
INTRODUCTION
The World Health Organization has warned that cancer is a perilous disease that can result in a high number of fatalities. GLOBOCAN’s 2020 report revealed that there were 19.3 million cancer cases globally, resulting in 10.0 million deaths. Among them, breast cancer in females (11.7%), lung cancer (11.4%), colorectal cancer (10.0%), prostate cancer (7.3%), and stomach cancer (5.6%) were the most-deadly types. Chemotherapeutic agents used to treat cancer can cause toxic and resistant effects [1], prompting researchers to develop potential compounds as anticancer agents.
Thiophene is a heterocyclic compound that shows promise as a drug component [2], and its bioactivity as an anticancer compound can be enhanced by combining it with other heterocyclic compounds [3,4]. Pyrazoline is a heterocyclic compound consisting of a five-ring structure with two nitrogen atoms joined by an endocyclic double bond and has also demonstrated broad bioactivity in drug discovery, particularly when it contains phenyl groups [5,6]. Pyrazoline derivatives have been shown to have significant potential as anticancer compounds [7]. Among the various methods for synthesizing pyrazoline derivatives, the cyclo-condensation reaction between chalcones and hydrazine derivatives is one of the most commonly used methods [8].
The molecular docking method is employed in drug discovery to explore the relationship between ligands and the active regions of targeted proteins [9]. The docking procedure entails projecting the ligand’s conformational arrangement and estimating the binding affinity via a docking score. Epidermal growth factor receptor (EGFR), a transmembrane protein, has been linked to multiple cancer types, such as ovarian, lung, breast, and colorectal cancer [10]. Nowadays, in silico approaches such as absorption, distribution, metabolism, and excretion (ADME) prediction are preferred over traditional methods [11]. One tool that can be used for this purpose is the pkCSM webtool [12]. This webtool provides important information about a molecule’s potential as a therapeutic candidate [13].
Studies conducted previously reported the effective cytotoxicity of N-phenyl pyrazolines against various cancer cell lines [5,14]. The present investigation centers on the synthesis of targeted thiophene-based N-phenyl pyrazoline (Scheme 1) and their evaluation for anticancer activity in vitro using colorectal (WiDr), breast (T47D and 4T1), cervix (HeLa), cancer cell lines, and normal (Vero) cell line. In addition, we validated our results through in silico assays, including molecular docking studies with EGFR receptors to determine their conformational pose and an ADME study to predict their pharmacokinetic properties.
MATERIALS AND METHODS
Synthesis of 1,5-diphenyl-3-thiophen-2-yl-4,5-dihydro-1H-pyrazole (pyrazoline 1)
The chemicals used in this study were pro-analysis grade and did not require further purification. The synthesis of thiophene-based N-phenyl pyrazoline is depicted in Scheme 1. Initially, 0.27 g (1 mmol) of Chalcone 1 (3-phenyl-1-thiophen-2-yl-2-propen-1-one) was dissolved in absolute ethanol. Subsequently, 20% (w/v) of NaOH and 1 mmol of phenylhydrazine were added, and the mixture was refluxed for 24 hours before being poured into ice-cold water. After filtration, the solid was dried using a vacuum desiccator. The final step of the process was purification through recrystallization in ethanol, which yielded pyrazoline 1 as a white solid, yield: 65.51%, purity: 93.59%, mp: 131.0°C–132.6°C. Fourier Transform Infra-Red (FTIR) (cm−1): 702 (C?S), 1,126 (C?N), 1,381 (Ar C?N), 1,504 (Ar C=C), 1,597 (C=N), 3,071 (Csp2?H). 1H-NMR (500 MHz, CDCl3, ppm): δ 3.13 (1H, CH2, dd, J = 7.50, 17.50 Hz), 3.83 (1H, CH2, dd, J = 12.50, 17.50), 5.25 (1CH, dd, J = 7.50, 12.50 Hz), 6.76 (1HAr, m), 7.01 (4HAr, m), 7.15 (2HAr, m), 7.26 (1HAr, m), 7.32 (5HAr, m). 13C-NMR (125 MHz, CDCl3, ppm): δ 29.80 (CH2), 44.42 (CH2), 64.70 (CHAr), 113.49 (2CHAr), 119.27 (CHAr), 125.96 (2CHAr), 126.64 (CHAr), 127.41 (CHAr), 127.74 (CHAr), 128.97 (2CHAr), 129.26 (2CHAr), 136.96, 142.39, 142.96 (3CAr), 144.72 (C). MS (EI) m/z: 304 (M+), 227, 212, 91, 77, 39.
