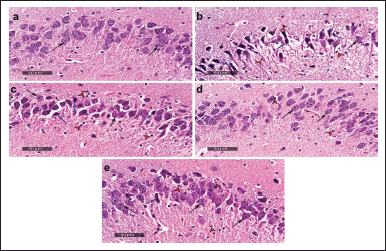 | Figure 5. Histological examination of hippocampal tissue sections of the different groups; (a) negative control group; (b) dementia group; (c) TMZ 20 group; (d) TMZ 40 group; and (e) memantine group (Scale bars: 50 μm). [Click here to view] |
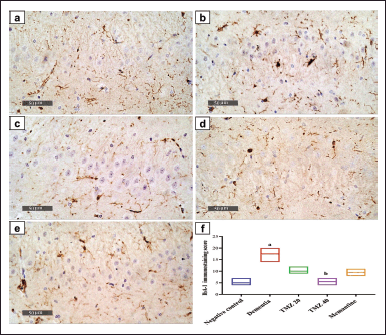 | Figure 6. IbA-1 immunopositive cells in the hippocampal tissue sections of the different groups; (a) negative control group; (b) dementia group; (c) TMZ 20 group; (d) TMZ 40 group and (e) memantine group (scale bars: 50 μm), (f) IbA-1 immunostaining score with K = 25.46 (colums = 5, total number = 30, and p < 0.0001). Data are presented as median ± IQR (n = 6). a Significance versus the negative control group and b Significance versus dementia group at p < 0.05. [Click here to view] |
DISCUSSION
Neuroinflammation is the common hallmark of many neurodegenerative diseases, including AD, Parkinson’s disease, and multiple sclerosis [32]. Abnormal aggregation of extracellular Aβ plaques in the brain is one of the pathological markers of AD. Aβ activates the inflammatory pathway and tau phosphorylation, resulting in neurotoxicity through free radicals and nitric oxide (NO) production. NO stimulates the secretion of excitatory glutamate, which ultimately triggers the “N-methyl-d-aspartate (NMDA) receptor,” which ultimately leads to neuronal cell death [33].
According to our findings and in comparison with the negative controls, dementia rats experienced a decline in cognitive functions, as indicated by more extended periods in the learning acquisition phase, longer times on the MWM probing test with a lesser ORT discriminating index, and a lower number of correct trials in the Y-maze. These findings are greatly supported by those of Lee et al. [34] who indicated impairment in cognitive memory as elucidated by a remarkable upsurge in the time for familiar object exploration and a decline in the DI. This supposes a great brain degeneration after exposure to memory-impairing events, leading to a decline in episodic memory and recognition ability [35]. ORT evaluates the reactions of animals to unfamiliar and familiar objects [36]. Rats are subjected to ORT to examine their ability to recognize a novel object as their natural habit and stimulus recognition is subjected to evaluation [37]. Memory consolidation is hippocampus-dependent but not persistent. During the ORT, spatial or contextual object features are moved in the brain [38]. When a memory is retrieved in the context of novelty, it becomes labile and needs stability. Reconsolidation reorganizes previously established memories to add new knowledge [39]. Spatial memory consolidation is linked to hippocampal neurogenesis [40]. The MWM test was initially devised to assess the spatial learning and memory of mice [41]. This approach is reliable for evaluating cognitive function and visual short-term memory [42]. Similarly, the Y-maze task has been demonstrated to indicate hippocampal injury, evaluate cognitive deficits, and assess the impact of different medications on cognition [43].
The data in the current approach indicated that the dementia group exhibited a substantial drop in Aβ1-42 serum level. Recent research has focused on a blood-based Aβ screening tool for AD diagnosis since it is less intrusive, cost-effective, and widely available than cerebrospinal fluid (CSF) analysis and amyloid PET scans. Lower Aβ 1–42 serum levels enhanced the risk of AD as they were linked to the decreased hippocampus volume in the elderly, suggesting that when Aβ 1–42 deposits in the brain, plasma Aβ levels drop, leading to hippocampal atrophy [44]. It has also been mentioned that Aβ42, as well as the Aβ42/Aβ40 ratio in plasma, were significantly lower in AD patients and inversely correlated with in vivo measures of fibrillar brain Aβ load measured by Pittsburgh Compound B [45]. Plasma Aβ levels have also been found to drop in those at risk for developing AD, and the low plasma levels of Aβ mirror the decline of Aβ in CSF to be indebted to amplified accumulation in the brain [46]. In addition, in the Tg2576 transgenic mouse model of AD, increased amyloid deposition in the brain was linked to concomitant decreases in A levels in CSF and plasma [47]. Sundelöf et al. [48] suggested that boosted accumulation of A in the CNS preceding the beginning of clinically evident cognitive impairment may account for the correlation between low plasma Aβ and the risk of AD.
In the current investigation, blood tau protein levels were significantly higher in the dementia group than in the negative control group. This outcome fits the study of Mattsson et al. [49] who found elevated plasma tau levels in individuals with AD dementia. Moreover, research showed a strong correlation between plasma tau and cognitive decline, brain shrinkage, and hypometabolism in AD patients during follow-up. Tau, which usually resides in the brain’s interstitial fluid, has been linked to extracellular leakage in AD. Plasma tau partly reflects brain pathology and high plasma tau reflects fast disease progression in the last stages. A recent study by Foiani et al. [50] indicated that plasma tau levels are raised in frontotemporal dementia, a neurodegenerative condition that causes personality changes or language difficulties.
The dementia group showed significantly higher serum P-Tau levels versus the normal control group. Serum P-Tau level is a portion of the brain’s CSF-secreted P-Tau and indicates brain pathophysiological changes. Thus, it is considered a promising AD diagnostic marker [51]. AD patients have higher plasma P-Tau181 than controls. Such elevation begins in the early AD stage and increases throughout later stages, which coincides with CSF P-Tau181 elevation [52]. In individuals with Lewy body dementia, plasma P-Tau217 is related to plasma P-Tau181 and tau-positron emission tomography (PET) signal in the temporal brain. Plasma P-Tau may be a marker for AD co-pathology in Lewy body dementia [53].
The current research indicates that serum neurogranin showed significantly lower levels in the dementia group than in the negative control group. Neurogranin, a postsynaptic protein in dendrites, is released by excitatory neurons in the neocortex and hippocampus. It regulates synaptic plasticity, such as long-term potentiation and depression, which are crucial to memory consolidation [54]. AD patients were reported to have greater CSF neurogranin concentrations than cognitively healthy people [55], indicating synaptic degradation, which is associated with cognitive impairment [56]. Recent research by He et al. [57] found that neurogranin in neuron-derived exosomes (NDEs) in plasma showed the opposite pattern, possibly owing to transit from plasma to CSF. Furthermore, it has been found that NDEs in plasma declined in AD patients during the course of the illness and were even repressed in the years leading up to the beginning of the disease [58].
Serum APL25, APL27, and APL28 levels were significantly lower in the dementia group than in the negative control group. Amyloid precursor protein (APP) is a type I transmembrane protein. It is implicated in AD because of its proteolytic processing via the amyloidogenic pathway, which generates neurotoxic Aβ peptides that provoke AD’s extracellular amyloid plaques [59]. APP belongs to a gene family including APLP1 and APLP2 [60]. It has been proposed that APLP1 also can be managed by γ- or β–secretase, such as APP, to produce simple Aβ-like peptides. These include some changes in amino acids “denoted APL1β25, APL1β27, and APL1β28, respectively” [61]. APLP1-based peptides may be a reliable alternative biomarker for Aβ 1-42 formation in the brain’s tissue [62]. Thus, the decreased serum levels of the three APL1 peptides in the dementia group may be due to overexpression of APP and competition between APP and APLP1 for β- and γ-secretase processing, leading to an increase in Aβ1-42 deposition in the brain and a reduction in Aβ1-42 and APLP1-derived peptides in the serum, as shown in the present study.
