
INTRODUCTION
The first case of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection was reported in Wuhan, Hubei Province, China, in December 2019, following an outbreak of people exhibiting pneumonia-like symptoms [1,2]. The virus rapidly spread globally, leading to its declaration as a pandemic. The virus continues to infect people and cause deaths, especially among older individuals with underlying health conditions [3]. In the Philippines, the first confirmed case was reported in January 2020 when two Chinese nationals traveling for vacation in the country developed fever, cough, and sore throat [4]. As of now, there have been over 2.84 million cases and 51,373 deaths in the Philippines, while globally, more than 287 million cases and 5.43 million deaths have been reported [5,6].
Although the respiratory system is primarily affected in COVID-19 patients, evidence of the virus’s detrimental effects on other organ systems has been reported [7]. Understanding the structure and characteristics of spike proteins is crucial for developing effective vaccines and treatments. The spike protein is a vital component of the SARS-CoV-2 virus. These proteins protrude from the surface of the virus and facilitate its entry into human cells. By binding to specific receptors on the cell surface, spike proteins enable the virus to invade and initiate infection. Researchers are currently mapping T-cell furin cleavage site epitopes within spike protein sequences to gain insights into the virus’s behavior and potential immune detection.
The furin cleavage site is one of the main virulence factors of SARS-CoV-2, distinguishing it from other betacoronaviruses. This site increases the virus’s infectivity in humans by facilitating the fusion of the viral membrane with host cells. Plasma samples that contain epitopes adjacent to the SARS-CoV-2 S1/S2 cleavage sites have been found to inhibit proteolysis mediated by furin [8]. Recently, a novel spike epitope, RSVASQSIIAYT, that maps to the furin cleavage site (i.e., 685–686) has been identified [9]. Given that this epitope is located at the furin cleavage site, we hypothesized that the corresponding antibody would react to the virus before it is cleaved and enters a host cell, potentially preventing infection. To the best of our knowledge, no studies have been conducted to map the furin cleavage site epitopes using the sequences of the SARS-CoV-2 variants identified in the Philippines. While previous research has explored this aspect, our study specifically focuses on analyzing the unique variants circulating within the country. By mapping the furin cleavage site epitopes of these Philippine-specific sequences, we aim to contribute valuable insights into the behavior and potential immune recognition of these variants. This research will provide a deeper understanding of the SARS-CoV-2 landscape in the Philippines and aid in the development of targeted interventions and strategies.
Although COVID-19 vaccines are available, new SARS-CoV-2 variants continue to emerge, hindering efforts to achieve herd immunity and potentially rendering current vaccines less effective [10]. To address this, diagnostic tools and vaccines should be developed while considering the variants’ genetic drift and impact on immune recognition [11,12].
This study aims to identify immunogenic epitopes that are both distinct and conserved to SARS-CoV-2 variants of concern and interest (VOCs and VOIs) in the spike protein that maps to the furin cleavage site using an in silico approach.
MATERIALS AND METHODS
Sequence retrieval
The SARS-CoV-2 sequences were retrieved from the NCBI virus database [13]. The search results for the SARS-CoV-2 sequences were refined by setting the virus of interest to SARS-CoV-2. The sequences for each variant were obtained by adding the Pango Lineage of each variant to the search filter. For this study, sequences from the Philippines were prioritized. Variants that did not have available sequences from the Philippines were obtained by widening the geographic filter to sequences from Southeast Asia, Asia, and then other continents. Only sequence IDs that have complete coding sequences were considered. The NCBI virus GenBank accession IDs of the SARS-CoV-2 spike sequences that were used in the study are listed in Table 1.
Epitope prediction
The immune epitope database (IEDB) T-cell epitope prediction tool for major histocompatibility complex (MHC) I and MHC II was used to predict T-cell epitopes of SARS-CoV-2 spike protein sequences [14]. This study focused on the two most frequent Southeast Asian human leukocyte antigen (HLA)-A, HLA-B, and HLA-C for MHC I and HLA-DRB for MHC II. Specifically, the HLA alleles studied were HLA-A11:01, HLA-A24:02, HLA-B40:01, HLA-B46:01, HLA-C01:02, HLA-C07:02, HLA-DRB109:01, and HLA-DRB112:02 [15]. Default prediction methods, NetMHCpan EL 4.1 for MHC I and NetMHCIIpan for MHC II were used to predict T-cell epitopes and 9-mer peptides were considered for MHC I and 15-mer peptides for MHC II. Predicted epitopes with a percentile rank of ≤1% for MHC I and ≤10% for MHC II were selected for further analysis [16].
Epitope modeling
The identified conserved epitope sequences were used for further analysis. The conserved epitopes were 3-D modeled in preparation for molecular docking using PEP-Fold3. The sequences of the conserved epitopes were entered into the server of PEP-Fold3 to create its most likely structure [17–19]. The model from the best cluster from the output models based on the Hidden Markov Model and sOPEP energy were used in molecular docking.
Epitope-MHC docking
The conserved epitope sequences were docked to their respective HLA allele proteins to determine their binding energies to the MHC molecules. The HLA protein molecules for docking were obtained from the Protein Data Bank (PDB) for HLA alleles that have available structures [20]. Only structures without mutation were used for the study. The HLA molecules that have available structures in PDB were HLA-A*11:01 (Accession No. 6JOZ), HLA-A*24:02 (Accession No. 7JYV), HLA-B*40:01 (Accession No. 6IEX), HLA-B*46:01 (Accession No. 4LCY), and HLA-C*07:02 (Accession No. 5VGE). The HLA alleles that did not have available downloadable structures (HLA-C*01:02, HLA-DRB1*09:01, and HLA-DRB1*12:02) were modeled using SWISS-MODEL using their sequences obtained from IPD-IMGT/HLA Database [21,22].