Synthesis of 5-(2-methoxyphenyl)-1-phenyl-3-thiophen-2-yl-4,5-dihydro-1H-pyrazole (pyrazoline 2)
Pyrazoline 2 was synthesized using the identical method as pyrazoline 1, starting by mixing chalcone 2 (3-(2-methoxyphenyl)-1-thiophen-2-yl-2-propen-1-one) (0.24 g, 1 mmol), 1 mmol of phenylhydrazine, and 20% (w/v) of NaOH. A light brown solid of pyrazoline 2 was obtained with a yield of 80.84%, purity: 100%, mp: 140.1°C–141.2°C. FTIR (cm−1): 702 (C?S), 1,234 and 1,026 (C?O asymm. and sym.), 1,134 (C?N), 1,381 (Ar C?N), 1,512 (Ar C=C), 1,597 (C=N), 3,032 (Csp2?H), 1H-NMR (500 MHz, CDCl3, ppm): δ 2.99 (1H, CH2, dd, J = 7.00, 17.50 Hz,), 3.86 (1H, CH2, dd, J = 12.00, 17.50 Hz,), 3.93 (3H, s, OCH3), 5.57 (1CH, dd, J = 7.00, 12.00 Hz), 6.76 (1HAr, d, J = 7.50 Hz), 6.83 (1HAr, d, J = 7.50 Hz), 6.93 (1HAr, d, J = 7.50 Hz), 6.99 (1HAr, dd, J = 5.00, 4.00 Hz), 7.01 (2HAr, m), 7.02 (1HAr, dd, J = 1.00, 4.00 Hz), 7.13 (1HAr, dd, J = 2.00, 7.50 Hz), 7.17 (2HAr, dd, J = 7.50, 2.00 Hz), 7.22 (1HAr, dd, J = 2.00, 7.50 Hz), 7.27 (1HAr, dd, J = 5.00, 1.00 Hz). 13C-NMR (125 MHz, CDCl3, ppm): δ 29.81 (CH2), 42.94 (OCH3), 55.56 (CH), 110.56 (CHAr), 113.23 (CHAr), 118.92 (2CHAr), 121.08 (CHAr), 125.87 (CHAr), 126.43 (CHAr), 126.69 (C), 127.36 (CHAr), 128.90 (2CHAr), 129.64 (CHAr), 137.01, 143.74 (2CAr), 144.68 (C), 156.04 (CAr). MS (EI) m/z: 334 (M+), 303, 227, 121, 91, 77, 51.
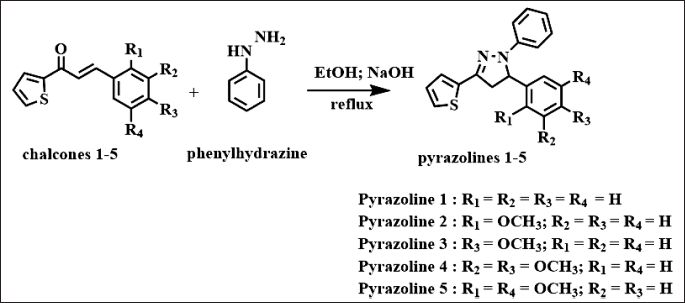 | Scheme 1. Synthesis of N-phenyl pyrazoline 1–5. [Click here to view] |
Synthesis of 5-(4-methoxyphenyl)-1-phenyl-3-thiophen-2-yl-4,5-dihydro-1H-pyrazole (pyrazoline 3)
Pyrazoline 3 was synthesized using the same method as described for pyrazoline 1, starting by mixing chalcone 3 (3-(4-methoxyphenyl)-1-thiophen-2-yl-2-propen-1-one) (0.24 g, 1 mmol), 1 mmol of phenylhydrazine, and 20% (w/v) of NaOH. A yellow solid of pyrazoline 3 was obtained with a yield of 60.37%, purity: 96.41%, mp: 132.5°C–134.1°C. FTIR (cm−1): 702 (C?S), 1,242 and 1,026 (C?O asymm. and sym.), 1,118 (C?N), 1,381 (Ar C?N), 1,504 (Ar C=C), 1,597 (C=N), 3,032 (Csp2?H). 1H-NMR (500 MHz, CDCl3, ppm): δ 3.10 (1H, CH2, dd, J = 7.00, 16.50 Hz), 3.77 (3H, s, OCH3), 3.80 (1H, CH2, dd, J = 12.50, 16.50 Hz), 5.21 (1CH, dd, J = 7.00, 12.50 Hz), 6.76 (1HAr, d, J = 7.00 Hz), 6.85 (2HAr, dd, J = 2.00, 7.00 Hz), 7.02 (4HAr, m), 7.16 (2HAr, m), 7.23 (2HAr, m), 7.29 (1HAr, m). 13C-NMR (125 MHz, CDCl3, ppm): δ 44.48 (CH2), 55.37 (OCH3), 64.24 (CH), 113.53 (2CHAr), 114.57 (2CHAr), 119.23 (CHAr), 125.93 (CHAr), 126.58 (CHAr), 127.16 (2CHAr), 127.40 (CHAr), 128.95 (2CHAr), 134.45, 136.81, 142.97 (3CAr), 144.77 (C), 159.08 (CAr). MS (EI) m/z: 334 (M+), 303, 227, 121, 91, 77, 39.