The current attempt demonstrated a reduced serum level of MHPG in the dementia group. This result is consistent with Dekker et al. [63], who recorded a significant decrease in the MHPG serum level of dementia patients as compared to healthy subjects. German et al. [64] reported a correlation between the severity of the clinical manifestations of AD and noradrenergic deficits. Noradrenaline (NA), produced from dopamine (DA), is the precursor of MHPG and is considered the primary neurotransmitter of the sympathetic branch of the peripheral nervous system and brain. MHPG can freely cross the blood–brain barrier in contrast to NA, reflecting the main NA activity. Thus, plasma MHPG levels have been utilized to indicate the noradrenergic metabolism in the brain [65]. It has been reported that the decreased NA concentrations in brain areas and serum lead to diminished blood levels of the freely diffusing MHPG [63].
The current investigation demonstrated a significant elevation of hippocampal TLR4 and NF-κB gene expression levels in dementia-induced rats conforms to the study of Zarezadeh et al. [5]. In addition, the hippocampal tissue of dementia rats revealed significant overexpression of TNF- α, iNOS, and COX-2 genes, which are in agreement with the study of Yeo et al. [2]. Toll-like receptors are essential in recognizing invading pathogens and initiating immune responses. TLR4 exists as a complex with the co-receptor myeloid differentiation protein-2 (MD-2), expressed on the microglia surface. It has been suggested that LPS could induce dementia via triggering TLR4–MD-2 complex dimerization, resulting in the induction of the downstream NF-κB signaling pathway mediating the expression of pro-inflammatory mediators, including COX-2, TNF-α, IL-1, IL-6, and iNOS, which are responsible for neuroinflammation and neurodegeneration [66].
The present attempt demonstrated that the dementia group showed a significant up-regulation of the hippocampal caspase-3 gene accompanied by a substantial down-expression of the BCL-2 gene. These results are in harmony with those of Zhang et al. [67] and Khan et al. [68]. It has been reported that dementia induced by LPS is initiated by neuroinflammation, which stimulates the mitochondrial apoptotic pathway and apoptotic neurodegeneration. The process of causing neuronal cell death by LPS via the TLR4 pathway results in AP-1 activation, which further activates the JNK signaling pathway to inhibit BCL-2 proteins and activate the mitochondrial apoptotic pathway [69]. The activation of the mitochondrial apoptotic pathway upon LPS injection is triggered by Bax/Bcl-2 signaling. Activated Bax/Bcl-2 provokes the activation and release of cytochrome c, which in turn stimulates the activation of caspase cascades, including caspase-3, which is known to play an essential role in apoptotic neurodegeneration. The activated caspase-3 induces neural cell apoptosis and cleaves poly(ADP-ribose) polymerase 1, which causes damage to the DNA of neurons [68,69].
Zarifkar et al. [70] reported that intraperitoneal injection of LPS provokes neuroinflammation, hippocampal apoptosis, and beta-amyloid plaque formation in the hippocampus, leading to cognitive impairment and learning deficits. This explains the neuronal cell death and the pathological alterations observed in the dementia-induced group upon the histological investigation of their hippocampal sections, which agrees with the result reported by Amraie et al. [71].
IbA-1 is a specific marker for microglia; the current study revealed a significant increase in the number of IbA-1-positive cells in dementia-induced rats. With this result, we agree with Ifuku et al. [72] who demonstrated a significant elevation in IbA-1-positive microglia in the hippocampus of LPS-injected rats. Microglia, the major brain macrophages, are known to play an essential role in the development of neuroinflammation, and microglial stimulation mainly contributes to central nervous system-related injuries. Under normal conditions, microglia phagocytize brain metabolic products, synaptic elements, and dead cells in the brain. Thus, once activated, microglia are recruited to the site of the lesion and eliminate cellular debris. Although microglial activation seems to be essential for host defense, persistent microglial activation can cause apoptosis of neural cells and the release of pro-inflammatory cytokines in the hippocampus. Aβ stimulates microglia and activates their production of pro-inflammatory mediators, such as TNF-α, NO, and interleukin (IL)-1β [32]. In LPS-induced dementia, the induction of macrophages and microglial cell activation occurs via stimulating the TLR4 signaling cascade, which in turn initiates inflammation as a result of the production of inflammatory mediators such as oxygen-free radical, NO, and pro-inflammatory cytokines, including TNF-α and IL-1β [5].
TMZ administration resulted in reduced cognitive impairment, which was demonstrated by shorter times in the learning acquisition phase and probing test of MWM with improvement in the DI of ORT accompanied by an increased number of correct trials in Y-maze. These results are in accordance with those of the study of Mohamed et al. [19] who declared the improvement of cognitive function in diabetic epileptic rats in response to TMZ intake. These investigators documented that TMZ reduced apoptotic markers and inflammatory cytokines and alleviated hippocampus neuronal damage-induced cognitive impairment. TMZ inhibits LPS-induced brain inflammation by activating antioxidant enzymes, promoting axonal regeneration and myelination, and preventing drug-induced hippocampal oxidative damage and experimental brain shrinkage [73]. In addition, TMZ enhances brain activity by protecting neuronal cells from intracellular acidosis, preserving the brain mitochondrial membrane, inhibiting lipid peroxidation, and regulating Na/Ca channels [74].
Neuronal excitement increases brain energy consumption from glucose metabolism, producing acetyl-coenzyme A, which synthesizes brain acetylcholine [75]. The cholinomimetic drugs reduced Aβ formation, which explains TMZs actions [76]. In addition, they slowed the AD progression and improved learning and memory functions in the Tg2576 AD mice model that overexpressed human APP [77].
Furthermore, the treatment of dementia groups with TMZ brought about a significant amelioration in the serum biochemical markers of dementia represented by serum Aβ, tau protein, P-tau, and neurogranin levels in addition to APL1β25, APL1β27, APL1β28, and MHPG serum levels. TMZ has been reported to restore energy metabolism in mitochondria [78], regulating the apoptosis rate in brain injury [79]. Moreover, TMZ has been found to be anti-inflammatory and block cytokine release to restore blood–brain barrier integrity [80]. The capacity of TMZ to easily pass the blood–brain barrier and disperse across significant brain regions to impact numerous processes of brain damage is a crucial factor in the drug’s neuroprotective effect [81]. Earlier studies demonstrated that TMZ possesses anti-oxidative and anti-inflammatory properties and plays cytoprotective roles in various tissues, including nervous tissue [82]. A more recent study reported that the antioxidant enzymes superoxide dismutase and catalase were both stimulated by TMZ, and malondialdehyde levels were lowered [8]. Moreover, Hassanzadeh et al. [73] reported that TMZ could increase seladin-1 mRNA levels in the hippocampus of the rat model of sporadic AD. Seladin-1 has been demonstrated to be resistant to β-amyloid deposition-induced toxicity and OS [83]. All of this evidence could explain the almost normal level of serum Aβ 1-42 and the reduced level of tau protein and P-Tau in the serum of the dementia group upon treatment with TMZ, as shown in the current study.
The pathogenesis of AD involves Aβ deposition that is responsible for stimulating synaptic impairment and neurodegeneration. The susceptibility of the brain to OS is deliberated to be a critical damaging issue in AD. Aβ and OS are closely related to one another since Aβ stimulates OS and the contrary. Evidence demonstrates that prolonged oxidative damage leads to the manifestation of AD symptoms, comprising Aβ buildup, formation of the neurofibrillary tangle, metabolic disorder, and cognitive dysfunction [84]. The significant recovery of neurogranin serum level to almost normal level in the dementia group treated with TMZ could be able to be accounted for TMZ’s ability to reduce lipid peroxidation by tempering β-oxidation via hindering of β-keto thiolase (3- KAT) coenzyme and ensuring glucose uptake in the brain [78]. This antioxidant activity contributes to the neuroprotective action exerted by TMZ in neurodegenerative diseases triggered by pro-inflammatory mediators via lipid peroxidation [85]. This neuroprotective activity of TMZ is endorsed due to its potential to reduce fatal oxidation of mitochondria, motivate p-ERK 1/2 and p-AMPK signaling cascades, and reduce adenosine triphosphate-dependent energy disruptions. This was all displayed as a decline in neuronal death and conserved neuronal integrity [86]. Interestingly, mitochondria of rat brains have been found to contain binding sites for TMZ [87]. Previous in vitro investigations indicated that TMZ could hinder mitochondrial permeability transition pores’ opening [88] and, thus, prevent apoptotic neuronal loss following brain injury. Therefore, one can postulate that the antioxidant, anti-inflammatory, and anti-apoptotic properties of TMZ enable it to reduce synaptic degeneration and restore neurogranin serum levels.