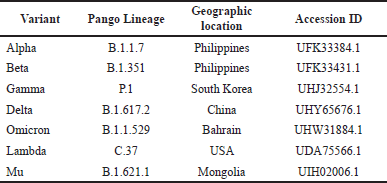 | Table 1. NCBI virus GenBank accession IDs of SARS-CoV-2 spike sequences used and their geographic origins. [Click here to view] |
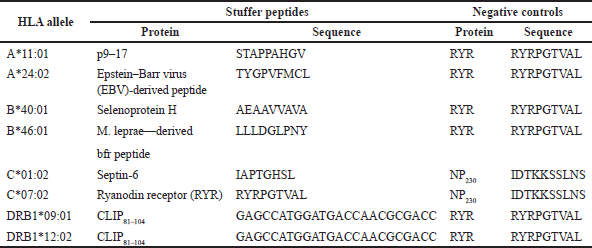 | Table 2. Stuffer and negative control peptides used in molecular docking of predicted SARS-CoV-2 spike protein epitopes on HLA molecules. [Click here to view] |
Stuffer and negative control peptides were also docked to the MHC molecules to serve as the positive and negative controls. The stuffer peptides are protein sequences that bind to the MHC molecule that was used in this study were p9–17, an EBV-derived peptide, selenoprotein H, M. leprae—derived bacterioferritin (bfr) peptide, septin-6, RYR, and CLIP81-104 [23–29]. On the other hand, the negative control peptides that do not bind to the HLA molecules that were used in this study were an HLA-C*07:01 restricted epitope (RYR) and NP230 [30]. Table 2 lists the stuffer and negative control peptides that were used in the study.
The ligands and proteins were prepared for docking using Schrödinger PyMOL 2.5.1 and Scripps Research AutoDock Tools 1.5.6. Using PyMOL 2.5.1, the ligands available from the protein obtained from PDB were removed. Then, using AutoDock Tools 1.5.6, the water molecules from the protein were removed, and then the polar hydrogens and Kollman charges were added to the protein molecule [31]. A grid box that spans the active site was prepared for each MHC molecule [31].
After the preparation of the ligands and MHC proteins, the predicted epitopes, stuffer peptides, and negative controls were docked to their respective HLA molecules using Scripps Research AutoDock Vina v1.2.3 using default parameters (exhaustiveness: 8) [32]. The binding energies from the molecular docking using AutoDock Vina v1.2.3 of the conserved epitopes were compared to the binding energies of the stuffer and negative control peptides. Epitopes whose complexes have lower binding energies than the stuffer peptides with the corresponding HLA molecule were considered promising candidate epitopes for vaccine design [33]. Ligands that have lower binding energies may displace docked ligands with higher binding energies [34]. A reference protein complex from PDB (Accession No. 6IEX) was used to validate the docked complexes. The original ligand and HLA molecule were each isolated and then redocked using AutoDock Vina. The resulting docked ligand and the original ligand structure were validated by superimposing the corresponding structures. An root mean square deviation (RMSD) value of 1.5 Å was used as the threshold for validation [35].
Prediction of peptide-MHC (pMHC)-T-cell receptors (TCR) binding
After docking the epitopes to the MHC proteins, the potential TCRs that were able to recognize the pMHC complexes were determined. TCRs that can identify epitopes with the greatest similarity to the predicted epitopes in the study were selected for pMHC-TCR docking. A percent similarity of the IEDB predicted epitopes and TCR epitopes was calculated using Sequence Manipulation Suite under default parameters (similar amino acids considered) [36]. TCRs that were used were identified from VDJdb [37]. The TCR sequences were obtained from the TCRmodel web server [38]. The TCR, ligand, and MHC sequences were uploaded to TCRpMHCmodels to precisely model each complex based on homology.
The docked pMHC complex using AutoDock Vina v1.2.3 and PyMOL 2.5.1 replaced the pMHC obtained from the output from TCRpMHCmodels by first removing the pMHC part of the modeled complex from TCRpMHCmodels using PyMOL 2.5.1, then docked on the corresponding prospect TCR using ClusPro 2.0 [39]. The new pMHC-TCR complex was refined using the GalaxyRefineComplex web server [40]. To determine the binding energy and interactions of the pMHC complex to the prospect TCR, the PRODIGY web server was used for the prediction [41].
Postdock analysis
The conserved and distinct predicted T-cell epitopes using IEDB were determined using the aligned sequences of the SARS-CoV-2 variants. The complexes obtained from molecular docking were visualized using PyMOL 2.5.1. After visualizing the complexes, the interactions between the ligand and the MHC proteins were identified using BIOVIA, Dassault Systèmes Discovery Studios. The interactions were identified using the classifications available in Discovery Studios: hydrogen bonds, charge interactions, hydrophobic, unfavorable, and others for the ones not included in the classifications. The binding interactions present in TCR-pMHC complexes were identified using the PRODIGY web server.
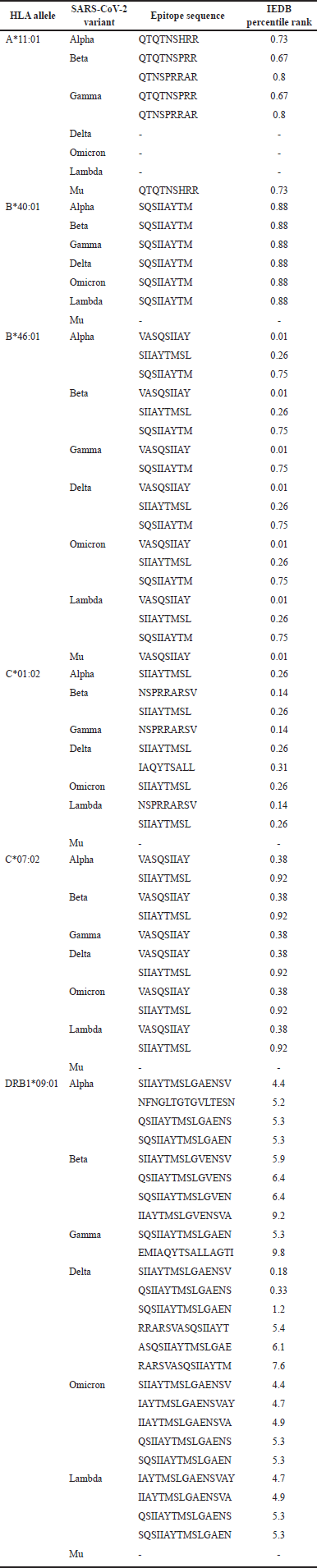 | Table 3. IEDB-predicted SARS-CoV-2 spike protein T-cell epitopes based on the upstream and downstream residues of the furin cleavage site (S1/S2) on the spike protein. [Click here to view] |
RESULTS
Epitope prediction
The T-cell epitopes of SARS-CoV-2 spike protein sequences were predicted using the IEDB T-cell epitope prediction tool for MHC-I and MHC-II servers, selecting peptides with percentile ranks of ≤1.0 for MHC-I and ≤10.0 for MHC-II [16]. The predicted epitope sequences that map to the furin cleavage site of SARS-CoV-2, along with their respective IEDB scores for each variant and HLA allele, are presented in Table 3. Notably, no T-cell epitopes within the threshold values were mapped to the furin cleavage site for HLA alleles HLA-A24:02 and HLA-DRB112:02.