Synthesis of 5-(3,4-dimethoxyphenyl)-1-phenyl-3-thiophen-2-yl-4,5-dihydro-1H-pyrazole (pyrazoline 4)
Following the synthesis procedure of pyrazoline 1, pyrazoline 4 was produced by reacting chalcone 4 (3-(3,4-dimethoxyphenyl)-1-thiophen-2-yl-2-propen-1-one) (0.27 g, 1 mmol) with 1 mmol of phenylhydrazine, and 20% (w/v) of NaOH. An orange solid of pyrazoline 4 was obtained with a yield of 74.85%, purity: 100%, mp: 139.2°C–140.4°C. FTIR (cm−1): 702 (C?S), 1,257 and 1,026 (C?O asymm. and sym.), 1,134 (C?N), 1,381 (Ar C?N), 1,512 (Ar C=C), 1,597 (C=N), 3,032 (Csp2?H). 1H-NMR (500 MHz, CDCl3, ppm): δ 3.13 (1H, CH2, dd, J = 7.50, 16.50 Hz), 3.79 (1H, CH2, dd, J = 12.50, 16.50 Hz), 3.81 (3H, s, OCH3), 3.85 (3H, s, OCH3), 5.17 (1H, CH, dd, J = 7.50, 12.50 Hz), 6.78 (1HAr, d, J = 8.00 Hz), 6.81 (1HAr, d, J = 5.00), 6.82 (1HAr, d, J = 4.00 Hz), 6.88 (1HAr, d, J = 8.00 Hz), 7.01 (1HAr, d, J = 5.00 Hz), 7.04 (3HAr, m), 7.16 (2HAr, d, J = 8.00 Hz), 7.30 (1HAr, d, J = 5.00 Hz). 13C-NMR (125 MHz, CDCl3, ppm): δ 44.58 (CH2), 55.99, 56.04 (OCH3), 64.93 (CH), 108.61 (CHAr), 111.51 (CHAr), 113.67 (2CHAr), 118.14 (CHAr), 119.43 (CHAr), 126.04 (CHAr), 126.68 (CHAr), 127.44 (CHAr), 128.95 (2CHAr), 135.02, 136.71, 143.20 (3CAr), 145.08 (C), 148.46, 149.68 (2CAr). MS (EI) m/z: 364 (M+), 333, 227, 151, 121, 91, 77, 39.
Synthesis of 5-(2,5-dimethoxyphenyl)-1-phenyl-3-thiophen-2-yl-4,5-dihydro-1H-pyrazole (pyrazoline 5)
Pyrazoline 5 was obtained by synthesizing it using the method described for pyrazoline 1. The synthesis involved using a mixture of chalcone 5 (3-(2,5-dimethoxyphenyl)-1-thiophen-2-yl-2-propen-1-one) (0.27 g, 1 mmol), 1 mmol of phenylhydrazine, and 20% (w/v) of NaOH. The pyrazoline 5 resulted as a yellow solid, yield: 93.14%, purity: 98.96%, mp: 148.2°C–149.3°C. FTIR (cm−1): 702 (C?S), 1,219 and 1,046 (C?O asymm. and sym.), 1,119 (C?N), 1,381 (Ar C?N), 1,520 (Ar C=C), 1,597 (C=N), 3,102 (Csp2?H). 1H-NMR (500 MHz, CDCl3, ppm): δ 3.00 (1H, CH2, dd, J = 7.00, 17.00 Hz,), 3.61 (3H, s, OCH3), 3.85 (1H, CH2, dd, J = 12.00, 17.00 Hz), 3.89 (3H, s, OCH3), 5.54 (1H, dd, J = 7.00, 12.00 Hz, CH), 6.74 (2HAr, dd, J = 4.00, 1.00 Hz), 6.77 (1HAr, d, J = 7.50 Hz), 6.86 (1HAr, dd, J = 4.00, 5.00 Hz), 7.01 (4HAr, m), 7.17 (2HAr, dd, J = 7.50, 2.00 Hz), 7.28 (1HAr, d, J = 2.00 Hz). 13C-NMR (125 MHz, CDCl3, ppm): δ 43.02 (CH2), 55.76, 56.06 (OCH3), 58.98 (CH), 111.66 (CHAr), 112.65 (2CHAr), 112.97 (CHAr), 113.36 (CHAr), 119.08 (CHAr), 125.92 (CHAr), 126.47 (CHAr), 127.36 (CHAr), 128.96 (2CHAr), 131.04, 136.93, 143.74 (3CAr), 144.85 (C), 150.16, 154.06 (2CAr). MS (EI) m/z: 364 (M+), 333, 227, 200, 121, 91, 77, 39.