Treatment of the dementia groups with TMZ yielded a substantial improvement in the blood levels of APL25, APL27, and APL28, as well as MHPG. It has been reported that there is an equilibrium between Aβ formation and destruction via self-regulatory pathways [89]. However, the impairment of this balance leads to Aβ accumulation in the brain [90]. Essential pathways that interfere with this equilibrium are free radical injury and neuron inflammation [91]. Thus, the overproduction of reactive oxygen species (ROS) is considered a crucial factor of synaptic dysfunction and ultimately provokes senile plaque formation [92].
Furthermore, in vitro study by Muche et al. [93] indicated that oxidative damage gradually motivated APP expression levels in endothelial cells originating from cerebral cortical tissue of transgenic Tg2576 AD mice. In addition, oxidizing agents have been reported to surge the translation of APP [94] and escalate secreted Aβ levels [95]. Many antioxidants have been employed to decrease AD-related OS and defend cells against Aβ-dependent neurotoxicity [96].
Regarding the role of neuroinflammation in dementia, the “family pyrin domain containing 3” (NLRP3), an inflammasome, is a new cytosolic-multi-protein implicated with neuron inflammation and the body’s natural defenses [97]. Thus, Aβ deposition in a transgenic AD mice model may be reduced by blocking the NLRP3 [98–100]. In addition, inflammatory mediators promote APP expression and the development of Aβ [101]. Anti-inflammatory medications may, thus, prevent or cure AD by preventing neuroinflammation, which reduces the generation of anti-inflammatory medications may thus prevent or cure AD by preventing neuroinflammation, which reduces the generation of Aβ [102,103]. Hence, the underlying mechanism by which TMZ could ameliorate the serum levels of APL25, APL27, APL28, and MHPG in the dementia group relies on the antioxidant and anti-inflammatory potential of this drug, which reduces the overproduction of APP and Aβ deposition in the brain with consequent protection of central neurons including noradrenergic neurons.
In the current study, TMZ administration produced a significant down-regulation of hippocampal TNF-α TLR-4 and NF-κB genes. These data are greatly supported by the findings of Su et al. [104] which demonstrated the decline of inflammatory markers, such as NF-κB and TNF-α, following TMZ treatment of coronary artery micro-embolism pig model. Furthermore, Ozturk et al. [105] commented that TMZ ameliorated the ovary ischemia-reperfusion injury via down-regulating the expression of TLR-4 and NF-κB proteins, attenuating OS and inflammation. TMZ has been reported to possess anti-inflammatory effects owing to its potential to have a major impact on lowering the concentrations of inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, in the bloodstream [80]. Moreover, TMZ reduces the release of pro-inflammatory mediators from macrophages induced by ROS, including C-reactive protein, TNF-α, IL-1, and interleukin 8 during both inflammation and ischemia [106].
In the present investigation, the hippocampal tissue of TMZ-treated rats illustrated a significant down expression of iNOS and COX-2 genes, which comes in line with the study of Natarajan et al. [107] who reported the decrease in the intestinal expression of iNOS, COX-2 in methotrexate-induced injury following TMZ treatment. Moreover, our findings revealed that TMZ treatment had reversed the mitochondrial apoptotic pathway and neurodegeneration by up-regulating BCL-2 transcribing genes and reducing the activated caspase-3 transcriptional activity in the hippocampal region of the dementia-induced rat brain. These findings are consistent with those of the study of Zhang et al. [108] which indicated that TMZ attenuated cardiac damage by hindering programmed cell death through decreasing Bax/Bcl-2 proportion and deactivating caspase-3 expression, sliced PARP, and reducing cytochrome c levels in the rats’ myocardium. In addition, TMZ inhibited oxidation strain and cardiac programmed cell death via activating Nrf2/HO-1 and suppressing NF-κB signal transduction. These outcomes could be attributed to the ability of TMZ to decrease the TLR4 expression, which in turn down-regulates the downstream signaling of NF-κB, resulting in inhibition of the pro-inflammatory mediators, including COX-2, TNF-α, IL-1, IL-6, and iNOS, which are the culprit for neuroinflammation and neurodegeneration. This is because dementia induced by LPS is commenced through neuroinflammation, causing apoptotic neurodegeneration via the TLR4 signaling cascade. Such signaling activates AP-1, stimulating JNK to interfere with BCL-2 and activate mitochondrial apoptosis [69]. Therefore, the mechanism underlying the over-expression of BCL-2 following TMZ treatment in the dementia group could be ascribed to the down-regulation of TLR4 and its downstream signaling pathway.
Histological examination of hippocampal sections obtained from the TMZ 40 group displayed little neuronal degenerative changes and intact neurons, indicating its notable neuroprotective efficacy. This discovery is as per the observations of Hassanzadeh et al. [73] who revealed the neuroprotective effect of TMZ against sporadic AD as confirmed by the attenuation of hippocampal CA3 neuron loss upon histological examination. Moreover, these investigators indicated that this beneficial effect of TMZ could be attributed to the free-radical scavenging activity and restoration of intracellular antioxidant activities. In our study, TMZ treatment displayed such a protective effect against neural damage, which may be due to its scavenging ability of ROS released upon LPS administration, resulting in ameliorating the OS and the inflammatory response induced by LPS.
In the present approach, TMZ administration resulted in the inhibition of the microglial activation, as validated by the substantial drop in IbA-1 immunopositive cells in the hippocampal regions of rat brains. This could be explained by the anti-inflammatory effect of TMZ responsible for hindering the TLR-4/NF-κB pathway [105], resulting in the deactivation of microglia and hence suppressing the inflammatory response.
Memantine is regarded as a neuroprotective agent for treating several dementias [109]. Memantine has been known as the antagonist of the NMDA receptor since it exerts its neuroprotective action against AD by blocking the N-methyl-D-aspartate receptor channel and hence alleviating glutamate-mediated neurotoxicity [33].
The present study indicated that dementia rats treated with memantine experienced a remarkable enhancement in novel object recognition time, DI in ORT, decreased training time latency on the fourth and fifth training days of the MWM, and a more extended exploration time in the probing test. Moreover, memantine treatment in dementia rats showed an improved alternating memory as indicated by the higher number of correct trials of Y-maze. Higaki et al. [42] reported that memantine treatment reduced age-related memory loss in LPS-induced neuroinflammation in rats.
The current approach demonstrated that dementia groups treated with memantine exhibited a significant recovery of the serum levels of Aβ, neurogranin, APL1β25, APL1β27, APL1β28, and MHPG together with a significant decrease in tau protein and P-tau serum levels. It has been stated that the supplementation of rat cortical neuronal cultures with memantine reduced the secretion of Aβ1–42. Moreover, APP/presenilin-1 (PS1) transgenic mice treated with memantine exhibited significantly reduced cortical levels of soluble Aβ 1–42. Thus, memantine could reduce amyloid plaque formation and might inhibit the accumulation of fibrillogenic Aβ in mammalian brains [110]. Memantine is a cationic amphiphilic drug with a similar structure to lysosomotropic drugs that are known to accumulate in acidic cellular compartments and inhibit phospholipases [111]. The possible mechanism by which memantine could reduce the secretion of APP and Aβ peptides into the conditioned media of cell cultures could be attributed to the accumulation of memantine in lysosomes. Moreover, it has been suggested that memantine could alter the activity of the γ-secretase complex so that the majority of the Aβ produced is the shorter, less amyloidogenic Aβ1–40 [110]. All of this evidence supports the reason behind the recovery of Aβ1–42 serum levels in memantine-treated groups.