Epitope conservation
To identify conserved and distinct epitopes, we conducted a multiple-sequence alignment using ClustalW2. Epitopes with at least an 80% identity threshold were considered conserved [42]. For vaccine design, we focused on epitopes that were potential targets for other variants while remaining conserved across different variant sequences. We only considered distinct epitopes that fell within the IEDB prediction threshold. However, we found that some of the distinct epitopes specific to certain variants were partially or fully conserved across other variants, even though they were not initially identified as potential T-cell epitope targets. Despite these limitations, we performed further analysis of the identified conserved and distinct epitopes. Table 4 presents the different conserved and distinct SARS-CoV-2 spike protein T-cell epitopes and their corresponding target variants.
pMHC docking
To determine the potentially immunogenic properties of the selected epitopes, molecular docking was conducted to model the interaction between the peptides and HLA alleles [33,43]. A higher predicted binding energy (kcal/mol) of the HLA allele to the selected epitopes compared to the stuffer and negative control peptides makes it more likely for the epitopes to replace the stuffer peptides and be involved in antigen presentation [33].
Table 5 displays the predicted binding energies of the docked complexes of the selected epitope sequences. Among the epitope sequences, only one of each in HLA allele B40:01 (SQSIIAYTM), C01:02 (IAQYTSALL), and C07:02 (VASQSIIAY) showed a higher binding energy than the stuffer and negative control peptides. In contrast, two epitope sequences were observed in B46:01 (VASQSIIAY and SIIAYTMSL), and four epitope sequences for DRB1*09:01 (SQSIIAYTMSLGAEN, ASQSIIAYTMSLGAE, EMIAQYTSALLAGTI, and NFNGLTGTGVLTESN).
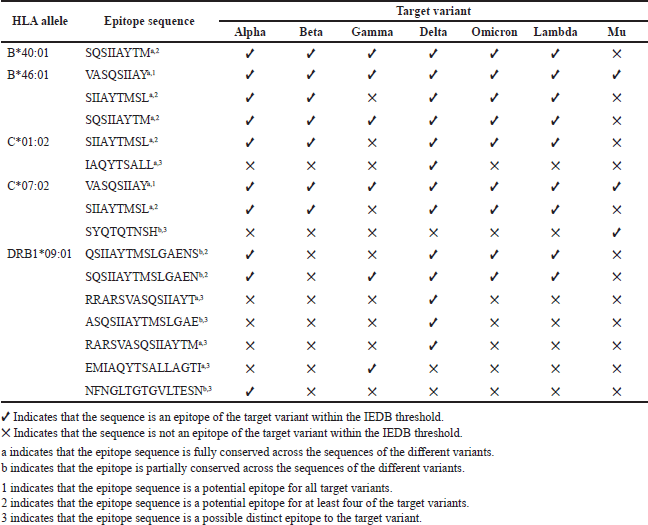 | Table 4. Conserved and distinct SARS-CoV-2 spike protein T-cell epitopes based on the upstream and downstream residues of the furin cleavage site (S1/S2) on the spike protein. [Click here to view] |
 | Table 5. Predicted binding energies (?Gbinding) of docked complexes of predicted SARS-CoV-2 spike epitopes with the stuffer and negative control peptides. [Click here to view] |
Studies suggest that shorter peptides bind to MHC-I epitopes that have buried extremes and a raised center ligand conformation, while relatively longer and larger peptides bind to MHC-II epitopes that have a more linear and extended ligand conformation [44]. The predicted epitopes’ conformations when bound to the selected HLA alleles showed a similarity to this claim, as shown in Figures 1–5. The buried amino acid residues that were deep in the pocket of the binding groove may have contributed to higher binding energy than the stuffer and negative control peptides.
For validation of the docked complexes, a reference protein complex from PDB (Accession No. 6IEX) was used. The original ligand and HLA molecule were isolated and then redocked. The new docked complex from the isolated molecules was compared to the original structure, resulting in an RMSD value of 1.410 after superimposing the two complexes.