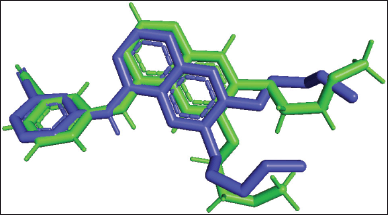 | Figure 1. Superimpose native ligand (green) into native ligand re-docking (blue). [Click here to view] |
Anticancer activity of pyrazolines 1–5
To culture cancer cells, Roswell Park Memorial Institute (RPMI) and Dubelccos’s Modified Eagle’s Medium (DMEM) media with 10% Fetal Bovine Serum (FBS) were used, and the cells were exposed to a water-saturated flow of 5% CO2. A cell-containing solution of 100 μl was added to each well of a 96-well plate, with each well containing 104 cells. Pyrazoline samples and commercial drugs (100 μl) were added to each 96-well plate, with seven concentrations tested and each sample tested four times. After 24 hours of incubation, a 1 ml aliquot of 3-(4,5-dimethyl thiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution in Phosphate Buffer Saline (PBS) (5 mg/ml) was prepared by dissolving it in 9.5 ml of culture medium. Subsequently, 100 μl of this mixture was added to each well and incubated for 4 hours. Then, 100 μl of sodium dodecyl sulfate (SDS) stopper solution was added, and the plate was incubated overnight. Finally, the absorbance of the solution was measured at 595 nm using an Enzyme-linked immunosorbent assay (ELISA) reader.
 | Figure 2. The 3-D and 2-D interaction of EGFR with (a) erlotinib and (b) pyrazoline 2. [Click here to view] |
Molecular docking study of pyrazolines 1–5
Pyrazoline derivatives were sketched in 2-D and then converted to 3-D structures using Chem 3-D Professional 16 software. The geometry optimization was then performed using the Density-functional theory (DFT) method basis set B3YLP 6-31G with Gaussian 09 software. All tested compounds were docked into EGFR with a PDB ID of 1m17 using a grid box size of 18 × 18 × 18 Å, suitable for validated protocol docking. The Root-mean-square deviation (RMSD) value was ensured to be lower than 2 Å before conducting the docking protocol. The binding energy and chemical interaction were analyzed to assess the compounds’ inhibitory activity compared to erlotinib, a native ligand.
ADME study of pyrazolines 1–5
Pyrazolines 1-5 underwent in silico analysis to predict their adsorption, distribution, metabolism, and excretion (ADME) using the pkCSM webtool (http://biosig.unimelb.edu.au/pkcsm/prediction). The SMILES notations generated during the ligand preparation process were uploaded to the server to evaluate ADME.
RESULTS AND DISCUSSION
Synthesis of N-phenyl pyrazolines
Pyrazolines 1-5 (Scheme 1) were synthesized from chalcones 1–5 and phenylhydrazine via a cyclocondensation reaction, utilizing the method described by Suma et al. [15]. The physical data for pyrazolines 1–5 are presented in Table 1. The characterization of the synthesized products was carried out using FTIR, Gas chromatography-mass spectrometry (GC-MS), and 1H- and 13C-NMR spectrometers. The formation of N-phenyl pyrazoline products was confirmed by the appearance of three peaks in the δ 3, 4, and 5 ppm region as a doublet of a doublet, which comes from the methylene proton on the pyrazoline rings due to the diastereotopic effect [16]. GC-MS was employed to verify the molecular weight and purity of the pyrazoline products.
The formation of N-phenyl pyrazoline was further confirmed by FTIR spectra showing the C=N stretching at 1,597 cm−1 and vibration of C–N aromatic and aliphatic stretching at 1,381 and 1,126 cm−1, respectively, and 13C-NMR spectra, which revealed the existence of carbon atoms within the pyrazoline ring.
In vitro anticancer activity
To evaluate the anticancer activity of pyrazolines 1–5, the MTT assay was used to perform in vitro cytotoxicity testing against cervical (HeLa), breast (T47D and 4T1), colorectal (WiDr), cancer cell lines, and a normal cell line (Vero). The percentage of cell growth inhibition was transformed into the probit numbers and plotted against log C to determine the IC50 value. Three commercial drugs, doxorubicin for breast cancer [17], cisplatin for cervical cancer [18], and 5-fluorouracil for colorectal cancer [19], were used as positive controls.
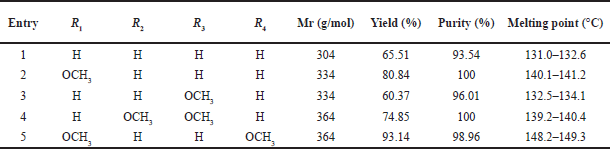 | Table 1. Physical data of N-phenyl pyrazoline derivatives. [Click here to view] |
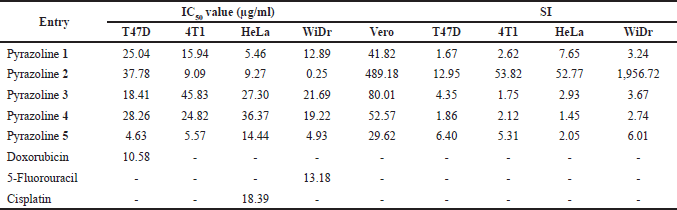 | Table 2. IC50 and SI values of N-phenyl pyrazoline derivatives. [Click here to view] |
The resulting IC50 and selectivity index (SI) values of pyrazolines 1–5 and commercial drugs are recorded in Table 2. The cytotoxic activity of a compound is classified into three categories, i.e., active (IC50 < 20 μg/ml), moderate (IC50 20–100 μg/ml), and inactive (IC50 > 100 μg/ml) [20].
