
INTRODUCTION
Terminalia myriocarpa (East Indian Almond) is a tall evergreen tree growing up to 40 m, native to eastern Asia, southern China, northeast India, Nepal, Bhutan, Myanmar, Thailand, Malaysia, Indonesia, Laos, and Vietnam [1]. Several compounds were previously reported from the leaves of T. myriocarpa, including cinnamic acid, trans-ferulic acid, syringic acid, gallic acid, methyl gallate, ethyl gallate, 2,3-(S)-HHDP-D-glucose, ellagic acid, flavogallonic acid, methyl-(S)-flavogallonate, (α/β)-punicalagin, epigallocatechin gallate, vitexin, isovitexin, orientin, iso-orientin, kaempferol-3-O-β-D-rutinoside, rutin, neosaponarin, quercetin, and myricetin [2–4]. Moreover, β-sitosterol, β-amyrin, oleanolic acid, betulinic acid, maslinic acid, and arjunolic acid were separated from the bark of T. myriocarpa [5]. From the standpoint of bioactivity, ellagic acid and methyl-(S)-flavogallonate, isolated from T. myriocarpa leaves, were found to possess antioxidant and anti-inflammatory activities [3].
Diabetes mellitus is a widespread metabolic disorder, which affects about 422 million people globally [6]. The most prevalent form of diabetes, type 2 (noninsulin-dependent diabetes mellitus, or T2DM), is characterized by a relative insulin shortage caused by the concomitant presence of insufficient insulin production, tissue insulin resistance, and insufficient compensatory mechanisms [7]. Ineffective glycemic control in T2DM patients can lead to serious retinal, renal, and cardiovascular problems, as well as a sharp decline in life expectancy [8]. Limiting postprandial hyperglycemia by inhibiting polysaccharide-digesting enzymes in the proximal small intestine, including α-glucosidase, and thus reducing glucose absorption from the gut, is a treatment approach for T2DM [9]. The development of safer α-glucosidase inhibitors from natural sources for the treatment of T2DM has received a lot of attention due to the unpleasant side effects associated with the currently available α-glucosidase inhibitors on the market [6]. Some Terminalia species were reported to contain several α-glucosidase inhibitory constituents, including 23-O-galloylarjunolic acid, 23-O-galloylarjunolic acid-28-O-β-D-glucopyranosyl ester, 1,2,3,6-tetra-O-galloyl-4-O-cinnamoyl-β-D-glucose, and 4-O-(2’’,4’’-di-O-galloyl-α-L-rhamnosyl) ellagic acid isolated from Terminalia chebula fruits, alongside rutin, narcissin, chebulagic acid, and corilagin isolated from Terminalia macroptera leaves [10–12]. To the best of our knowledge, however, there have not been any additional discoveries about the α-glucosidase inhibitory capacity of T. myriocarpa leaf extract.
As a part of our continuous interest in exploring bioactive phytoconstituents, we investigated T. myriocarpa leaf extract aiming at identifying its α-glucosidase inhibitory constituents.
MATERIALS AND METHODS
Plant material
The leaves of T. myriocarpa Van Heurck & Müll. Arg. were collected from the Zoo Garden, Giza, Egypt, in March 2020, and identified by Ms. Therese Labib, the taxonomical consultant at Al-Orman and Al-Qubba Botanical Gardens. A voucher specimen, with the identifier M165, was deposited in the herbarium of the National Research Centre, Giza, Egypt.
General experimental procedures
Column chromatography (CC) was performed using silica gel 60 (E-Merck, Darmstadt, Germany), Sephadex LH-20 (Pharmacia Fine Chemicals AB Uppsala, Sweden), and Diaion HP-20 (Sigma-Aldrich, St. Louis, MO). Preparative thin-layer chromatography (PTLC) and analytical thin-layer chromatography (TLC) were carried out using silica gel and polyamide (E-Merck, Darmstadt, Germany). Chromatograms were first observed under ultraviolet (UV) light, before being treated with ferric chloride reagent, or sulfuric acid reagent (20% in methanol). UV spectra were recorded in methanol on a Jasco V-730 UV-visible spectrophotometer (Tokyo, Japan). Nuclear magnetic resonance (NMR) spectra were obtained in deuterated methanol (CD3OD) or deuterated chloroform (CDCl3) using a Bruker High-Performance Digital FT-NMR-Spectrophotometer Avance III HD (1H-NMR: 400 MHz, 13C-NMR: 100 MHz, Bremen, Germany). Tetramethylsilane was used as an internal standard.
Extraction and isolation of the leaf constituents
Fresh T. myriocarpa leaves (10 kg) were air-dried and powdered to yield 2.2 kg of leaf powder (22%). The obtained dry powder was thoroughly extracted thrice, at room temperature, by maceration with 100% methanol (MeOH), followed by 70% aqueous MeOH twice. A portion (200 g) of the brownish–green residue (258 g), resulting from solvent evaporation of the combined leaf extract, was suspended in distilled water (2 l) and then partitioned with dichloromethane (CH2Cl2) (2l × 5) followed by ethyl acetate (EtOAc) (2l × 5). The solvent in each fraction was separately evaporated under vacuum to dryness.
An aliquot (20 g) of the dried CH2Cl2 fraction (24 g) was subjected to silica gel CC (800 g). The gradient elution system consisted of a mixture of n-hexane and EtOAc (93:7, 90:10, 85:15, 80:20, 75:25, 50:50, 20:80, and 0:100, v/v), yielding fractions D1–D8, respectively. Fraction D5 (345 mg) was subjected to Sephadex LH-20 CC (50 g) and eluted with CH2Cl2–MeOH (3:2, v/v) to afford subfractions D5.1–D5.4. Subfraction D5.3 (86 mg) was chromatographed using silica gel PTLC plates [chloroform (CHCl3)–MeOH, 15:1, v/v, triple development] to give crude alphitolic acid (1) (42 mg) which was further purified on a Sephadex LH-20 column (30 g) eluted with MeOH to yield pure 1 (37 mg).