Memantine can repress the tau neuronal forma via the cap-independent triggered pathway. This means that memantine can be used as a potential tau internal ribosome entry site (IRES)-reliant on inhibition of translation as it inhibits the ability of tau-IRES, resulting in decreased tau expression. As a result, memantine’s effect on AD might be attributable to its ability to inhibit both NMDA receptor-induced excitotoxicity and IRES-mediated translation [112]. The impairment of phosphoseryl/phosphothreonyl protein phosphatase (PP)-2A, known to regulate tau phosphorylation in the AD brain, is believed to be the causative factor of abnormal tau accumulation and hyper-phosphorylation. It has been reported that the NMDA receptor forms a complex with PP-2A. However, NMDA receptor overactivation in chronic neurodegenerative conditions leads to PP-2A dissociation from that complex, resulting in the reduction of PP-2A activity. It has been suggested that memantine could suppress the abnormal tau hyperphosphorylation and neurofibrillary degeneration in AD via modulating PP-2A signaling in addition to its effect as an NMDA receptor antagonist [113].
AD is characterized by several neuropathological hallmarks, including amyloid plaque deposition, loss of acetylcholine in the hippocampus, and generalized synaptic loss [114]. It has been reported the neuroprotective effect of memantine on neuronal cell culture models and in a transgenic AD mouse model is manifested by improved neuronal morphology and synaptic transmission in degenerating primary neurons, in addition to preserving synaptic markers. Memantine was shown to alter APP processing in AD animal models. APP and Aβ are associated with neurite and synapse regulation. In addition to lowering synaptic noise, memantine has been shown in preclinical studies to restore the normal physiological function of NMDA receptors. This means memantine restores impaired synaptic plasticity [115]. It is considered a neuroprotective agent by reducing glutamate-dependent toxicity and was found to boost the levels of “brain-derived neurotrophic factor,” hence affecting rodent neuroplasticity [116]. Thus, memantine possesses the ability to recover synaptic plasticity and support the treatment of diseases interfering with cognitive function [117]. The potency of memantine to stimulate synaptic transmission in the hippocampus is in harmony with the described positive impact on cognition dysfunction in humans [118]. These findings indicate that memantine enhances neurogranin through its potent effect on the restoration of synaptic impairment.
Regarding the effect of treatment of the dementia groups with memantine on APL25, APL27, and APL28 serum levels, it has been established that memantine administration reduced the brain levels of soluble and insoluble Aβ peptides inTg2576 mice, suggesting that memantine acts via reducing Aβ production through the regulation of intracellular APP trafficking [119]. Memantine treatment in the 5XFAD model of AD significantly reduced amyloid plaque pathology [120]. In addition, the study of Liu et al. [121] observed that the over-expressions of Aβ1-42 and APP were decreased in the brains of APP/PS1 mice upon treatment with memantine. A combination of mechanisms for this action of memantine may bypass the NMDA receptor and act on γ-secretase [110].
Concerning the effect of memantine on MHPG, it has been reported that away from its major action at the NMDA receptors, memantine may affect extracellular targets in the brain, e.g., affect dopaminergic and noradrenergic transmission or stimulate the expression of neurotrophic factors [122]. Treatment with memantine has been found to significantly increase the extracellular DA, NA, and their metabolites in the cortical regions [123]. Nerve growth factor (NGF) is a neurotrophin known to stimulate neuron differentiation, survival, and plasticity. Its deficiency in the brain induces neuron dysfunction and apoptosis, as well as accelerating Aβ deposits and Aβ-induced toxicity [124]. Memantine elicited beneficial neuroprotective effects on amyloidosis and cognitive improvement, possibly by increasing the endogenous NGF [121]. Scott et al. [125] suggested that NGF may be responsible for the regenerative growth of noradrenergic neurons following axonal damage in rat brains. Moreover, NGF signaling is implicated in elevating NA content and noradrenergic neuron-specific enzymes [126]. One can postulate that memantine could normalize the serum level of MHPG via regulating NGF signaling.
The current findings indicated that memantine treatment abrogated the neuroinflammation in dementia, as shown by the significant down-regulation of TNF-α, COX-2, iNOS, TLR-4, and NF-κB genes. These results are in line with those of Alomar et al. [127] who showed that memantine alleviated diabetic neuropathic pain via downregulating the TLR-4/NF-κB inflammatory axis, resulting in downregulating the pro-inflammatory cytokines such as TNF-α.
The present study also showed that dementia rats treated with memantine displayed down-regulation of caspase 3 with concurrent up-regulation of the BCL-2 gene. These findings are supported by Song et al. [33] who indicated that the AD cell model treated with memantine revealed a significant down-regulation of caspase-3 protein expression and over-expression of BCL-2, suggesting its anti-apoptotic activity against neuronal cell death.
Furthermore, the current data indicated a remarkable improvement in the pathological aberrations of hippocampal tissue of dementia rats treated with memantine. This observation conforms with that of Eldeeb et al. [128]. Such favorable neuroprotective effect of memantine against dementia may be attributed to its anti-inflammatory and anti-apoptotic activity through NMDA receptor modulation, inhibiting the microglial activation and hence blocking the inflammatory cascade activation [129–131].
The current study revealed that memantine treatment resulted in mild IbA-1 immunostaining in the hippocampal tissue of dementia rats. This result fits Mei et al. [132], who revealed that memantine treatment attenuated microglial activation after repetitive mild traumatic brain injury in mice. This was validated by the significant decrease of IbA-1-positive cells in the hippocampus. Moreover, an earlier study by Rosi et al. [133] proved that treatment with memantine dramatically decreased the number of activated microglia during chronic neuroinflammation without affecting the number of resident microglia.
CONCLUSION
This study provides evidence for the neuroprotective effects of TMZ against lipopolysaccharide-induced dementia in a rat model, likely through inhibiting neuroinflammation via the TLR4/NF-κB pathway. TMZ treatment attenuated the behavioral and cognitive deficits, improved hippocampal pathology, modulated biochemical markers, and beneficially altered gene expression patterns related to dementia pathophysiology. These preclinical findings reveal the potential of TMZ as a promising therapeutic agent against neurodegenerative dementia. Further clinical studies are warranted to translate these results to humans and investigate the efficacy of TMZ for mitigating mitochondrial damage and neuroinflammation in patients with dementia.
The preclinical evidence from this study raises important implications for potential clinical applications of TMZ to treat neuroinflammation and mitochondrial dysfunction associated with dementia in humans. As a clinically approved drug for angina treatment, TMZ has a known safety profile in humans. The dosage used in this rat study (20–40 mg/kg) would translate to a human equivalent dose of 1.6–3.2 mg/kg based on body surface area conversion between species. This is within the clinical dosage range for TMZ. Therefore, repurposing TMZ as an adjuvant therapeutic in dementia patients could be a promising translational approach.
Clinical trials are needed to evaluate the efficacy of TMZ for modulating biomarkers, cognitive outcomes, and disease progression in patients with mild cognitive impairment and early AD. TMZ could be tested alone or in combination with standard dementia medications like cholinesterase inhibitors. Studies are also warranted to examine if TMZ may have a preventive effect to reduce dementia risk in susceptible populations. In addition, research should explore optimal dosing, safety, and pharmacokinetics of chronic TMZ administration in older adults.
Overall, this study provides a strong rationale for further investigation of TMZ in human clinical trials to assess its potential as a therapeutic agent that targets neuroinflammation and mitochondrial dysfunction relevant to dementia pathophysiology. The findings open promising avenues for developing effective adjuvant treatment strategies for neurodegenerative dementias.
ACKNOWLEDGMENT
The authors would like to give special thanks to Dr. Mohamed Abdelrazik (Assistant Professor of Veterinary Cytology and Histology, Faculty of Veterinary Medicine, Cairo University, Egypt) for this work’s histological and immunohistochemical investigation. In addition, the authors would like to thank Science Shake Inc. for conducting proofreading and English language editing (https://www.science-shake.com/).
AUTHORS’ CONTRIBUTIONS
Nadia S Mahmoud: Conceptualization, molecular analysis, statistical analysis, writing—original draft, data visualization. Marawan A. Elbaset: Conceptualization, animal experiments, and behavioral assessment, statistical analysis, writing—review and editing, data visualization. Ola A.M. Mohawed: molecular analysis, statistical analysis, writing—original draft. Hanaa H Ahmed: Conceptualization, biochemical assessment, data validation, writing—review and editing. All authors wrote, revised, and approved the final manuscript.