pMHC interactions
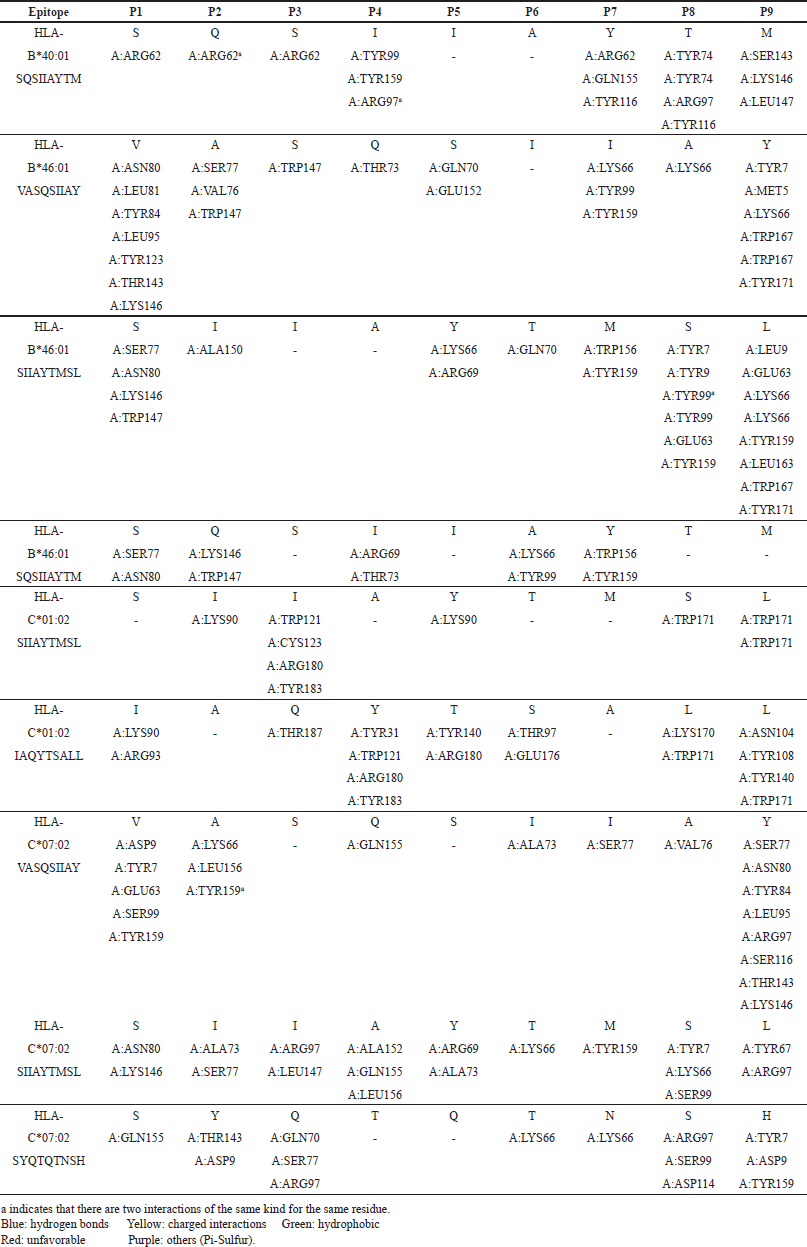
To visualize the interactions between the MHC-I and MHC-II complexes and the chosen epitopes, BIOVIA, Dassault Systèmes Discovery Studio was utilized. The resulting interactions are presented in Tables 6 and 7 for MHC-I and MHC-II, respectively.
In the MHC-I interactions, hydrogen bonds (blue) and hydrophobic bonds (green) dominate the epitopes, with favorable charged interactions (yellow) observed at P9 of HLA-B46:01 VASQSIIAY and P1 of HLA-C07:02 VASQSIIAY. However, unfavorable interactions (red) were identified at P1, P8, and P9 of HLA-B46:01 SIIAYTMSL, P1 and P6 of HLA-C01:02 IAQYTSALL, P1 and P8 of HLA-C07:02 SIIAYTMSL, and P3 of HLA-C07:02 SYQTQTNSH. The unfavorable amino acid residues are lysine (K) and arginine (R) for P1, serine (S) for P3, threonine (T) for P6, tyrosine (Y) and lysine (K) for P8, and lysine (K) for P9.
For MHC-II interactions, hydrogen bonds (blue) and hydrophobic bonds (green) also dominate, with favorable charged interactions (yellow) observed at P14 of HLA-DRB109:01 SQSIIAYTMSLGAEN and P1 of both HLA-DRB109:01 RARSVASQSIIAYTM and EMIAQYTSALLAGTI. However, unfavorable interactions (red) are observed in all epitope sequences for HLA-DRB1*09:01, specifically at P1, P2, P4, P6, P7, P10, P14, and P15. The unfavorable amino acid residues are glutamic acid (E) and arginine (R) for P1, serine (S) for P2, arginine (R) for P4, histidine (H) for P6, serine (S) and glutamine (Q) for P7, glutamine (Q) and asparagine (N) for P10, and arginine (R), glutamic acid (E), histidine (H), and tryptophan (W) for P14, and Asn (N) for P15.
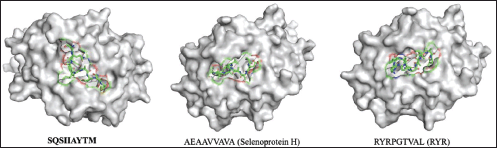 | Figure 1. Conformations of the predicted epitopes, when bound to B*40:01. Epitope sequences that have higher binding affinity than the stuffer and negative control peptides, are indicated in bold. [Click here to view] |
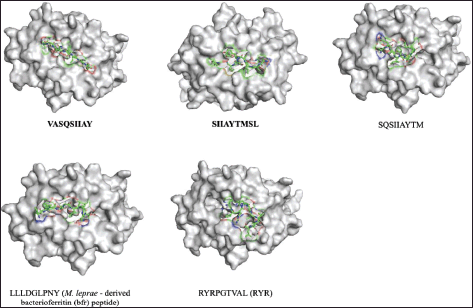 | Figure 2. Conformations of the predicted epitopes, when bound to B*46:01. Epitope sequences that have higher binding affinity than the stuffer and negative control peptides, are indicated in bold. [Click here to view] |
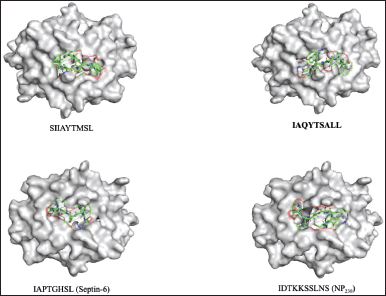 | Figure 3. Conformations of the predicted epitopes, when bound to C*01:02. Epitope sequences that have higher binding affinity than the stuffer and negative control peptides, are indicated in bold. [Click here to view] |
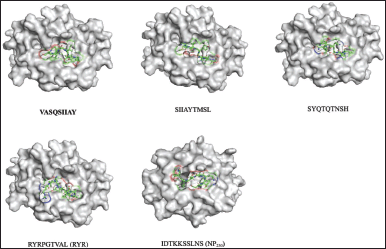 | Figure 4. Conformations of the predicted epitopes, when bound to C*07:02. Epitope sequences that have higher binding affinity than the stuffer and negative control peptides, are indicated in bold. [Click here to view] |
TCR-pMHC docking
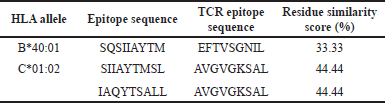
To identify potential TCR molecules that may interact with the pMHC complexes of the identified epitopes, we first docked the complexes to their respective MHC molecules. We then used the VDJdb database to prospect for TCR molecules that have been experimentally shown to recognize similar epitope sequences as those we predicted. However, we were unable to find any corresponding TCRs for HLA-B46:01, HLA-C07:02, and HLA-DRB109:01 in the database, so the TCR-pMHC docking was only carried out for HLA-B40:01 and HLA-C01:02 epitopes. To select the most relevant TCR molecules, we compared the predicted epitope sequences with the TCR epitope sequences in the database using Sequence Manipulation Suite. We then chose the TCR epitopes with the highest residue similarity scores for docking. Unfortunately, the similarity scores between the predicted epitopes and the selected TCR epitopes were low, at only 33.33% for the SQSIIAYTM epitope of HLA-B40:01 and 44.44% for both SIIAYTMSL and IAQYTSALL epitopes of HLA-C*01:02. Table 8 summarizes the similarity scores between the predicted epitope sequences and the available TCR epitope sequences.