A portion (25 g) of the EtOAc fraction (36 g) was chromatographed using Diaion HP-20 CC (200 g) and eluted with a mixture of MeOH—distilled water (H2O) (0:100, 25:75, 40:60, 50:50, 60:40, 70:30, 80:20, and 100:0, v/v) to give fractions E1–E8, respectively. Fraction E3 (2.5 g) was subjected to a Sephadex LH-20 column (50 g) eluted with MeOH to give subfractions E3.1–E3.5. Using polyamide PTLC plates developed with MeOH–H2O (15:1, v/v), subfraction E3.2 (84 mg) afforded crude isovitexin (2) (25 mg) which was further purified on a Sephadex LH-20 column (30 g) eluted with MeOH to provide pure 2 (20 mg). Subfraction E3.5 (104 mg) was loaded on a Sephadex LH-20 column (50 g) eluted with MeOH to afford flavogallonic acid (3) (77 mg). Fraction E4 (1.7 g) was subjected to Sephadex LH-20 CC (50 g) eluted with MeOH to give subfractions E4.1–E4.6. Separation of subfraction E4.2 (301 mg) using repeated silica gel PTLC with CHCl3–MeOH (2:1, v/v) yielded crude nigaichigoside F1 (4) (74 mg) which was further purified using a Sephadex LH-20 column (30 g) eluted with MeOH to afford pure 4 (65 mg). Subfraction E5 (2.6 g) was subjected to a Sephadex LH-20 column (50 g) and eluted with MeOH to give subfractions E5.1–E5.5. Subfraction E5.4 (144 mg) was chromatographed on a silica gel column (50 g) using a gradient mixture of CH2Cl2–MeOH (10:1 and 7:1, v/v) to yield crude quercetin (5) (24 mg) which was further purified by Sephadex LH-20 CC (30 g) using MeOH as eluent to afford pure 5 (19 mg). Fraction E6 (2.3 g) was loaded on a Sephadex LH-20 column (100 g) and eluted with MeOH to afford subfractions E6.1–E6.3. Subfraction E6.2 (270 mg) was subjected to repeated silica gel PTLC using CHCl3–MeOH (5:1, v/v, double development) to give crude quadranoside IV (6) (73 mg), rosamultin (7) (78 mg), and 19α-hydroxyasiatic acid (8) (27 mg). Each compound was separately loaded on a Sephadex LH-20 column (30 g) eluted with MeOH to afford pure 6 (65 mg), 7 (68 mg), and 8 (22 mg). Fraction E7 (370 mg) was subjected to CC on a Sephadex LH-20 column (50 g) eluted with MeOH to give subfractions E7.1–E7.4. Repeated chromatography of subfraction E7.2 (197 mg) using silica gel PTLC plates developed with CHCl3–MeOH (5:1, v/v) yielded crude asiatic acid (9) (56 mg) and arjunic acid (10) (44 mg) which were individually purified using Sephadex LH-20 CC (30 g) eluted with MeOH to give pure 9 (48 mg) and 10 (39 mg).
Identification of the isolated compounds
The structures of compounds 1–10 (Fig. 1) were established, based on their spectral data, as follows.
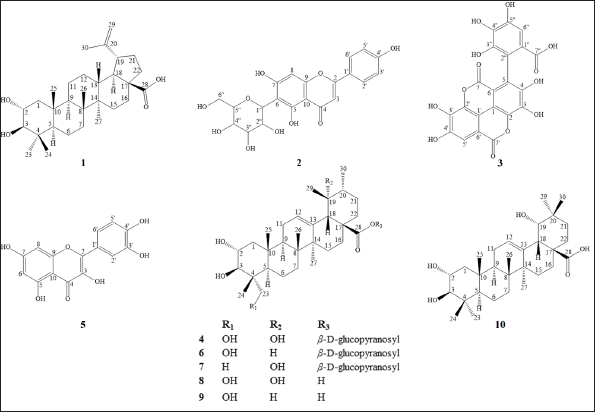 | Figure 1. Structures of compounds isolated from T. myriocarpa leaves. [Click here to view] |
Alphitolic acid (1)
1H -NMR (CD3OD, ppm); 4.69 (1H, d, J = 2.6 Hz, H-29a), 4.54 (1H, br s, H-29b), 3.60 (1H, m, H-2), 2.90 (1H, d, J = 9.6 Hz, H-3), 1.68 (3H, s, Me-30), 1.01 (3H, s, Me-27), 0.99 (3H, s, Me-23), 0.98 (3H, s, Me-26), 0.91 (3H, s, Me-25), and 0.78 (3H, s, Me-24). 13C-NMR (APT) (CD3OD, ppm); 153.4 (C, C-20), 109.5 (CH2, C-29), 84.6 (CH, C-3), 69.9 (CH, C-2), 59.1 (C, C-17), 57.0 (CH, C-5), 52.3 (CH, C-9), 51.1 (CH, C-18), 43.8 (C, C-14), 42.2 (C, C-8), 40.7 (C, C-4), 39.6 (C, C-10), 39.5 (CH, C-13), 39.3 (CH2, C-22), 35.7 (CH2, C-7), 33.2 (CH2, C-16), 32.4 (CH2, C-21), 30.9 (CH2, C-15), 29.3 (CH3, C-23), 27.2 (CH2, C-12), 22.5 (CH2, C-11), 19.9 (CH3, C-30), 19.7 (CH2, C-6), 18.1 (CH3, C-25), 17.4 (CH3, C-24), 17.1 (CH3, C-26), and 15.2 (CH3, C-27).