FINANCIAL SUPPORT
This work did not receive any financial support.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study was conducted according to the guidelines for laboratory animal care and use and was approved by the “Ethical Committee of the Medical Researches of the National Research Centre, Egypt” (Approval No. 3415062021), complying with the “ARRIVE guidelines and was performed according to the National Institutes of Health Guide for the Care and Use of Experimental Animals (NIH Publications No. 8023, revised 1978).”
DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Ravindranath V, Sundarakumar JS. Changing demography and the challenge of dementia in India. Nat Rev Neurol. 2021;17:747–58. CrossRef
2. Yeo IJ, Yun J, Son DJ, Han SB, Hong JT. Antifungal drug miconazole ameliorated memory deficits in a mouse model of LPS-induced memory loss through targeting iNOS. Cell Death Dis. 2020;11:623. CrossRef
3. Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396:413–46. CrossRef
4. Elshahidi M, Elhadidi M, Sharaqi A, Mostafa A, Elzhery M. Prevalence of dementia in Egypt: a systematic review. Neuropsychiatr Dis Treat. 2017;13:715–20. CrossRef
5. Zarezadeh M, Baluchnejadmojarad T, Kiasalari Z, Afshin-Majd S, Roghani M. Garlic active constituent s-allyl cysteine protects against lipopolysaccharide-induced cognitive deficits in the rat: possible involved mechanisms. Eur J Pharmacol. 2017;795:13–21. CrossRef
6. Jin Y, Peng J, Wang X, Zhang D, Wang T. Ameliorative effect of ginsenoside Rg1 on lipopolysaccharide-induced cognitive impairment: role of cholinergic system. Neurochem Res. 2017;42:1299–307. CrossRef
7. Amber KI, Hadi NR, Muhammad-Baqir BM, Jamil DA, Al-Aubaidy HA. Trimetazidine attenuates the acute inflammatory response induced by Novolimus eluting bioresorbable coronary scaffold implantation. Int J Cardiol. 2016;220:514–9. CrossRef
8. Martins M, Silva R, Pinto MMM, Sousa E. Marine natural products, multitarget therapy and repurposed agents in Alzheimer’s disease. Pharmaceuticals. 2020;13:242. CrossRef
9. Abdelbaset M, Safar MM, Mahmoud SS, Negm SA, Agha AM. Red yeast rice and coenzyme Q10 as safe alternatives to surmount atorvastatin-induced myopathy in hyperlipidemic rats. Can J Physiol Pharmacol. 2014;92:481–9. CrossRef
10. Yassa NW, Khalil S, Saleh SR, Ghareeb DA, El Demellawy MA, El-Sayed MM. Ipriflavone and ipriflavone loaded albumin nanoparticles reverse lipopolysaccharide induced neuroinflammation in rats. PLos One. 2020;15:e0237929. CrossRef
11. Erba? O, Akseki HS, Eliküçük B, Ta?k?ran D. Antipsychotic-like effect of trimetazidine in a Rodent model. Sci World J. 2013;2013:1–5. CrossRef
12. Chen A, Li W, Chen X, Shen Y, Dai W, Dong Q, et al. Trimetazidine attenuates pressure overload-induced early cardiac energy dysfunction via regulation of neuropeptide Y system in a rat model of abdominal aortic constriction. BMC Cardiovasc Disord. 2016;16:225. CrossRef
13. Ahmed H, Zaazaa A, El-Motelp BA. Zingiber officinale and Alzheimer’s disease: evidences and mechanisms. Int J Pharm Sci Rev Res. 2014;27:142–52.
14. El-Marasy SA, El-Shenawy SM, El-Khatib AS, El-Shabrawy OA, Kenawy SA. Effect of Nigella sativa and wheat germ oils on scopolamine-induced memory impairment in rats. Bull Fac Pharm Cairo Univ. 2012;50:81–8. CrossRef
15. Ogaly HA, Abdel-Rahman RF, Mohamed MAE, Ahmed-Farid OA, Khattab MS, Abd-Elsalam RM. Thymol ameliorated neurotoxicity and cognitive deterioration in a thioacetamide-induced hepatic encephalopathy rat model; involvement of the BDNF/CREB signaling pathway. Food Funct. 2022;13:6180–94. CrossRef
16. Contet C. A comparison of 129S2/SvHsd and C57BL/6JOlaHsd mice on a test battery assessing sensorimotor, affective and cognitive behaviours: implications for the study of genetically modified mice. Behav Brain Res. 2001;124:33–46. CrossRef
17. Deacon RMJ, Bannerman DM, Kirby BP, Croucher A, Rawlins JNP. Effects of cytotoxic hippocampal lesions in mice on a cognitive test battery. Behav Brain Res. 2002;133:57–68. CrossRef
18. Hussien YA, Mansour DF, Nada SA, Abd El-Rahman SS, Abdelsalam RM, Attia AS, et al. Linagliptin attenuates thioacetamide-induced hepatic encephalopathy in rats: modulation of C/EBP-β and CX3CL1/Fractalkine, neuro-inflammation, oxidative stress and behavioral defects. Life Sci. 2022;295:120378. CrossRef
19. Mohamed MAE, Abdel-Rahman RF, Mahmoud SS, Khattab MM, Safar MM. Metformin and trimetazidine ameliorate diabetes-induced cognitive impediment in status epileptic rats. Epilepsy Behav. 2020;104:106893. CrossRef
20. Mahmoud NS, Ahmed HH, Mohamed MR, Amr KS, Aglan HA, Ali MAM, et al. Role of nanoparticles in osteogenic differentiation of bone marrow mesenchymal stem cells. Cytotechnology. 2020;72:1–22. CrossRef
21. Liu F, Wen Y, Kang J, Wei C, Wang M, Zheng Z, et al. Regulation of TLR4 expression mediates the attenuating effect of erythropoietin on inflammation and myocardial fibrosis in rat heart. Int J Mol Med. 2018;42:1436–44. CrossRef
22. He J, Han S, Li XX, Wang QQ, Cui Y, Chen Y, et al. Diethyl blechnic exhibits anti-inflammatory and antioxidative activity via the TLR4/MyD88 signaling pathway in LPS-stimulated RAW264.7 cells. Molecules. 2019;24:4502. CrossRef
23. Hussien HM, Abd-Elmegied A, Ghareeb DA, Hafez HS, Ahmed HEA, El-moneam NA. Neuroprotective effect of berberine against environmental heavy metals-induced neurotoxicity and Alzheimer’s-like disease in rats. Food Chem Toxicol. 2018;111:432–44. CrossRef
24. Wu XH, Liu CP, Xu KF, Mao XD, Zhu J, Jiang JJ, et al. Reversal of hyperglycemia in diabetic rats by portal vein transplantation of islet-like cells generated from bone marrow mesenchymal stem cells. World J Gastroenterol. 2007;13:3342–9. CrossRef
25. McCormick DL, Phillips JM, Horn TL, Johnson WD, Steele VE, Lubet RA. Overexpression of cyclooxygenase-2 in rat oral cancers and prevention of oral carcinogenesis in rats by selective and nonselective COX inhibitors. Cancer Prev Res (Phila. Pa.). 2010;3:73–81. CrossRef
26. Saini U, Gumina RJ, Wolfe B, Kuppusamy ML, Kuppusamy P, Boudoulas KD. Preconditioning mesenchymal stem cells with caspase inhibition and hyperoxia prior to hypoxia exposure increases cell proliferation. J Cell Biochem. 2013;114:2612–23. CrossRef
27. Thangarajan S, Ramachandran S, Krishnamurthy P. Chrysin exerts neuroprotective effects against 3-nitropropionic acid induced behavioral despair—mitochondrial dysfunction and striatal apoptosis via upregulating Bcl-2 gene and downregulating Bax—Bad genes in male wistar rats. Biomed. Pharmacother. 2016;84:514–25. CrossRef
28. Xu K, Tian Y, Weng X, Hu X, Heng D, Xia G, et al. Effect of thyroid dysfunction on NOS expression in the female rat. Cell Tissue Res. 2020;379:291–300. CrossRef
29. Ibrahim WW, Abdel Rasheed NO. Diapocynin neuroprotective effects in 3-nitropropionic acid Huntington’s disease model in rats: emphasis on Sirt1/Nrf2 signaling pathway. Inflammopharmacology. 2022;30:1745–58. CrossRef
30. Abbas H, Gad HA, Khattab MA, Mansour M. The tragedy of Alzheimer’s disease: towards better management via resveratrol-loaded oral bilosomes. Pharmaceutics. 2021;13:1635. CrossRef
31. Abdel-Salam OME, Sleem AA, Abd El Baset M, Sayed M, Khadrawy YA, Morsy FA. Cannabis sativa increases seizure severity and brain lipid peroxidation in pentylenetetrazole-induced kindling in rats. Biomed Pharmacol J. 2018;11:1187–97. CrossRef
32. Zhao J, Bi W, Xiao S, Lan X, Cheng X, Zhang J, et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci Rep. 2019;9:5790. CrossRef
33. Song G, Li Y, Lin L, Cao Y. Anti-autophagic and anti-apoptotic effects of memantine in a SH-SY5Y cell model of Alzheimer’s disease via mammalian target of rapamycin-dependent and -independent pathways. Mol Med Rep. 2015;12:7615–22. CrossRef