To obtain the TCR complex sequences for TCR-pMHC docking, the prospective TCR molecules identified earlier were retrieved. Table 9 presents the prospective TCRs used in the study, along with their corresponding V and J gene segments. These TCR molecules were selected based on their ability to successfully recognize and dock with the recorded epitope sequences and MHC molecules in previous experimental studies.
To model the TCR-pMHC complexes, we used TCRpMHC models for the identified TCR sequences, which were then docked with the pMHC complexes obtained from AutoDock Vina. ClusPro was used for protein–protein docking to improve the accuracy of the complex formation. The resulting complexes were further refined using the GalaxyRefine Complex web server. The final TCR-pMHC complexes are shown in Figure 6.
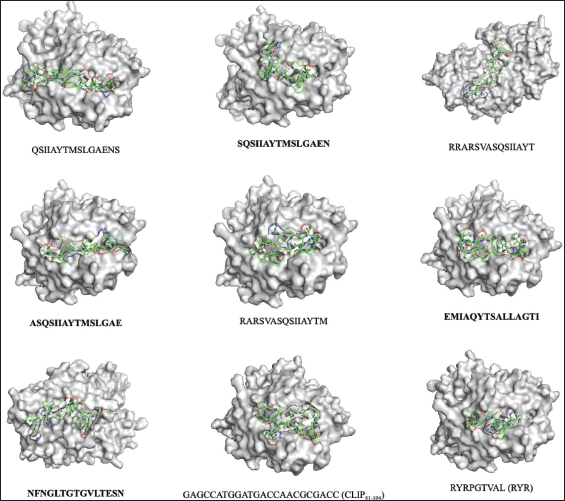 | Figure 5. Conformations of the predicted epitopes, when bound to DRB1*09:0. Epitope sequences that have higher binding affinity than the stuffer and negative control peptides, are indicated in bold. [Click here to view] |
 | Table 6. Predicted noncovalent interactions of MHC-I docked complexes. PΩ refers to the position of the amino acid in the epitope. [Click here to view] |
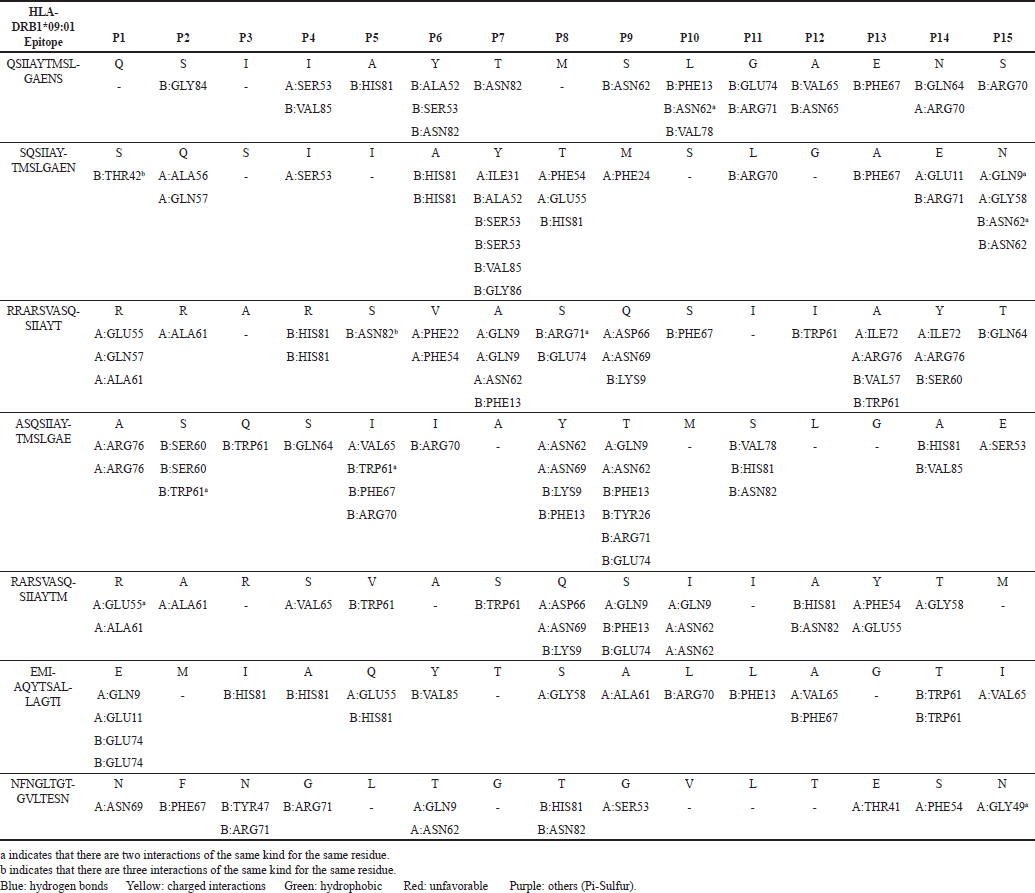 | Table 7. Predicted ligand interactions of MHC-II docked complexes. PΩ refers to the position of the amino acid in the epitopes. [Click here to view] |
 | Table 8. Similarity scores of the predicted epitope sequences and the available TCR epitope sequences. [Click here to view] |
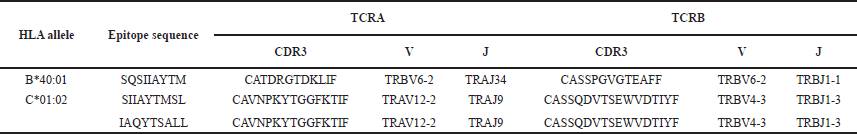 | Table 9. TCRs are used for TCR-pMHC modeling. [Click here to view] |
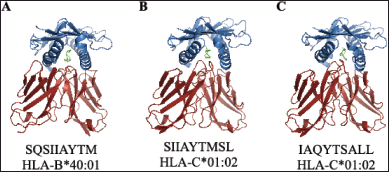 | Figure 6. Conformations of the following TCR-pMHC complexes: (A) SQSIIAYTM of HLA-B*40:01, (B) SIIAYTMSL of HLA-C*01:02, and (C) IAQYTSALL of HLA-C*01:02. [Click here to view] |
 | Table 10. Predicted binding energies (?Gbinding) of SARS-CoV-2 pMHC complexes to TCR (TCR-pMHC). [Click here to view] |
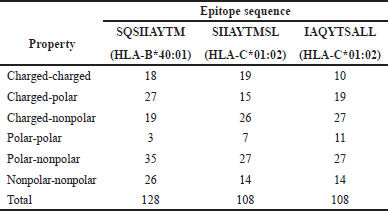 | Table 11. Number of binding interactions of the TCR-pMHC complexes. [Click here to view] |
The binding energies obtained from TCR-pMHC docking can be used as an indicator of the immunogenicity of the identified epitopes [33]. Therefore, to further evaluate the potential immunogenicity of the identified epitopes, the binding energies of the TCR-pMHC complexes were predicted using the PRODIGY web server. Epitope SQSIIAYTM of HLA-B40:01 exhibited the lowest binding energy among the three complexes at −16.0 kcal/mol, followed by SIIAYTMSL and IAQYTSALL of HLA-C01:02. Table 10 presents the predicted binding energies of the TCR-pMHC complexes.
To further evaluate the TCR-pMHC complexes, the number of different interfacial contacts per property at the binding cleft for each complex was recorded. The epitope SQSIIAYTM of HLA-B40:01 had the highest number of interfacial contacts, with a total of 128 binding interactions. Both epitopes of HLA-C01:02 were predicted to have 108 binding interactions, but SIIAYTMSL had more charged-charged interactions compared to IAQYTSALL. Table 11 summarizes the number of binding interactions for each property present in each TCR-pMHC complex.