Isovitexin (2)
UV spectral data (CH3OH, nm); 272, 335; 1H-NMR (CD3OD, ppm); 7.85 (2H, d, J = 8.8 Hz, H-2’,6’), 6.93 (2H, d, J = 8.8 Hz, H-3’,5’), 6.61 (1H, s, H-3), 6.52 (1H, s, H-8), 4.90 (1H, d, J = 10.0 Hz, H-1’’), 4.16 (1H, t, J = 9.1 Hz, H-2’’), 3.88 (1H, dd, J = 12.1, 2.3 Hz, H-6’’a), 3.74 (1H, dd, J = 12.1, 5.2 Hz, H-6’’b), 3.49–3.48 (2H, m, H-3’’,4’’), and 3.47 (1H, m, H-5’’). 13C-NMR (CD3OD, ppm); 183.9 (C-4), 166.3 (C-2), 166.1 (C-7), 163.0 (C-4’), 162.1 (C-5), 158.9 (C-9), 129.5 (C-2’,6’), 123.1 (C-1’), 117.2 (C-3’,5’), 109.5 (C-6), 104.9 (C-10), 103.8 (C-3), 95.7 (C-8), 82.7 (C-5’’), 80.4 (C-3’’), 75.5 (C-1’’), 72.6 (C-2’’), 71.9 (C-4’’), and 62.9 (C-6’’).
Flavogallonic acid (3)
UV spectral data (CH3OH, nm); 213, 257, 269sh., 354; 1H-NMR (CD3OD, ppm); 7.52 (1H, s, H-5’), 7.24 (1H, s, H-6’’).13C-NMR (CD3OD, ppm); 169.2 (C-7’’), 161.9 (C-7’), 160.5 (C-7), 149.4 (C-4’), 147.6 (C-4), 145.7 (C-5’’), 144.7 (C-3’’), 141.9 (C-3’), 141.0 (C-3), 139.4 (C-4’’), 137.9 (C-2’), 137.2 (C-2), 125.9 (C-5), 121.1 (C-1’’), 119.3 (C-2’’), 114.8 (C-1’), 114.4 (C-1), 111.5 (C-6’’), 111.4 (C-5’), 109.4 (C-6’), and 108.6 (C-6).
Nigaichigoside F1 (4)
1H-NMR (CD3OD, ppm); 5.33 (1H, d, J = 8.1 Hz, H-1’), 5.32 (1H, br s, H-12), 3.82 (1H, m, H-6’a), 3.68 (1H, m, H-2), 3.56 (1H, m, H-6’b), 3.51 (1H, d, J = 11.2 Hz, H-23a), 3.40 (1H, d, J = 9.6 Hz, H-3), 3.37–3.33 (4H, m, H-2’,3’,4’,5’), 3.27 (1H, d, J = 10.8 Hz, H-23b), 2.52 (1H, br s, H-18), 1.34 (3H, s, CH3-27), 1.21 (3H, s, CH3-29), 1.04 (3H, s, CH3-25), 0.93 (3H, d, J = 6.6 Hz, CH3-30), 0.78 (3H, s, CH3-24), and 0.70 (3H, s, CH3-26). 13C-NMR (APT) (CD3OD, ppm); 178.6 (C, C-28), 139.8 (C, C-13), 129.6 (CH, C-12), 95.9 (CH, C-1’), 78.6 (CH, C-3), 78.4 (CH, C-5’), 78.4 (CH, C-3’), 73.9 (CH, C-2’), 73.6 (C, C-19), 71.2 (CH, C-4’), 69.8 (CH, C-2), 66.6 (CH2, C-23), 62.4 (CH2, C-6’), 55.0 (CH, C-18), 49.6 (C, C-17), 49.2 (CH, C-9), 48.6 (CH, C-5), 47.2 (CH2, C-1), 44.2 (C, C-8), 43.0 (CH, C-20), 42.9 (C, C-14), 41.4 (C, C-4), 39.1 (C, C-10), 38.4 (CH2, C-22), 33.6 (CH2, C-7), 29.7 (CH2, C-15), 27.3 (CH2, C-21), 27.2 (CH3, C-29), 26.6 (CH2, C-16), 24.9 (CH2, C-11), 24.9 (CH3, C-27), 19.4 (CH2, C-6), 17.8 (CH3, C-25), 17.8 (CH3, C-26), 16.8 (CH3, C-30), and 14.0 (CH3, C-24).
Quercetin (5)
UV spectral data (nm); 256, 272sh., 372 (CH3OH), 1H-NMR (CD3OD, ppm); 7.73 (1H, d, J = 2.1 Hz, H-2’), 7.62 (1H, dd, J = 8.5, 2.1 Hz, H-6’), 6.88 (1H, d, J = 8.5 Hz, H-5’), 6.37 (1H, d, J = 2.0 Hz, H-8), and 6.17 (1H, d, J = 1.9 Hz, H-6). 13C-NMR (APT) (CD3OD, ppm); 177.4 (C, C-4), 165.7 (C, C-7), 162.6 (C, C-5), 158.3 (C, C-9), 148.9 (C, C-4’), 148.1 (C, C-2), 146.3 (C, C-3’), 137.3 (C, C-3), 124.3 (C, C-1’), 121.8 (CH, C-6’), 116.4 (CH, C-5’), 116.1 (CH, C-2’), 104.6 (C, C-10), 99.4 (CH, C-6), and 94.6 (CH, C-8).