34. Lee B, Shim I, Lee H. Gypenosides attenuate lipopolysaccharide-induced neuroinflammation and memory impairment in rats. Evid Based Complement Alternat Med. 2018;2018:1–10. CrossRef
35. Lee B, Sur B, Park J, Kim SH, Kwon S, Yeom M, et al. Ginsenoside Rg3 alleviates lipopolysaccharide-induced learning and memory impairments by anti-inflammatory activity in rats. Biomol Ther. 2013;21:381–90. CrossRef
36. Ennaceur A. One-trial object recognition in rats and mice: methodological and theoretical issues. Behav Brain Res. 2010;215:244–54. CrossRef
37. Baxter MG. “I’ve seen it all before”: explaining age-related impairments in object recognition. Theoretical comment on Burke et al. (2010). Behav Neurosci. 2010;124:706–9. CrossRef
38. Oliveira AMM, Hawk JD, Abel T, Havekes R. Post-training reversible inactivation of the hippocampus enhances novel object recognition memory. Learn Mem. 2010;17:155–60. CrossRef
39. Clarke JR, Cammarota M, Gruart A, Izquierdo I, Delgado-García JM. Plastic modifications induced by object recognition memory processing. Proc Natl Acad Sci. 2010;107:2652–7. CrossRef
40. Sarkisyan G, Hedlund PB. The 5-HT7 receptor is involved in allocentric spatial memory information processing. Behav Brain Res. 2009;202:26–31. CrossRef
41. Morris RG. Spatial localization does not require the presence of local cues. Learn Motiv. 1981;12:239–60. CrossRef
42. Higaki A, Mogi M, Iwanami J, Min LJ, Bai HY, Shan BS, et al. Predicting outcome of Morris water maze test in vascular dementia mouse model with deep learning. PLos One. 2018;13:e0191708. CrossRef
43. Kraeuter AK, Guest PC, Sarnyai Z. The Y-Maze for assessment of spatial working and reference memory in mice. Methods Mol Biol. 2019;1916:105–11. CrossRef
44. Hilal S, Wolters FJ, Verbeek MM, Vanderstichele H, Ikram MK, Stoops E, et al. Plasma amyloid-β levels, cerebral atrophy and risk of dementia: a population-based study. Alzheimers Res Ther. 2018;10:63. CrossRef
45. Devanand DP, Schupf N, Stern Y, Parsey R, Pelton GH, Mehta P, et al. Plasma A and PET PiB binding are inversely related in mild cognitive impairment. Neurology. 2011;77:125–31. CrossRef
46. Mayeux R, Honig LS, Tang MX, Manly J, Stern Y, Schupf N, et al. Plasma A 40 and A 42 and Alzheimer’s disease: relation to age, mortality, and risk. Neurology. 2003;61:1185–90. CrossRef
47. Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:372–81. CrossRef
48. Sundelöf J, Giedraitis V, Irizarry MC, Sundström J, Ingelsson E, Rönnemaa E, et al. Plasma β amyloid and the risk of Alzheimer disease and dementia in elderly men: a prospective, population-based cohort study. Arch Neurol. 2008;65(2):256–63. CrossRef
49. Mattsson N, Zetterberg H, Janelidze S, Insel PS, Andreasson U, Stomrud E, et al. Plasma tau in Alzheimer disease. Neurology. 2016;87:1827–35. CrossRef
50. Foiani MS, Woollacott IO, Heller C, Bocchetta M, Heslegrave A, Dick KM, et al. Plasma tau is increased in frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2018;89:804–7. CrossRef
51. Palmqvist S, Tideman P, Cullen N, Zetterberg H, Blennow K, Stomrud E, et al. Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Alzheimers Dement. 2021;17:1034–42. CrossRef
52. Janelidze S, Mattsson N, Palmqvist S, Smith R, Beach TG, Serrano GE, et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med. 2020;26:379–86. CrossRef
53. Hall S, Janelidze S, Londos E, Leuzy A, Stomrud E, Dage JL, et al. Plasma phospho-Tau identifies Alzheimer’s Co-pathology in patients with Lewy body disease. Mov Disord. 2021;36:767–71. CrossRef
54. Casaletto KB, Elahi FM, Bettcher BM, Neuhaus J, Bendlin BB, Asthana S, et al. Neurogranin, a synaptic protein, is associated with memory independent of Alzheimer biomarkers. Neurology. 2017;89:1782–8. CrossRef
55. Tarawneh R, D’Angelo G, Crimmins D, Herries E, Griest T, Fagan AM, et al. Diagnostic and prognostic utility of the synaptic marker neurogranin in Alzheimer disease. JAMA Neurol. 2016;73:561. CrossRef
56. Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W, et al. Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J Alzheimers Dis. 2005;7:103–17. CrossRef
57. He M, Sun L, Cao W, Yin C, Sun W, Liu P, et al. Association between plasma exosome neurogranin and brain structure in patients with Alzheimer’s disease: a protocol study. BMJ Open. 2020;10:e036990. CrossRef
58. Goetzl EJ, Kapogiannis D, Schwartz JB, Lobach IV, Goetzl L, Abner EL, et al. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 2016;30:4141–8. CrossRef
59. Cousins SL, Dai W, Stephenson FA. APLP1 and APLP2, members of the APP family of proteins, behave similarly to APP in that they associate with NMDA receptors and enhance NMDA receptor surface expression. J Neurochem. 2015;133:879–85. CrossRef
60. Shariati SAM, De Strooper B. Redundancy and divergence in the amyloid precursor protein family. FEBS Lett. 2013;587:2036–45. CrossRef
61. Yanagida K, Okochi M, Tagami S, Nakayama T, Kodama TS, Nishitomi K, et al. The 28-amino acid form of an APLP1-derived Aβ-like peptide is a surrogate marker for Aβ42 production in the central nervous system. EMBO Mol Med. 2009;1:223–35. CrossRef
62. Portelius E, Soininen H, Andreasson U, Zetterberg H, Persson R, Karlsson G, et al. Exploring Alzheimer molecular pathology in down’s syndrome cerebrospinal fluid. Neurodegener Dis. 2014;14:98–106. CrossRef
63. Dekker AD, Coppus AMW, Vermeiren Y, Aerts T, van Duijn CM, Kremer BP, et al. Serum MHPG strongly predicts conversion to Alzheimer’s disease in behaviorally characterized subjects with down syndrome. J Alzheimers Dis. 2014;43:871–91. CrossRef
64. German DC, Manaye KF, White CL, Woodward DJ, McIntire DD, Smith WK, et al. Disease-specific patterns of locus coeruleus cell loss. Ann Neurol. 1992;32:667–76. CrossRef
65. Herrmann N, Lanctôt KL, Khan LR. The role of norepinephrine in the behavioral and psychological symptoms of dementia. J Neuropsychiatry Clin Neurosci. 2004;16:261–76. CrossRef
66. Zusso M, Lunardi V, Franceschini D, Pagetta A, Lo R, Stifani S, et al. Ciprofloxacin and levofloxacin attenuate microglia inflammatory response via TLR4/NF-kB pathway. J Neuroinflammation. 2019;16:148. CrossRef
67. Zhang XY, Cao JB, Zhang LM, Li YF, Mi WD. Deferoxamine attenuates lipopolysaccharide-induced neuroinflammation and memory impairment in mice. J Neuroinflammation. 2015;12:20. CrossRef
68. Khan A, Ali T, Rehman SU, Khan MS, Alam SI, Ikram M, et al. Neuroprotective effect of quercetin against the detrimental effects of LPS in the adult mouse brain. Front Pharmacol. 2018;9:1383. CrossRef
69. Badshah H, Ali T, Kim MO. Osmotin attenuates LPS-induced neuroinflammation and memory impairments via the TLR4/NFκB signaling pathway. Sci Rep. 2016;6:24493. CrossRef
70. Zarifkar A, Choopani S, Ghasemi R, Naghdi N, Maghsoudi AH, Maghsoudi N, et al. Agmatine prevents LPS-induced spatial memory impairment and hippocampal apoptosis. Eur J Pharmacol. 2010;634:84–8. CrossRef
71. Amraie E, Pouraboli I, Rajaei Z. Neuroprotective effects of Levisticum officinale on LPS-induced spatial learning and memory impairments through neurotrophic, anti-inflammatory, and antioxidant properties. Food Funct. 2020;11:6608–21. CrossRef
72. Ifuku M, Katafuchi T, Mawatari S, Noda M, Miake K, Sugiyama M, et al. Anti-inflammatory/anti-amyloidogenic effects of plasmalogens in lipopolysaccharide-induced neuroinflammation in adult mice. J Neuroinflammation. 2012;9:197. CrossRef
73. Hassanzadeh G, Hosseini A, Pasbakhsh P, Akbari M, Ghaffarpour M, Takzare N, et al. Trimetazidine prevents oxidative changes induced in a rat model of sporadic type of Alzheimer’s disease. Acta Med Iran. 2015;53:17–24.