Among the tested pyrazolines, pyrazoline 1 showed activity against 4T1, HeLa, and WiDr, with moderate activity against T47D. Pyrazoline 2 was also active against 4T1, HeLa, and WiDr, with moderate activity against T47D. Pyrazoline 3 exhibited activity against T47D and moderate activity against 4T1, HeLa, and WiDr. Pyrazoline 4 was active against WiDr, and moderate activity against T47D, 4T1, and HeLa. Among all the tested pyrazolines, pyrazoline 5 demonstrated the lowest IC50 values and exhibited activity against all cancer cell lines tested, including T47D, 4T1, HeLa, and WiDr. However, despite its broad-spectrum anticancer activity, pyrazoline 5 showed low selectivity against the normal (Vero) cell line (IC50 29.62 μg/ml).
This study considers the compound’s selectivity against the normal (Vero) cell line to identify the best candidate for an anticancer agent. According to Amin et al. [21], a compound with a SI value greater than 6 is considered highly selective. Compounds with SI values ranging from 3 to 6 are moderately selective, while those with SI values below 3 are considered to be non-selective. Based on Table 2, pyrazoline 2 is the most selective among the tested compounds against the normal (Vero) cell line (IC50 489.18 μg/ml) with a SI of 12.94, 53.81, 52.77, and 1956.72 against T47D, 4T1, HeLa, and WiDr, respectively. Additionally, pyrazoline 2 exhibits high activity in inhibiting the cervical (HeLa), breast (4T1), and colorectal (WiDr) cancer cell lines. According to the research, pyrazoline 2 has been identified as the most effective candidate for fighting cancer compared to the other candidates. The variation in their effectiveness can be attributed to the location of the methoxy group within the pyrazoline structure. Pyrazoline 2, which contains a methoxy substituent at position 2 on the benzene ring, demonstrated greater potency. The current findings agree with Ciupa et al.’s [5] report, which suggested that adding methoxy groups to pyrazoline derivatives could affect their ability to inhibit cancer cells.
Molecular docking
Table 3 presents the results of a molecular docking study conducted on synthesized pyrazoline 1–5 against EGFR. To ensure accurate results, it is essential to validate the docking protocol. The superimpose of erlotinib, a native ligand, into re-docking native ligand produced an RMSD of 1.304 Å (Fig. 1). In this study, erlotinib was used as a reference inhibitor for the synthetic compounds. The molecular docking of erlotinib showed a binding affinity of −7.9 kcal/mol. The chemical interaction between erlotinib and the amino acid residue Met769, which is involved in the catalytic reaction, was visualized (Fig. 2) [22]. Furthermore, erlotinib is bound to Cys773 through the oxygen atom of the ether moiety, contributing to the inhibition of EGFR growth [23].
Upon inspection of the chemical interaction, it was observed that the nitrogen atom of quinazoline acted as an acceptor hydrogen bond to Met769. Erlotinib is bound to the amino acid residue Lys721, which connects the α- and β-phosphate of the N-lobe to stabilize catalytic activity through hydrophobic interaction. Additionally, erlotinib also formed a pi-donor hydrogen bond with Thr830 and it has a role in the activation loop for the conformation of the phosphorylation process [24]. Furthermore, the hydrophobic II areas of EGFR, where the catalytic protein kinase is located [25], were also bound by erlotinib through hydrophobic interactions with amino acid residues such as Leu694 and Leu820. The quinazoline ring facilitated this interaction with these hydrophobic residues. The quinazoline ring facilitated this interaction with these hydrophobic residues.
The molecular docking study of pyrazoline derivatives containing a thiophene ring demonstrated good binding affinity. Compound 5 exhibited a slightly lower binding affinity (−7.6 kcal/mol) than erlotinib, while compound 4 was bound to EGFR with a similar affinity as the native ligand. Compounds 3, 1, and 2 showed strong binding affinity of −8.4, −8.8, and −8.8 kcal/mol, respectively. All compounds were found to bind within the binding pocket of EGFR, with the N-phenyl substituent of the pyrazoline ring binding to the hydrophobic area and some residue amino acids, such as Leu694 and Leu820. Compounds 1, 2, and 3 contained an aromatic ring B that bound to Lys721 through hydrophobic interaction. This interaction destabilized the side chain of the N-lobe, resulting in a decrease in catalytic activity.
Although compounds 4 and 5 exhibited the lowest binding affinity among synthetic compounds, they both interacted with Met769. Compound 5 bound to Met769 through hydrophobic interaction on methoxy moiety may explain its lower binding affinity than compound 4. Similarly, compound 4 interacted with Met769 through a carbon-hydrogen bond, which is also a catalytic site of EGFR. Furthermore, this compound is bound to Cys773 through a hydrogen bond with a bond length of 3.19 Å. The methoxy moiety of aromatic ring B also contributed to the interaction with Leu694. The thiophene ring of this compound was linked to Lys721, resulting in the destabilization of catalytic activity.