Quadranoside IV (6)
1H-NMR (CD3OD, ppm); 5.35 (1H, d, J = 8.0 Hz, H-1’), 5.26 (1H, br t, J = 3.7 Hz, H-12), 3.69 (1H, m, H-2), 3.68 (1H, dd, J = 11.2, 2.0 Hz, H-6’a), 3.59 (1H, dd, J = 11.2, 4.8 Hz, H-6’b), 3.51 (1H, d, J = 11.3 Hz, H-23a), 3.39–3.33 (5H, m, H-3,2’,3’,4’,5’), 3.27 (1H, d, J = 11.0 Hz, H-23b), 2.24 (1H, d, J = 11.3 Hz, H-18), 1.13 (3H, s, CH3-27), 1.05 (3H, s, CH3-25), 0.97 (3H, br s, CH3-30), 0.90 (3H, d, J = 6.4 Hz, CH3-29), 0.84 (3H, s, CH3-26), and 0.70 (3H, s, CH3-24). 13C-NMR (APT) (CD3OD, ppm); 178.0 (C, C-28), 139.4 (C, C-13), 127.1 (CH, C-12), 95.8 (CH, C-1’), 78.8 (CH, C-3), 78.4 (CH, C-5’), 78.3 (CH, C-3’), 74.0 (CH, C-2’), 71.3 (CH, C-4’), 69.8 (CH, C-2), 66.6 (CH2, C-23), 62.6 (CH2, C-6’), 54.3 (CH, C-18), 49.5 (C, C-17), 49.1 (CH, C-9), 48.4 (CH, C-5), 48.1 (CH2, C-1), 44.2 (C, C-4), 43.5 (C, C-14), 41.1 (C, C-8), 40.5 (CH, C-20), 40.4 (CH, C-19), 39.1 (C, C-10), 37.6 (CH2, C-22), 33.8 (CH2, C-7), 31.7 (CH2, C-21), 29.3 (CH2, C-15), 25.4 (CH2, C-16), 24.6 (CH2, C-11), 24.2 (CH3, C-27), 21.7 (CH3, C-30), 19.2 (CH2, C-6), 18.1 (CH3, C-26), 17.9 (CH3, C-25), 17.8 (CH3, C-29), and 14.1 (CH3, C-24).
Rosamultin (7)
1H-NMR (CD3OD, ppm); 5.33 (1H, d, J = 8.0 Hz, H-1’), 5.32 (1H, br s, H-12), 3.80 (1H, dd, J = 12.1, 2.0 Hz, H-6’a), 3.68 (1H, dd, J = 11.9, 4.2 Hz, H-6’b), 3.62 (1H, m, H-2), 3.42 - 3.33 (4H, H-2’,3’,4’,’5’), 2.92 (1H, d, J = 9.5 Hz, H-3), 2.52 (1H, s, H-18), 1.33 (3H, s, CH3-27), 1.20 (3H, s, CH3-29), 1.01 (3H, s, CH3-25), 1.00 (3H, s, CH3-23), 0.93 (3H, d, J = 6.4 Hz, CH3-30), 0.81 (3H, s, CH3-24), and 0.78 (3H, s, CH3-26). 13C-NMR (APT) (CD3OD, ppm); 178.7 (C, C-28), 139.8 (C, C-13), 129.6 (CH, C-12), 95.9 (CH, C-1’), 84.6 (CH, C-3), 78.6 (CH, C-5’), 78.4 (CH, C-3’), 74.1 (CH, C-2’), 73.9 (C, C-19), 71.2 (CH, C-4’), 69.6 (CH, C-2), 62.6 (CH2, C-6’), 56.8 (CH, C-5), 55.0 (CH, C-18), 49.6 (C, C-17), 48.7 (CH2, C-1), 48.7 (CH, C-9), 43.0 (CH, C-20), 42.8 (C, C-14), 40.6 (C, C-8), 39.3 (C, C-10), 39.3 (C, C-4), 38.4 (CH2, C-22), 34.2 (CH2, C-7), 29.7 (CH2, C-15), 29.5 (CH3, C-23), 27.3 (CH2, C-21), 27.2 (CH3, C-29), 26.6 (CH2, C-16), 24.9 (CH2, C-11), 24.8 (CH3, C-27), 19.8 (CH2, C-6), 17.8 (CH3, C-25), 17.6 (CH3, C-24), 17.3 (CH3, C-26), and 16.8 (CH3, C-30).
19α-hydroxyasiatic acid (8)
1H-NMR (CD3OD, ppm); 5.28 (1H, t, J = 3.6 Hz, H-12), 3.68 (1H, m, H-2), 3.51 (1H, d, J = 11.2 Hz, H-23a), 3.37 (1H, d, J = 9.6 Hz, H-3), 3.28 (1H, d, J = 11.2 Hz, H-23b), 2.61 (1H, br s, H-18), 1.32 (3H, s, CH3-27), 1.20 (3H, s, CH3-29), 1.03 (3H, s, CH3-25), 0.92 (3H, d, J = 6.4 Hz, CH3-30), 0.86 (3H, s, CH3-26), 0.71 (3H, s, CH3-24).13C-NMR (APT) (CD3OD, ppm); 180.6 (C, C-28), 141.2 (C, C-13), 128.7 (CH, C-12), 78.7 (CH, C-3), 74.2 (C, C-19), 69.8 (CH, C-2), 66.9 (CH2, C-23), 55.8 (CH, C-18), 50.1 (C, C-17), 48.8 (CH, C-9), 48.6 (CH, C-5), 47.7 (CH2, C-1), 44.2 (C, C-8), 43.2 (CH, C-20), 42.9 (C, C-14), 41.2 (C, C-4), 39.4 (CH2, C-22), 39.2 (C, C-10), 33.6 (CH2, C-7), 29.2 (CH2, C-15), 27.9 (CH2, C-21), 27.5 (CH3, C-29), 27.2 (CH2, C-16), 24.9 (CH2, C-11), 24.4 (CH3, C-27), 19.5 (CH2, C-6), 17.7 (CH3, C-26), 17.5 (CH3, C-25), 16.9 (CH3, C-30), and 14.0 (CH3, C-24).