74. Mironova OP, Zarubina IV, Krivoruchko BI, Smirnov AV. Antioxidant effects of amtizol and trimetazidine in brain ischemia. Patol Fiziol Eksp Ter. 2001;3:13–6.
75. Al-Kuraishy H, Al-Gareeb A. Central beneficial effects of trimetazidine on psychomotor performance in normal healthy volunteers. Adv Biomed Res. 2017;6:69. CrossRef
76. Basun H, Nilsberth C, Eckman C, Lannfelt L, Younkin S. Plasma levels of Aβ42 and Aβ40 in Alzheimer patients during treatment with the acetylcholinesterase inhibitor tacrine. Dement Geriatr Cogn Disord. 2002;14:156–60. CrossRef
77. Dong H, Csernansky CA, Martin MV, Bertchume A, Vallera D, Csernansky JG. Acetylcholinesterase inhibitors ameliorate behavioral deficits in the Tg2576 mouse model of Alzheimer’s disease. Psychopharmacology (Berl.). 2005;181:145–52. CrossRef
78. Onay-Besikci A, Özkan SA. Trimetazidine revisited: a comprehensive review of the pharmacological effects and analytical techniques for the determination of trimetazidine. Cardiovasc Ther. 2008;26:147–65. CrossRef
79. Christophe M, Nicolas S. Mitochondria: a target for neuroprotective interventions in cerebral ischemia-reperfusion. Curr Pharm Des. 2006;12:739–57. CrossRef
80. Zhou X, Li C, Xu W, Chen J. Trimetazidine protects against smoking-induced left ventricular remodeling via attenuating oxidative stress, apoptosis, and inflammation. PLoS One. 2012;7:e40424. CrossRef
81. Iqbal S, Baziany A, Hussain M, James S, Wright S, Hemmings S, et al. Trimetazidine as a potential neuroprotectant in transient global ischemia in gerbils: a behavioral and histological study. Brain Res. 2002;928:1–7. CrossRef
82. Atilgan D, Parlaktas BS, Uluocak N, Erdemir F, Markoc F, Saylan O, et al. The effects of trimetazidine and sildenafil on bilateral cavernosal nerve injury induced oxidative damage and cavernosal fibrosis in rats. Sci World J. 2014;2014:1–6. CrossRef
83. Greeve I, Hermans-Borgmeyer I, Brellinger C, Kasper D, Gomez-Isla T, Behl C, et al. The human DIMINUTO/DWARF1 homolog seladin-1 confers resistance to Alzheimer’s disease-associated neurodegeneration and oxidative stress. J Neurosci. 2000;20:7345–52. CrossRef
84. Tamagno E, Guglielmotto M, Vasciaveo V, Tabaton M. Oxidative stress and beta amyloid in Alzheimer’s disease. Which comes first: the chicken or the egg? Antioxidants. 2021;10:1479. CrossRef
85. Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7:22–30. CrossRef
86. Al-Shorbagy MY, Wadie W, El-Tanbouly DM. Trimetazidine modulates mitochondrial redox status and disrupted glutamate homeostasis in a rat model of epilepsy. Front Pharmacol. 2021;12. CrossRef
87. Morin D, Sapena R, Elimadi A, Testa B, Labidalle S, Le Ridant A, et al. [3 H]-Trimetazidine mitochondrial binding sites: regulation by cations, effect of trimetazidine derivatives and other agents and interaction with an endogenous substance. Br J Pharmacol. 2000;130:655–63. CrossRef
88. Argaud L, Gomez L, Gateau-Roesch O, Couture-Lepetit E, Loufouat J, Robert D, et al. Trimetazidine inhibits mitochondrial permeability transition pore opening and prevents lethal ischemia–reperfusion injury. J Mol Cell Cardiol. 2005;39:893–9. CrossRef
89. Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med. 2012;2:a006270. CrossRef
90. Yu H, Wu J. Amyloid-β: a double agent in Alzheimer’s disease? Biomed Pharmacother. 2021;139:111575. CrossRef
91. Chang YT, Chang WN, Tsai NW, Huang CC, Kung CT, Su YJ, et al. The roles of biomarkers of oxidative stress and antioxidant in alzheimer’s disease: a systematic review. BioMed Res Int. 2014;2014:182303. CrossRef
92. Karisetty BC, Bhatnagar A, Armour EM, Beaver M, Zhang H, Elefant F. Amyloid-β peptide impact on synaptic function and neuroepigenetic gene control reveal new therapeutic strategies for Alzheimer’s disease. Front Mol Neurosci. 2020;13:577622. CrossRef
93. Muche A, Arendt T, Schliebs R. Oxidative stress affects processing of amyloid precursor protein in vascular endothelial cells. PLos One. 2017;12:e0178127. CrossRef
94. McCarty MF, DiNicolantonio JJ, Lerner A. A fundamental role for oxidants and intracellular calcium signals in Alzheimer’s pathogenesis—and how a comprehensive antioxidant strategy may aid prevention of this disorder. Int J Mol Sci. 2021;22:2140. CrossRef
95. Butterfield DA, Castegna A. Proteomic analysis of oxidatively modified proteins in Alzheimer’s disease brain: insights into neurodegeneration. Cell Mol Biol (Noisy--Gd. Fr). 2003;49:747–51.