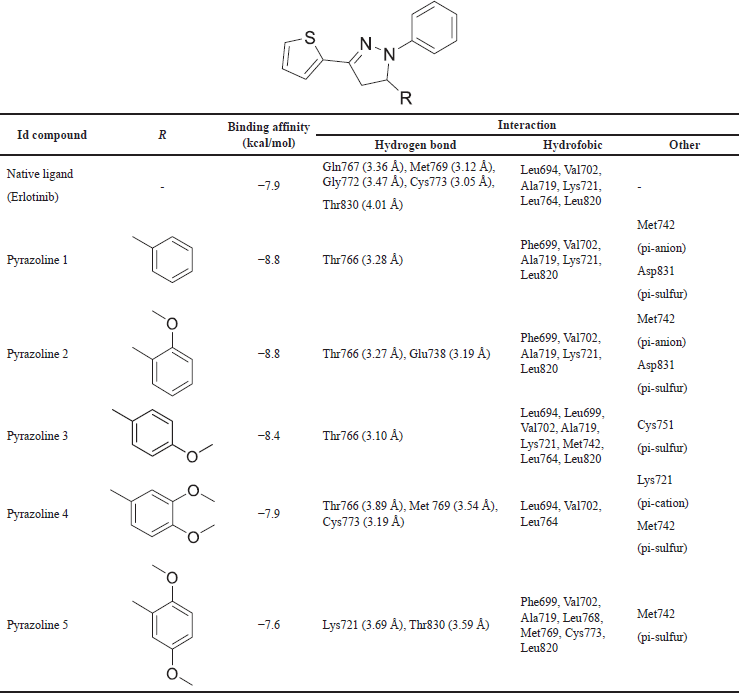 | Table 3. The binding affinity of chemical interaction by N-phenyl pyrazoline derivatives. [Click here to view] |
ADME study
The ADME properties of pyrazolines 1–5 were further evaluated using the pkCSM webtool for ADME prediction. Table 4 presents the ADME properties results of N-phenyl pyrazoline derivatives. The absorption of drugs orally relies on two crucial factors, human intestinal absorption (HIA) and CaCo-2 permeability. HIA predicts the number of compounds that the human small intestine can absorb, whereas CaCo-2 serves as an in vitro model that stimulates the human intestinal mucosa and predicts oral drug absorption. A compound with an HIA value greater than 80% is considered highly absorbed, while the permeability of CaCo-2 is high if the predicted value is above 0.90 [26]. The analysis showed that pyrazolines 1–5 have an HIA value above 90% and a CaCo-2 permeability value higher than 0.9, indicating that these compounds have an excellent absorption capacity.
The steady-state volume of distribution (VDss) represents the capacity to distribute a complete blood plasma dose. According to Lombardo et al. [27], VDss values below −0.15 are considered low, while values above 0.45 are considered good. Blood–brain barrier (BBB) permeability refers to a drug’s capability to traverse the barrier membrane of the brain, whereas central nervous system (CNS) permeability indicates a drug’s ability to penetrate the CNS. Pires et al. [12] stated that a BBB permeability value is considered good to penetrate if log BB > 0.3 and poor to penetrate if log BB < −1, while CNS permeability is high if log PS > −2 and low if log PS < −3. Pyrazolines 1–3 have VDss value above 0.45 and log BB > 0.3, which suggests that these compounds are uniformly distributed in blood plasma and can cross the brain barrier membrane. Conversely, pyrazolines 4 and 5 have VDss values below 0.45 and log BB < 0.3. Nonetheless, all pyrazoline derivatives have a log PS > −2, indicating that these compounds exhibit a robust capacity to infiltrate the CNS.
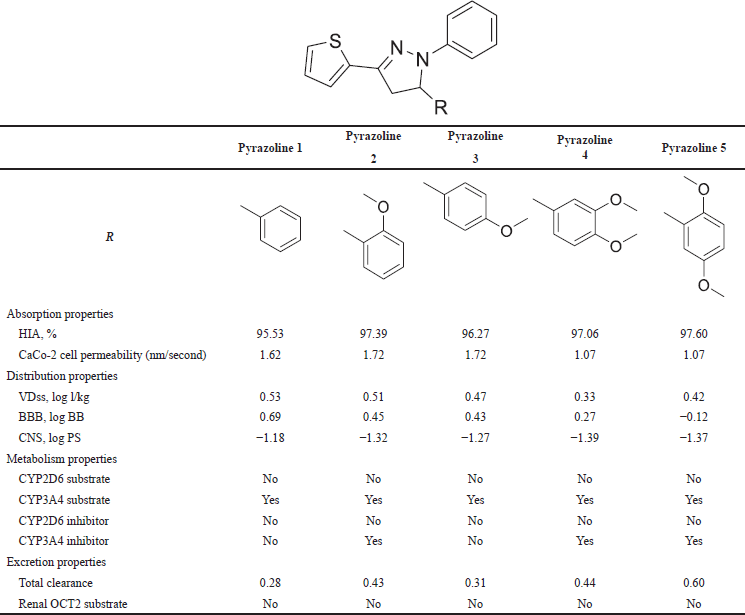 | Table 4. ADME property values of N-phenyl pyrazoline. [Click here to view] |
The liver is a common site for the key enzyme Cytochrome P450, which plays a crucial role in the body by oxidizing xenobiotics to render drug compounds inactive. Cytochromes have isoform models, such as CYP3A4 and CYP2D6, and these processes are essential for drug metabolism and xenobiotic activities [28]. Compounds that become substrates indicate that they can be metabolized by CYP450, while compounds that act as inhibitors can suppress the enzyme’s metabolic activity. Table 4 shows that all compounds did not act as inhibitors of CYP2D6, whereas pyrazolines 2, 4, and 5 acted as inhibitors of the CYP3A4 enzyme. It can be concluded that pyrazolines 2, 4, and 5 exhibit poor metabolism with Cytochrome P450.