DISCUSSION
Epitope prediction
Based on the furin cleavage site threshold, HLA-A11:01, HLA-A24:02, and HLA-DRB112:02 alleles are not viable targets for vaccine development. The following alleles were further used in the study: HLA-B40:01, HLA-B46:01, HLA-C01:02, HLA-C07:02, and HLA-DRB109:01. In selecting predicted epitopes for vaccine development, it is crucial to choose those with the highest binding affinity as the binding of the antigen to both MHC and TCR is necessary for its immunogenicity [33]. MHC-I molecules have close-ended binding clefts on both ends and typically bind to peptides with lengths of 8–10 amino acids containing appropriate anchor residues. Longer peptides are more likely to be trimmed [33,45,46]. A recent study found that 9 residue length (9-mer) is the optimal epitope length for MHC-I peptides, and this length was used in our study [47]. On the other hand, MHC-II molecules have an open binding groove and can accommodate peptides of 13–25 amino acid lengths with more flexibility [45]. Therefore, we used 15 residues (15-mer) as the length for MHC-II peptides in this study.
Epitope conservation
Although some epitopes were found to be distinct from their target variants, these epitope sequences were still present in the genomic sequence of other variants, indicating that they are not potential diagnostic indicators for the corresponding variants. Conserved epitopes are regions of pathogen sequences that evolve slowly and have very low variability in their amino acid residues, making them crucial for preserving the protein functions of the pathogens [48]. However, these epitopes are considered good targets for the development of epitope-based vaccines, which can provide broad-spectrum protection against the pathogen [49]. It is important to note that the flanking residues beside the antigen epitopes are also processed for presentation by antigen-presenting cells and recognition by TCRs [49]. Hence, there may be differences in the amount of recognition and reactivity of the immune system to the epitopes.
pMHC complex
The change in Gibbs free energy, measured as binding energy in kcal/mol, is negative in peptide-ligand complexes since binding only occurs when there is a decrease in the total Gibbs energy of the system at equilibrium while keeping the temperature and pressure constant [50]. Epitopes with higher binding affinity compared to the stuffer and negative control peptides have a favorable potential in designing vaccines for SARS-CoV-2 [33]. The following epitopes have a higher binding affinity for their respective allotypes: SQSIIAYTM for HLA-B40:01, VASQSIIAY and SIIAYTMSL for HLA-B46:01, IAQYTSALL for HLA-C01:02, and VASQSIIAY, SQSIIAYTMSLGAEN, ASQSIIAYTMSLGAE, EMIAQYTSALLAGTI, and NFNGLTGTGVLTESN for HLA-DRB109:01. Epitopes with lower binding energy can displace stuffer peptides during antigen presentation. The magnitude of negative Gibbs free energy change determines the stability and binding affinity of the protein-ligand complex [51]. With an RMSD value of 1.410 Å, the predicted complexes are expected to be close to the conformation of experimentally determined docking.