Asiatic acid (9)
1H-NMR (CD3OD, ppm); 5.21 (1H, br s, H-12), 3.69 (1H, m, H-2), 3.51 (1H, d, J = 11.0 Hz, H-23a), 3.36 (1H, d, J = 9.9 Hz, H-3), 3.27 (1H, d, J = 11.0 Hz, H-23b), 2.26 (1H, d, J = 11.2 Hz, H-18), 1.15 (3H, s, CH3-27), 1.11 (3H, s, CH3-25), 1.04 (3H, br s, CH3-30), 0.89 (3H, d, J = 9.7 Hz, CH3-29), 0.86 (3H, s, CH3-26), and 0.70 (3H, s, CH3-24). 13C-NMR (APT) (CD3OD, ppm); 180.7 (C, C-28), 141.0 (C, C-13), 125.7 (CH, C-12), 78.5 (CH, C-3), 69.8 (CH, C-2), 66.7 (CH2, C-23), 55.2 (CH, C-18), 50.2 (C, C-17), 49.2 (CH, C-9), 48.5 (CH, C-5), 48.2 (CH2, C-1), 44.2 (C, C-4), 43.6 (C, C-14), 41.1(CH, C-19), 40.9 (C, C-8), 40.7 (CH, C-20), 39.2 (C, C-10), 38.8 (CH2, C-22), 34.0 (CH2, C-7), 31.9 (CH2, C-21), 29.7 (CH2, C-15), 24.6 (CH2, C-16), 24.6 (CH2, C-11), 24.4 (CH3, C-27), 22.0 (CH3, C-30), 19.3 (CH2, C-6), 18.0 (CH3, C-26), 17.9 (CH3, C-29), 17.7 (CH3, C-25), and 14.1 (CH3, C-24).
Arjunic acid (10)
1H-NMR (CD3OD, ppm); 5.31 (1H, t, J = 3.8 Hz, H-12), 3.63 (1H, m, H-2), 3.26 (1H, d, J = 3.9 Hz, H-19), 3.16 (1H, br s, H-18), 2.92 (1H, d, J = 9.6 Hz, H-3), 1.28 (3H, s, CH3-27), 1.02 (3H, s, CH3-23), 0.99 (3H, s, CH3-25), 0.98 (3H, s, CH3-30), 0.93 (3H, s, CH3-29), 0.82 (3H, s, CH3-24), and 0.81 (3H, s, CH3-26). 13C-NMR (APT) (CD3OD, ppm); 180.6 (C, C-28), 145.8 (C, C-13), 124.2 (CH, C-12), 84.7 (CH, C-3), 83.3 (CH, C-19), 69.6 (CH, C-2), 57.0 (CH, C-5), 49.5 (CH, C-9), 48.2 (CH2, C-1), 47.6 (C, C-17), 46.0 (CH, C-18), 42.8 (C, C-14), 40.9 (C, C-8), 40.7 (C, C-4), 39.6 (C, C-10), 36.2 (C, C-20), 34.4 (CH2, C-22), 34.1 (CH2, C-7), 30.1 (CH2, C-15), 29.9 (CH2, C-21), 29.4 (CH3, C-23), 29.2 (CH2, C-16), 29.1 (CH3, C-29), 25.6 (CH3, C-30), 25.3 (CH3, C-27), 25.1 (CH2, C-11), 19.9 (CH2, C-6), 18.3 (CH3, C-26), 17.6 (CH3, C-24), and 17.1 (CH3, C-25).
General method of acid hydrolysis
Each isolated triterpene glycoside (10 mg) dissolved in a 10 ml 2 N hydrochloric acid-methanol mixture (1:1, v/v) was heated under reflux for 2 hours. The reaction mixture was left to cool and then vacuum-evaporated to dryness. The residue suspended in 5 ml of distilled water was extracted with EtOAc (5 ml × 3). The residual acidity of the aqueous layer was eliminated by repeated addition of methanol and evaporation. TLC analysis (Isopropanol-H2O, 7:1, v/v) of the residues against authentic material revealed the presence of D-glucose in each of the triterpene glycosides 4, 6, and 7 [13].
Evaluation of the α-glucosidase inhibitory activity
In phosphate buffer saline (pH 6.8), α-glucosidase from Saccharomyces cerevisiae (SIGMA G5003-100UN) was prepared at a concentration of 0.2 U/ml. α-Glucosidase (60 µl, 0.2 U/ml) was combined with each sample (10 µl) at different concentrations (0.3–700 ppm in the final volume). The mixture was then incubated for 20 minutes at 37°C in a 96-well plate. Subsequently, p-nitrophenyl-D-glucopyranoside (p-NPG) (SIGMA N1377) (150 µl, 1.25 mM) was added to each mixture and incubated for 20 minutes at 37°C, then 50 µl of 2 g/l sodium hydroxide (NaOH) was added to terminate the reaction. The amount of bright yellow p-nitrophenol released from the colorless p-NPG was measured spectrophotometrically at 405 nm to evaluate the activity of the α-glucosidase enzyme. Acarbose was utilized as a positive control, and a reaction mixture with 10 μl of buffer solution in place of the test entity was utilized as a negative control. For blank, p-nitrophenyl-α-D-glucopyranoside with buffer solution was added instead of the enzyme [14,15].
Docking study
The chemical structure of the screened compound was sketched using ChemBioDraw Ultra 14.0 software (CambridgeSoft corporation), and then energy was minimized by MMFF94x force field in the gas phase to a gradient of 0.01 kcal/mol.Å and saved in PDBQT format (A modification of the protein data bank format especially developed to hold the information needed by the protein-ligand docking software AutoDock, including the assigned charges). Co-crystal structures for human N-terminal maltase-glucoamylase ntMGAM (PDB:2QMJ), human C-terminal maltase-glucoamylase ctMGAM (PDB:3TOP), and human N-terminal sucrase-isomaltase ntSI (PDB:3LPP) were downloaded from the protein data bank (https://www.rcsb.org). All target receptors were prepared using MGL tools v1.5.7 to perform the deletion of water molecules and other hetatoms, the addition of polar hydrogens, and the addition of Kollman Charges, then saved in PDBQT format. Grid boxes were centered at the co-crystalized ligands with dimensions 30 × 30 × 30 Å to accommodate the whole binding sites of the target receptors. All docking calculations were implemented with the aid of the open-source software AutoDock vina v1.1.2. The docking poses were ranked according to their docking scores, and the best energy pose was selected. The interactions between the screened compound and the target proteins were analyzed using Discovery Studio Visualizer v21.1.0.20298 [16].