Proximal tubule cells contain a basolateral surface where it is located; the predominant human OCT transporter is OCT2. Shen et al. [29] established that OCT2 is crucial in removing and eliminating cationic drugs and endogenous substances. Additionally, administering OCT2 inhibitors with OCT2 substrates may result in unfavorable interactions. The total clearance value represents the characteristics of drug excretion, with the lowest total clearance indicating that the drug is susceptible to elimination. The data presented in Table 4 shows that all pyrazoline derivatives were not OCT2 substrates and had high total clearance values.
CONCLUSION
The novel thiophene-based N-phenyl pyrazolines 1–5 were successfully synthesized by reacting chalcones and phenylhydrazine. As determined by the MTT assay, Pyrazoline 2 displayed remarkable effectiveness as an anticancer agent targeting colorectal (WiDr), breast (4T1), and cervical (HeLa) cancer cell lines. It exhibited a high level of selectivity against the normal cell line (Vero). The molecular study revealed that N-phenyl pyrazoline 2 exhibited the lowest binding energy, with a value of 8.8 kcal/mol. Chemical interaction studies showed that this compound was located within the binding pocket and interacted with the essential amino acid of EGFR. The ADME study using the pkCSM program demonstrated that the N-phenyl pyrazoline derivatives had favorable pharmacokinetic properties.
ACKNOWLEDGMENT
The Indonesian Endowment Fund for Education (LPDP) Scholarship provided by the Ministry of Finance of the Republic of Indonesia is gratefully acknowledged by the authors for supporting Istna Chunaifah. Additionally, this study was supported by research funding through the Rekognisi Tugas Akhir (RTA-UGM) Project 2022.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. Patel K, Karthikeyan C, Raja Solomon V, S. Hari Narayana Moorthy N, Lee H, Sahu K, et al. Synthesis of some coumarinyl chalcones and their antiproliferative activity against breast cancer cell lines. Lett Drug Des Discov. 2011;8(4):308–11. CrossRef
2. Murugavel S, Ravikumar C, Jaabil G, Alagusundaram P. Synthesis, computational quantum chemical study, in silico ADMET and molecular docking analysis, in vitro biological evaluation of a novel sulfur heterocyclic thiophene derivative containing 1,2,3-triazole and pyridine moieties as a potential human topoisomerase IIα inhibiting anticancer agent. Comput Biol Chem. 2019;79:73–82. CrossRef
3. Semenyuta I, Kovalishyn V, Tanchuk V, Pilyo S, Zyabrev V, Blagodatnyy V, et al. 1,3-Oxazole derivatives as potential anticancer agents: computer modeling and experimental study. Comput Biol Chem. 2016;65:8–15. CrossRef
4. Gulipalli KC, Bodige S, Ravula P, Endoori S, Vanaja GR, Suresh Babu G, et al. Design, synthesis, in silico and in vitro evaluation of thiophene derivatives: a potent tyrosine phosphatase 1B inhibitor and anticancer activity. Bioorg Med Chem Lett. 2017;27:3558–64. CrossRef
5. Ciupa A, De Bank PA, Mahon MF, Wood PJ, Caggiano L. Synthesis and antiproliferative activity of some 3-(pyrid-2-yl)-pyrazolines. Med Chem Comm. 2013;4:956–61. CrossRef
6. Xu W, Pan Y, Wang H, Li H, Peng Q, Wei D, et al. Synthesis and evaluation of new pyrazoline derivatives as potential anticancer agents in HepG-2 cell line. Molecules. 2017;22(3):467. CrossRef
7. Lu Z-H, Gu X-J, Shi K-Z, Li X, Chen D-D, Chen L. Accessing anti-human lung tumor cell line (A549) potential of newer 3,5-disubstituted pyrazoline analogs. Arab J Chem. 2017;10(5):624–30. CrossRef
8. Pacheco DJ, Prent L, Trilleras J, Quiroga J. Facile sonochemical synthesis of novel pyrazolyne derivates at ambient conditions. Ultrason Sonochem. 2013;20(4):1033–6. CrossRef
9. McConkey BJ, Sobolev V, Edelman M. The performance of current methods in ligand-protein docking. Curr Sci. 2002;83(7):845–56.