MHC-I has a single α-chain with a binding groove with closed ends, limiting the possible residues to bind to 8–10 amino acids. Studies indicate that the C-terminal end or P9 at the F pocket region of the MHC-I plays a crucial role in presenting stable pMHC complexes [45]. The SQSIIAYTM epitope sequence has the highest binding energy as it matches the binding motif of HLA-B40:01, favoring the residue leucine (L) at the C-terminal (P9) and tyrosine (Y) at P4, P7, and P8. This is consistent with the preference of leucine as the C-terminal residue and tyrosine as one of the critical amino acids found in the hydrophobic F pocket of HLA-B40:02, a closely related allotype of HLA-B40:01 [28]. Hydrogen bonds and hydrophobic interactions are abundant in most positions of HLA-B40:01, in line with the presence of hydrogen bonds and hydrophobic side chains.
Both epitope sequences VASQSIIAY and SIIAYTMSL exhibit the highest binding energy, maybe due to their matching the preferential binding motif of HLA-B46:01, which favors residues tyrosine (Y) at P1 and tryptophan (W) and tyrosine (Y) at the C-terminal. However, the epitope sequence VASQSIIAY of HLA-B46:01 has a higher binding affinity as compared to SIIAYTMSL, possibly due to the absence of preferred residues at the P1 position in the latter.
HLA-C allotypes generally exhibit a preference for hydrophobic amino acid residues at the C-terminal anchor position [33,52]. This preference is observed in both epitope sequences SIIAYTMSL and IAQYTSALL of HLA-C01:02. However, only the epitope sequence IAQYTSALL of HLA-C01:02 contains tyrosine (Y) residues, which are one of the restricted aromatic amino acids found at the C-terminal of HLA-C allotypes [52].
HLA-C07:02 has a preference for lysine (L) or tyrosine (Y) as anchor residues in P1 and P2, respectively [53]. The epitope sequence VASQSIIAY matches the binding motif of HLA-C07:02, with a preference for both lysine (L) and tyrosine (Y) residues at P1 and P2, respectively. In addition, the presence of a tyrosine (Y) residue at the C-terminal position of the epitope sequence VASQSIIAY is the only residue accepted by HLA-C07:02 [52]. MHC-II has two chains, mainly α-chain and β-chain, with an open binding groove that exhibits a more flexible accommodation of 13–25 amino acid length. The P1 and P2 regions have a dominant effect on the presentation of stable pMHC complexes [45]. The epitope sequence ASQSIIAYTMSLGAE has the highest binding energy, which could be attributed to its matching the binding motif of HLA-DRB109:01, which favors the residue arginine (R) at P1 and serine (S) and tryptophan (W) at P2.
TCR-pMHC complex
In this study, we successfully modeled three TCR-pMHC complexes in silico: SQSIIAYTM of HLA-B40:01, SIIAYTMSL and IAQYTSALL of HLA-C01:02. The SQSIIAYTM epitope has been extensively studied previously, which was found to be effective in producing clonotypes in HLA-A02:01 COVID patients as well as in healthy individuals [54]. Based on this, it is reasonable to assume that people with the HLA-B40:01 allele could also produce positive clonotypes against SARS-CoV-2. Another epitope investigated in this study was SIIAYTMSL, which was found to be nontoxic and to have a high population coverage across several regions [55,56]. Although IAQYTSALL has been studied mainly as a core epitope for HLA-DRB101:01 and HLA-DRB115:01 [57,58], we identified it as a distinct epitope to the Delta variant that maps to the furin cleavage site.
Our results showed that the TCR-pMHC complex of SQSIIAYTM had the lowest binding energy among the three complexes. This could be due to the greater number of binding interactions in this complex compared to the other two. However, we also observed that, even with the same number of binding interactions, the SIIAYTMSL epitope had lower binding energy than IAQYTSALL, possibly because of the greater number of charged-charged interactions in SIIAYTMSL. Charge-pair interactions play a crucial role in the assembly of the complex [59].
CONCLUSION
This in silico study successfully predicted distinct and conserved MHC-I and MHC-II epitopes in the furin cleavage site of SARS-CoV-2 VOCs and VOIs spike protein. Only alleles that are potential targets for epitope vaccines were analyzed for pMHC docking, and fully and partially conserved epitopes were identified as important for the development of epitope-based vaccines with broad-spectrum protection against the pathogen. Epitopes with better binding affinities than stuffer and negative peptides were further analyzed for docking with their corresponding MHC alleles, as they have the potential to replace stuffer peptides during antigen presentation. The study found that hydrogen bonds and hydrophobic interactions were the dominant interactions that influenced the binding energy and contributed to the structural stability of the protein-ligand interaction, which is an important factor for predicting the immunogenicity of the identified epitopes. The pMHC complexes with corresponding TCRs (HLA-B40:01 and HLA-C01:02 epitopes) were also docked to prospect TCR molecules. The study identified the epitope SQSIIAYTM of HLA-B*40:01 as the candidate immunogenic epitope with the lowest binding energy (−16.0 kcal/mol) and the greatest number of binding interactions to TCR (128) from the furin cleavage site of the spike protein.
This in silico study has provided significant findings and opened up avenues for future research in the field of epitope-based vaccines for SARS-CoV-2. The predicted epitopes should be experimentally validated to confirm their binding affinity and immunogenicity. Conducting in vitro and in vivo studies will be crucial to validate the effectiveness of these epitopes in inducing an immune response. The docking of pMHC complexes with TCRs provides insights into the interaction between T cells and epitopes. Future studies can delve deeper into understanding the binding interactions and dynamics of TCRs with the identified epitopes to enhance our understanding of the immune recognition process. Overall, this study provides valuable insights into the development of epitope-based vaccines for SARS-CoV-2 by identifying potential candidate epitopes from the furin cleavage site of the spike protein. By exploring the identified epitopes, conducting experimental studies, and advancing vaccine design, future investigations can contribute to the development of effective and broad-spectrum vaccines against SARS-CoV-2 and its variants.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included in this research article.