RESULTS AND DISCUSSION
The CH2Cl2 and EtOAc fractions derived from T. myriocarpa methanolic leaf extract were found to exhibit α-glucosidase inhibitory activity (Table 1). The EtOAc fraction exhibited potent activity with an half-maximal inhibitory concentration (IC50) value of 0.49 ± 0.03 µg/ml. The CH2Cl2 and EtOAc fractions were subjected to further phytochemical analysis, which led to the isolation of compounds 1 through 10 due to their separation using variable chromatographic procedures. The structures of the isolates were determined by spectral means (UV, 1H-, and 13C-NMR) and acid hydrolysis. Among these, compounds 1 [17,18], 4 [19,20], 6 [21], and 8 [22,23] are recorded herein for the first time from the genus Terminalia. In addition, this report is the first to mention the occurrence of compounds 7 [24,25], 9 [21], and 10 [26] in T. myriocarpa. Compounds 7, 9, and 10 were previously isolated from Terminalia arjuna bark [27], Terminalia catappa leaves [28], and Terminalia chebula fruits [29], respectively. On the other hand, compounds 2 [30,31], 3 [32], and 5 [31,33] were previously reported from the leaves of T. myriocarpa [2]. Results of evaluation of the α-glucosidase inhibitory potential of the isolated compounds (Table 2) revealed that quercetin and flavogallonic acid with IC50 values equal to 7.5 ± 0.09 and 21.0 ± 1.4 µM, respectively, might be responsible for the activity of the EtOAc fraction. This was in agreement with earlier reports on the ability of quercetin to inhibit α-glucosidase, with an IC50 value of 7 µM, through the formation of hydrogen bond interactions with the active site pocket of the enzyme [34,35]. Furthermore, former studies indicated that arjunic acid [11], nigaichigoside F1, rosamultin, and 19α-hydroxyasiatic acid [36] do not significantly inhibit α-glucosidase enzyme, which agreed with our findings. Moreover, isovitexin and asiatic acid were reported to exert α-glucosidase inhibitory effects with IC50 values of 266.2 µM [37] and 100.2 ± 4.2 µM [38], respectively, which confirmed our results on the weak α-glucosidase inhibitory effects of these compounds. On the other hand, nothing could be traced in the literature concerning the α-glucosidase inhibitory activity of flavogallonic acid, alphitolic acid, and quadranoside IV. Therefore, to predict the mode of binding of flavogallonic acid to human α-glucosidase, a docking study with human C-terminal maltase-glucoamylase ctMGAM (PDB:3TOP), human N-terminal maltase-glucoamylase ntMGAM (PDB:2QMJ), and human N-terminal sucrase-isomaltase ntSI (PDB:3LPP) was pursued. Results of the docking study revealed that flavogallonic acid was docked successfully to the same binding site of the co-crystalized inhibitor and possessed a binding affinity comparable with or even superior to acarbose (positive control) for the three receptors (Table 3). It adapted similar orientations in the catalytic site of the three receptors showing its gallic acid moiety inserted inside the binding cavity while the ellagic acid moiety heading to the outside. Flavogallonic acid-ntMGAM (PDB:2QMJ) complex was stabilized by six hydrogen bond interactions with asp203, asp327, trp406, asp443, met444, and arg526 in addition to two hydrophobic pi-pi interactions with tyr299 and phe575. Flavogallonic acid-ctMGAM (PDB:3TOP) complex demonstrated seven hydrogen bond interactions with asp1279, trp1355, asp1420, ser1425, lys1460, asp1526, and asp1555, a pi-pi interaction with phe1559, and a pi-alkyl interaction with met1421. For flavogallonic acid-ntSI (PDB:3LPP), seven hydrogen bond interactions with asp355, trp435, asp472, met473, lys509, arg555, a pi-pi interaction with trp327, and a pi-alkyl interaction with leu233 were observed (Fig. 2).
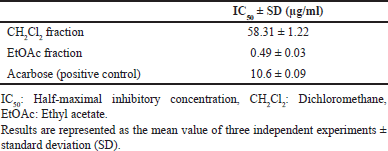 | Table 1. α-Glucosidase inhibitory activity of the CH2Cl2 and EtOAc fractions derived from T. myriocarpa methanolic leaf extract. [Click here to view] |
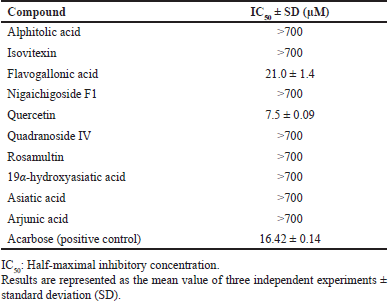 | Table 2. α-Glucosidase inhibitory activity of the isolated compounds from T. myriocarpa methanolic leaf extract. [Click here to view] |
 | Table 3. The binding affinities (kcal/mol) of flavogallonic acid with human α-glucosidase active sites, against those of acarbose. [Click here to view] |
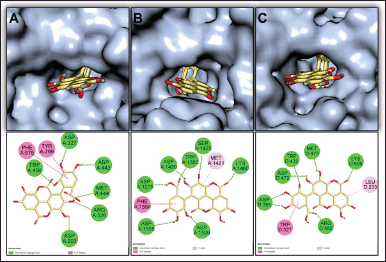 | Figure 2. 3-D presentations of the best docking poses and 2-D illustrations of hydrogen bond (green), Pi-Pi (dark pink), and Pi-alkyl (light pink) interactions between flavogallonic acid (yellow sticks), and (A) ntMGAM (PDB:2QMJ), (B) ctMGAM (PDB:3TOP), and (C) ntSI (PDB:3LPP). [Click here to view] |
CONCLUSION
The in-vitro and in-silico α-glucosidase inhibitory properties of quercetin and flavogallonic acid, which were isolated from the EtOAc fraction of T. myriocarpa methanolic leaf extract, point to their possible use as lead compounds to develop α-glucosidase inhibitors. This could only be implemented after confirming the obtained findings by assessing their efficacy and toxicity in-vivo.