10. Sunayana G, Shashikant B, Sandeep W. 2D, 3D, G-QSAR and docking studies of thiazolyl-pyrazoline analogues as potent (epidermal growth factor receptor-tyrosine kinase) EGFR-TK inhibitors. Lett Drug Des Discov. 2017;14(11):1228–38. CrossRef
11. Nazmi TKT, Aminudin NI, Hamzah N. Molecular docking and ADME profiling of xanthorrhizol derivatives as hyaluronidase inhibitors. Malays J Chem. 2022;24(2):120–30. CrossRef
12. Pires DE V, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58(9):4066–72. CrossRef
13. Tjitda PJP, Jumina, Wahyuningsih TD. Synthesis, molecular docking, and ADMET study of N1-hydrogen and N1-benzoyl pyrazoline as antibacterial agents. Chiang Mai Univ J Nat Sci. 2022;21(3):1–15. CrossRef
14. Yang W, Hu Y, Yang Y-S, Zhang F, Zhang Y-B, Wang X-L, et al. Design, modification and 3D QSAR studies of novel naphthalin-containing pyrazoline derivatives with/without thiourea skeleton as anticancer agents. Bioorg Med Chem. 2013;21(5):1050–63. CrossRef
15. Suma AAT, Wahyuningsih TD, Mustofa. Synthesis, cytotoxicity evaluation and molecular docking study of N-phenylpyrazoline derivatives. Indones J Chem. 2019;19(4):1081–90. CrossRef
16. Alt?ntop MD, Özdemir A, Turan-Zitouni G, Ilg?n S, Atl? Ö, Demirel R, et al. A novel series of thiazolyl–pyrazoline derivatives: synthesis and evaluation of antifungal activity, cytotoxicity and genotoxicity. Eur J Med Chem. 2015;92:342–52. CrossRef
17. Aghaee F, Islamian JP, Baradaran B, Mesbahi A, Mohammadzadeh M, Jafarabadi MA. Enhancing the effects of low dose doxorubicin treatment by the radiation in T47D and SKBR3 breast cancer cells. J Breast Cancer. 2013;16(2):164–70. CrossRef
18. Ismail I, Suppian R, Mohamad H, Radzi SAM, Abdullah H. In vitro cytotoxicity and apoptosis-inducing activity of Quercus infectoria extracts in HeLa cells. Pharmacogn J. 2021;13(2):401–10. CrossRef
19. Gilang Y, Hermawan A, Fitriasari A, Jenie RI. Hesperidin increases cytotoxic effect of 5-fluorouracil on WiDr cells. Indones J Cancer Chemoprevention. 2012;3(2):404–9. CrossRef
20. Tanamatayarat P, Limtrakul P, Chunsakaow S, Duangrat C. Screening of some Rubiaceous plants for cytotoxic activity against cervix carcinoma (KB-3-1) cell line J Pharm Sci. 2003;27(3–4):167–72.
21. Amin KM, Eissa AAM, Abou-Seri SM, Awadallah FM, Hassan GS. Synthesis and biological evaluation of novel coumarin-pyrazoline hybrids endowed with phenylsulfonyl moiety as antitumor agents. Eur J Med Chem. 2013;60:187–98. CrossRef
22. Eldeeb M, Sanad EF, Ragab A, Ammar YA, Mahmoud K, Ali MM, et al. Anticancer effects with molecular docking confirmation of newly synthesized isatin sulfonamide molecular hybrid derivatives against hepatic cancer cell lines. Biomedicines. 2022;10(3):1–21. CrossRef
23. Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702–11. CrossRef
24. Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277(48):46265–72. CrossRef
25. Singh P, Bast F. In silico molecular docking study of natural compounds on wild and mutated epidermal growth factor receptor. Med Chem Res. 2014;23(12):5074–85. CrossRef
26. Petrescu AM, Paunescu V, Ilia G. The antiviral activity and cytotoxicity of 15 natural phenolic compounds with previously demonstrated antifungal activity. J Environ Sci Heal Part B. 2019;54(6):498–504. CrossRef
27. Lombardo F, Bentzien J, Berellini G, Muegge I. In silico models of human PK parameters. prediction of volume of distribution using an extensive data set and a reduced number of parameters. J Pharm Sci. 2021;110(1):500–9. CrossRef
28. Basheer L, Kerem Z. Interactions between CYP3A4 and dietary polyphenols. Oxid Med Cell Longev. 2015;2015:1–15. CrossRef
29. Shen Q, Wang J, Yuan Z, Jiang Z, Shu T, Xu D, et al. Key role of organic cation transporter 2 for the nephrotoxicity effect of triptolide in rheumatoid arthritis. Int Immunopharmacol. 2019;77:105959. CrossRef