96. Ono K, Hamaguchi T, Naiki H, Yamada M. Anti-amyloidogenic effects of antioxidants: implications for the prevention and therapeutics of Alzheimer’s disease. Biochim Biophys Acta BBA Mol Basis Dis. 2006;1762:575–86. CrossRef
97. Van Zeller M, Dias D, Sebastião AM, Valente CA. NLRP3 inflammasome: a starring role in amyloid-β- and Tau-driven pathological events in Alzheimer’s disease. J Alzheimers Dis. 2021;83:939–61. CrossRef
98. Ray A. Cytokines and their role in health and disease: a brief overview. MOJ Immunol. 2016;4:00121. CrossRef
99. Walsh JG, Muruve DA, Power C. Inflammasomes in the CNS. Nat Rev Neurosci. 2014;15:84–97:e20190314. CrossRef
100. Chan AH, Schroder K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J Exp Med. 2020;217:e20190314. CrossRef
101. Zhan X, Cox C, Ander BP, Liu D, Stamova B, Jin LW, et al. Inflammation combined with ischemia produces myelin injury and plaque-like aggregates of myelin, amyloid-β and AβPP in adult rat brain. J Alzheimers Dis. 2015;46:507–23. CrossRef
102. McGeer PL, Rogers J, McGeer EG. Inflammation, antiinflammatory agents, and Alzheimer’s disease: the last 22 years. J Alzheimers Dis. 2016;54:853–7. CrossRef
103. Zhang C, Wang Y, Wang D, Zhang J, Zhang F. NSAID exposure and risk of Alzheimer’s disease: an updated meta-analysis from cohort studies. Front Aging Neurosci. 2018;10:83. CrossRef
104. Su Q, Li L, Zhao J, Sun Y, Yang H. Effects of trimetazidine on PDCD4/NF-κB/TNF-α pathway in coronary microembolization. Cell Physiol Biochem. 2017;42:753–60. CrossRef
105. Ozturk A, Topcu A, Deniz E, Duman Ozturk S, Arpa M, Kutlu Yilmaz E. The protective effects of trimetazidine against ovary ischemia–reperfusion injury via the TLR4/Nf-kB signal pathway. J Biochem Mol Toxicol. 2022;36(8):e23114. CrossRef
106. Dézsi CA. Trimetazidine in practice: review of the clinical and experimental evidence. Am J Ther. 2016;23:e871–9. CrossRef
107. Natarajan K, Abraham P, Kota R, Isaac B. NF-κB-iNOS-COX2-TNF α inflammatory signaling pathway plays an important role in methotrexate induced small intestinal injury in rats. Food Chem Toxicol. 2018;118:766–83. CrossRef
108. Zhang H, Liu M, Zhang Y, Li X. Trimetazidine attenuates exhaustive exercise-induced myocardial injury in rats via regulation of the Nrf2/NF-κB signaling pathway. Front Pharmacol. 2019;10:175. CrossRef
109. Kaengkan P, Kam KY, Do BR, Kang SG. Combination effect of memantine and mesenchymal stem cells on the recovery of brain damage in a rat model of brain ischemia. Anim Cells Syst. 2015;19:110–8. CrossRef
110. Alley GM, Bailey JA, Chen D, Ray B, Puli LK, Tanila H, et al. Memantine lowers amyloid-β peptide levels in neuronal cultures and in APP/PS1 transgenic mice: memantine lowers Aβ levels. J Neurosci Res. 2010;88:143–54. CrossRef
111. Hinkovska-Galcheva V, Treadwell T, Shillingford JM, Lee A, Abe A, Tesmer JJG, et al. Inhibition of lysosomal phospholipase A2 predicts drug-induced phospholipidosis. J Lipid Res. 2021;62:100089. CrossRef
112. Tasi YC, Chin TY, Chen YJ, Huang CC, Lee SL, Wu TY. Potential natural products for Alzheimer’s disease: targeted search using the internal ribosome entry site of Tau and amyloid-β precursor protein. Int J Mol Sci. 2015;16:8789–810. CrossRef
113. Li L, Sengupta A, Haque N, Grundke-Iqbal I, Iqbal K. Memantine inhibits and reverses the Alzheimer type abnormal hyperphosphorylation of tau and associated neurodegeneration. FEBS Lett. 2004;566:261–9. CrossRef
114. Lahiri DK, Bailey JA, Ray B, Tanila H, Banerjee P, Long J. P3-438: the effect of memantine and rivastigmine on synaptic markers and amyloid processing in cell culture and animal models of Alzheimer’s disease. Alzheimers Dement. 2010;6:S581–2. CrossRef
115. Parsons CG, Stöffler A, Danysz W. Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system—too little activation is bad, too much is even worse. Neuropharmacology. 2007;53;699–723. CrossRef
116. Flores ÉM, Cappelari SE, Pereira P, Picada JN. Effects of memantine, a non-competitive N-Methyl-d-aspartate receptor antagonist, on genomic stability: effects of memantine on genomic stability. Basic Clin Pharmacol Toxicol. 2011;109:413–7. CrossRef
117. Bello O, Blair K, Chapleau C, Larimore JL. Is memantine a potential therapeutic for Rett syndrome? Front Neurosci. 2013:7:245. CrossRef
118. Dimpfel W. Effects of memantine on synaptic transmission in the hippocampus in vitro. Arzneimittelforschung. 1995;45:1–5.
119. Ito K, Tatebe T, Suzuki K, Hirayama T, Hayakawa M, Kubo H, et al. Memantine reduces the production of amyloid-β peptides through modulation of amyloid precursor protein trafficking. Eur J Pharmacol. 2017;798:16–25. CrossRef
120. Jürgenson M, Zharkovskaja T, Noortoots A, Morozova M, Beniashvili A, Zapolski M, et al. Effects of the drug combination memantine and melatonin on impaired memory and brain neuronal deficits in an amyloid-predominant mouse model of Alzheimer’s disease. J Pharm Pharmacol. 2019;71:1695–705. CrossRef
121. Liu MY, Wang S, Yao WF, Zhang ZJ, Zhong X, Sha L, et al. Memantine improves spatial learning and memory impairments by regulating NGF signaling in APP/PS1 transgenic mice. Neuroscience. 2014;273:141–51. CrossRef
122. Kloc R, Luchowska E, Wielosz M, Owe-Larsson B, Urbanska EM. Memantine increases brain production of kynurenic acid via protein kinase A-dependent mechanism. Neurosci Lett. 2008;435:169–73. CrossRef
123. Shearman E, Rossi S, Szasz B, Juranyi Z, Fallon S, Pomara N, et al. Changes in cerebral neurotransmitters and metabolites induced by acute donepezil and memantine administrations: a microdialysis study. Brain Res Bull. 2006;69:204–13. CrossRef
124. Colafrancesco V. Targeting NGF-pathway for developing neuroprotective therapies for multiple sclerosis and other neurological diseases. Arch Ital Biol. 2011;149(2):183–92.
125. Scott SA, Liang S, Weingartner JA, Crutcher KA. Increased NGF-like activity in young but not aged rat hippocampus after septal lesions. Neurobiol Aging. 1994;15:337–46. CrossRef
126. Vogel HG. Drug effects on learning and memory. Drug discovery and evaluation. Berlin, Germany: Springer; 2007. 877–942 pp. CrossRef
127. Alomar SY, Gheit REAE, Enan ET, El-Bayoumi KS, Shoaeir MZ, Elkazaz AY, et al. Novel mechanism for memantine in attenuating diabetic neuropathic pain in mice via downregulating the spinal HMGB1/TRL4/NF-kB inflammatory axis. Pharmaceuticals. 2021;14:307. CrossRef
128. Eldeeb AA, Fathy AE, Elgendy SA. Differential potency of vitamin D3, folic acid and memantine in protecting against neurobehavioral alterations of scopolamine induced Alzheimer’s model in rats. Int J Basic Clin Pharmacol. 2021;10:471. CrossRef
129. Jantas D, Laso? W. Anti-apoptotic effect of memantine against staurosporine- and low-potassium-induced cell death in cerebellar granule cells: a development-dependent effect. Pharmacol Rep. 2009;61:827–37. CrossRef
130. Wu HM, Tzeng NS, Qian L, Wei SJ, Hu X, Chen SH, et al. Novel neuroprotective mechanisms of memantine: increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology. 2009;34:2344–57. CrossRef
131. Yick LW, Tang CH, Ma OKF, Kwan JSC, Chan KH. Memantine ameliorates motor impairments and pathologies in a mouse model of neuromyelitis optica spectrum disorders. J Neuroinflammation. 2020;17:236. CrossRef
132. Mei Z, Qiu J, Alcon S, Hashim J, Rotenberg A, Sun Y, et al. Memantine improves outcomes after repetitive traumatic brain injury. Behav Brain Res. 2018;340:195–204. CrossRef
133. Rosi S, Vazdarjanova A, Ramirez-Amaya V, Worley PF, Barnes CA, Wenk GL. Memantine protects against LPS-induced neuroinflammation, restores behaviorally-induced gene expression and spatial learning in the rat. Neuroscience. 2006;142:1303–15. CrossRef