ACKNOWLEDGMENTS
The authors are grateful to Ms. Therese Labib, the taxonomical consultant at Orman Botanical Garden and El Qubba Botanical Garden, for the identification and authentication of the plant material.
AUTHOR CONTRIBUTIONS
All authors have participated in designing the study, collection, and analysis of data, as well as writing the manuscript, which was approved by all authors.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
FINANCIAL SUPPORT
The National Research Centre, Giza, Egypt, provided this work’s funding.
ETHICAL APPROVALS
This study was carried out upon approval of the “Medical Research Ethics Committee” of the National Research Centre, Giza, Egypt (Registration number: 20115).
DATA AVAILABILITY
All the obtained and interpreted data are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
1. tropical.theferns.info [Internet]. Tropical plants database, Ken Fern [updated 2022 Jul 20]. 2022 [cited 2023 Sep 4]. Available from: https://tropical.theferns.info/viewtropical.php?id=Terminalia+myriocarpa
2. Zhang X-R, Kaunda JS, Zhu H-T, Wang D, Yang C-R, Zhang Y-J. The genus Terminalia (Combretaceae): an ethnopharmacological, phytochemical and pharmacological review. Nat Prod Bioprospect. 2019;9:357–92. CrossRef
3. Siraj MA, Islam Howlader MS, Islam MA, Irin T, Simal-Gandara J. Regulation of the redox signaling and inflammation by Terminalia myriocarpa leaves and the predictive interactions of its major metabolites with iNOS and NF-kB. J Ethnopharmacol. 2021;280:114459. CrossRef
4. Elmalah A, Abdel Khalik S, Abdelhady M, Taha K, Dawoud G. Phytochemical composition and antioxidant activity of Terminalia muelleri and Terminalia myriocarpa. Egypt J Chem. 2022;65(10):689–99. CrossRef
5. Majumdar K, Biswas M, Som UK, Das S. Chemical constituents of the bark of Terminalia myriocarpa. J Indian Chem Soc. 2005;82:673–4.
6. Singh A, Singh K, Sharma A, Kaur K, Kaur K, Chadha R, et al. Recent developments in synthetic α-glucosidase inhibitors: a comprehensive review with structural and molecular insight. J Mol Struct. 2023;1281:135115. CrossRef
7. Sakran N, Graham Y, Pintar T, Yang W, Kassir R, Willigendael EM, et al. The many faces of diabetes. Is there a need for re-classification? A narrative review. BMC Endocr Disord. 2022;22(1):9. CrossRef
8. Turner RC, Holman RR, Matthews DR, Oakes SF, Bassett RA, Stratton IM, et al. UK prospective diabetes study (UKPDS)—VIII. Study design, progress and performance. Diabetologia. 1991;34:877–90. CrossRef
9. Hermansen K, Mortensen LS, Hermansen ML. Combining insulins with oral antidiabetic agents: effect on hyperglycemic control, markers of cardiovascular risk and disease. Vasc Health Risk Manag. 2008;4(3):561–74. CrossRef
10. Lee DY, Kim HW, Yang H, Sung SH. Hydrolyzable tannins from the fruits of Terminalia chebula Retz and their α-glucosidase inhibitory activities. Phytochemistry. 2017;137:109–16. CrossRef
11. Lee DY, Yang H, Kim HW, Sung SH. New polyhydroxytriterpenoid derivatives from fruits of Terminalia chebula Retz. and their α-glucosidase and α-amylase inhibitory activity. Bioorg Med Chem Lett. 2017;27(1):34–9. CrossRef
12. Pham AT, Malterud KE, Paulsen BS, Diallo D, Wangensteen H. α-Glucosidase inhibition, 15-lipoxygenase inhibition, and brine shrimp toxicity of extracts and isolated compounds from Terminalia macroptera leaves. Pharm Biol. 2014;52(9):1166–9. CrossRef
13. Melek FR, El Zalabani SM, Ghaly NS, Sabry OM, Fayad W, Boulis AG. Phytoconstituents with cytotoxic activity from Ulmus pumila L. J Appl Pharm Sci. 2021;11(5):127–34. CrossRef
14. Elya B, Basah K, Mun’im A, Yuliastuti W, Bangun A, Septiana EK. Screening of α-glucosidase inhibitory activity from some plants of Apocynaceae, Clusiaceae, Euphorbiaceae, and Rubiaceae. J Biomed Biotechnol. 2012;2012:281078. CrossRef
15. Qaisar M, Chaudhary B, Sajid M, Hussain N. Evaluation of α-glucosidase inhibitory activity of dichloromethane and methanol extracts of Croton bonplandianum Baill. Trop J Pharm Res. 2014;13(11):1833–6. CrossRef
16. Raslan MA, Afifi AH. In vitro wound healing properties, antioxidant activities, HPLC–ESI–MS/MS profile and phytoconstituents of the stem aqueous methanolic extract of Dracaena reflexa Lam. Biomed Chromatogr. 2022;36(6):e5352. CrossRef
17. Raju R, Gunawardena D, Ahktar M, Low M, Reddell P, Münch G. Anti-inflammatory chemical profiling of the Australian rainforest tree Alphitonia petriei (Rhamnaceae). Molecules. 2016;21(11):1521. CrossRef
18. Kristanti AN, Aung EE, Aminah NS, Takaya Y, Aung HT, Ramadhan R. Bioactive triterpenoids from Indonesian medicinal plant Syzygium aqueum. Open Chem. 2022;20(1):204–11. CrossRef
19. Wu Z-J, Ouyang M-A, Wang C-Z, Zhang Z-K, Shen J-G. Anti-tobacco mosaic virus (TMV) triterpenoid saponins from the leaves of Ilex oblonga. J Agric Food Chem. 2007;55(5):1712–7. CrossRef
20. Reher G, Budešínský M. Triterpenoids from plants of the Sanguisorbeae. Phytochemistry. 1992;31(11):3909–14. CrossRef
21. Monti D, Candido A, Cruz Silva MM, Kren V, Riva S, Danieli B. Biocatalyzed generation of molecular diversity: selective modification of the saponin asiaticoside. Adv Synth Catal. 2005;347(7–8):1168–74. CrossRef
22. Zebiri I, Haddad M, Duca L, Sauvain M, Paloque L, Cabanillas B, et al. Biological activities of triterpenoids from Poraqueiba sericea stems. Nat Prod Res. 2017;31(11):1333–8. CrossRef
23. Lee D-Y, Jung L, Park J-H, Yoo K-H, Chung I-S, Baek N-I. Cytotoxic triterpenoids from Cornus kousa fruits. Chem Nat Compd. 2010;46(1):142–5. CrossRef
24. Enayati A, Salehi A, Alilou M, Stuppner H, Mirzaei H, Omraninava A, et al. Six new triterpenoids from the root of Potentilla reptans and their cardioprotective effects in silico. Nat Prod Res. 2022;36(10):2504–12. CrossRef
25. Yuan C, Huang L, Suh JH, Wang Y. Bioactivity-guided isolation and identification of antiadipogenic compounds in Shiya tea (leaves of Adinandra nitida). J Agric Food Chem. 2019;67(24):6785–91. CrossRef
26. Ponou BK, Teponno RB, Ricciutelli M, Nguelefack TB, Quassinti L, Bramucci M, et al. Novel 3-oxo- and 3,24-dinor-2,4-secooleanane-type triterpenes from Terminalia ivorensis A. Chev. Chem Biodivers. 2011;8(7):1301–9. CrossRef
27. Wang W, Ali Z, Shen Y, Li XC, Khan IA. Ursane triterpenoids from the bark of Terminalia arjuna. Fitoterapia. 2010;81(6):480–4. CrossRef
28. Fan YM, Xu LZ, Gao J, Wang Y, Tang XH, Zhao XN, et al. Phytochemical and antiinflammatory studies on Terminalia catappa. Fitoterapia. 2004;75(3–4):253–60. CrossRef
29. Kim MS, Lee DY, Sung SH, Jeon WK. Anti-cholinesterase activities of hydrolysable tannins and polyhydroxytriterpenoid derivatives from Terminalia chebula Retz. fruit. Rec Nat Prod. 2018;12(3):284–9. CrossRef
30. Hao B, Caulfield JC, Hamilton ML, Pickett JA, Midega CAO, Khan ZR, et al. Biosynthesis of natural and novel C-glycosylflavones utilising recombinant Oryza sativa C-glycosyltransferase (OsCGT) and Desmodium incanum root proteins. Phytochemistry. 2016;125:73–87. CrossRef
31. Kim SM, Kang K, Jho EH, Jung Y-J, Nho CW, Um B-H, et al. Hepatoprotective effect of flavonoid glycosides from Lespedeza cuneata against oxidative stress induced by tert-butyl hyperoxide. Phytother Res. 2011;25:1011–7. CrossRef
32. Pfundstein B, El Desouky SK, Hull WE, Haubner R, Erben G, Owen RW. Polyphenolic compounds in the fruits of Egyptian medicinal plants (Terminalia bellerica, Terminalia chebula and Terminalia horrida): characterization, quantitation and determination of antioxidant capacities. Phytochemistry. 2010;71(10):1132–48. CrossRef
33. Li X, Yang Y, Chen L, Zhang Y, Chen Y. Compounds from Lotus corniculatus. Chem Nat Compd. 2019;55(4):719–21. CrossRef
34. Tadera K, Minami Y, Takamatsu K, Matsuoka T. Inhibition of alpha-glucosidase and alpha-amylase by flavonoids. J Nutr Sci Vitaminol (Tokyo). 2006;52(2):149–53. CrossRef
35. Limanto A, Simamora A, Santoso AW, Timotius KH. Antioxidant, α-glucosidase inhibitory activity and molecular docking study of gallic acid, quercetin and rutin: a comparative study. Mol Cell Biomed Sci. 2019;3(2):67–74. CrossRef
36. Li W, Fu H, Bai H, Sasaki T, Kato H, Koike K. Triterpenoid Saponins from Rubus ellipticus var. obcordatus. J Nat Prod. 2009;72(10):1755–60. doi: 10.1021/np900237a
37. Chen YG, Li P, Li P, Yan R, Zhang XQ, Wang Y, et al. α-Glucosidase inhibitory effect and simultaneous quantification of three major flavonoid glycosides in Microctis folium. Molecules. 2013;18(4):4221–32. CrossRef
38. Zhang B-W, Xing Y, Wen C, Yu X-X, Sun W-L, Xiu Z-L, et al. Pentacyclic triterpenes as α-glucosidase and α-amylase inhibitors: structure-activity relationships and the synergism with acarbose. Bioorg Med Chem Lett. 2017;27(22):5065–70. CrossRef