PUBLISHER’S NOTE
This journal remains neutral about jurisdictional claims in published institutional affiliation.
REFERENCES
1. Yang S, Cao P, Du P, Wu Z, Zhuang Z, Yang L, et al. Early estimation of the case fatality rate of COVID-19 in mainland China: a data-driven analysis. Ann Transl Med. 2020;8(4):1–6.
2. De Soto J, Hakim S, Boyd F. The pathophysiology of virulence of the COVID-19. Preprint: 2020040077 [Preprint]. 2020 [cited 2021 Dec 6];31. Available from: https://www.preprints.org/manuscript/202004.0077/v2
3. Ciotti M, Ciccozzi M, Terrinoni A, Jiang WC, Wang CB, Bernardini S. The COVID-19 pandemic. Crit Rev Clin Lab Sci. 2020;57(6):365–88.
4. Edrada EM, Lopez EB, Villarama JB, Salva Villarama EP, Dagoc BF, Smith C, et al. First COVID-19 infections in the Philippines: a case report. Trop Med Health. 2020;48(1):1–7.
5. World Health Organization [Internet]. Coronavirus disease pandemic. Geneva, Switzerland: World Health Organization; 2020 [cited 2021 Dec 6]. Available from: https://covid19.who.int
6. World Health Organization [Internet]. Coronavirus disease pandemic. Geneva, Switzerland: World Health Organization; 2020 [cited 2021 Dec 6]. Available from: https://covid19.who.int/region/wpro/country/ph
7. Huang Y, Yang C, Xu XF, Xu W, Liu SW. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin. 2020;41(9):1141–9.
8. Farrera-Soler L, Daguer JP, Barluenga S, Vadas O, Cohen P, Pagano S, et al. Identification of immunodominant linear epitopes from SARS-CoV-2 patient plasma. PloS One. 2020;15(9):e0238089.
9. Haynes WA, Kamath K, Bozekowski J, Baum-Jones E, Campbell M, Casanovas-Massana A, et al. High-resolution epitope mapping and characterization of SARS-CoV-2 antibodies in large cohorts of subjects with COVID-19. Commun Biol. 2021;4(1):1–14.
10. Aschwanden C. Five reasons why COVID herd immunity is probably impossible. Nature. 2021 March 18;591(7851):520–2.
11. Roy B, Dhillon JK, Habib N, Pugazhandhi B. Global variants of COVID-19: current understanding. J Biomed Sci. 2021;8(1):8–11.
12. Koyama T, Weeraratne D, Snowdon JL, Parida L. Emergence of drift variants that may affect COVID-19 vaccine development and antibody treatment. Pathogens. 2020;9(5):324.
13. Hatcher EL, Zhdanov SA, Bao Y, Blinkova O, Nawrocki EP, Ostapchuck Y, et al. Virus variation resource–improved response to emergent viral outbreaks. Nucleic Acids Res. 2017;45(D1):D482–90.
14. Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2019;47(D1):D339–43.
15. Allelefrequencies.net [Internet]. Top 10 HLA allele frequencies by geographical region. 2021 [cited 2021 Nov 17]. Available from: http://www.allelefrequencies.net/top10freqs.asp
16. Immune Epitope Database and Analytics Resource [Internet]. Selecting thresholds (cut-offs) for MHC class I and II binding predictions. 2014 [cited 2022 Jan 13]. Available from: https://help.iedb.org/hc/en-us/articles/114094151811-Selecting-thresholds-cut-offs-for-MHC-class-I-and-II-binding-predictions
17. Lamiable A, Thévenet P, Rey J, Vavrusa M, Derreumaux P, Tufféry P. PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016;44(W1):W449–54.
18. Shen Y, Maupetit J, Derreumaux P, Tufféry P. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J Chem Theory Comput. 2014;10(10):4745–58.
19. Thévenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tuffery P. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012;40(W1):W288–93.
20. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–42.
21. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–303.
22. Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh SG. Ipd-imgt/hla database. Nucleic Acids Res. 2020;48(D1):D948–55.
23. Doménech N, Henderson RA, Finn OJ. Identification of an HLA-A11-restricted epitope from the tandem repeat domain of the epithelial tumor antigen mucin. J Immunol. 1995;155(10):4766–74.
24. Vollers SS, Stern LJ. Class II major histocompatibility complex tetramer staining: progress, problems, and prospects. Immunology. 2008;123(3):305–13.
25. Matsushita M, Otsuka Y, Tsutsumida N, Tanaka C, Uchiumi A, Ozawa K, et al. Identification of novel HLA-A*24:02-restricted epitope derived from a homeobox protein expressed in hematological malignancies. PloS One. 2018;11(1):e0146371.
26. Hilton HG, McMurtrey CP, Han AS, Djaoud Z, Guethlein LA, Blokhuis JH, et al. The intergenic recombinant HLA-B*46:01 has a distinctive peptidome that includes KIR2DL3 ligands. Cell Rep. 2017;19(7):1394–405.
27. Marcilla M, Alpízar A, Lombardía M, Ramos-Fernandez A, Ramos M, Albar JP. Increased diversity of the HLA-B40 ligandome by the presentation of peptides phosphorylated at their main anchor residue. Mol Cell Proteomics. 2014;13(2):462–74.
28. Kaur G, Gras S, Mobbs JI, Vivian JP, Cortes A, Barber T, et al. Structural and regulatory diversity shape HLA-C protein expression levels. Nat Commun. 2017;8:15924.
29. Wauquier N, Petitdemange C, Tarantino N, Maucourant C, Coomber M, Lungay V, et al. HLA-C-restricted viral epitopes are associated with an escape mechanism from KIR2DL2+ NK cells in Lassa virus infection. EBioMedicine. 2019;40:605–13.
30. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–91.
31. Nas JS, Enriquez JG, Villa-Ignacio AJ, Bungay AA, Salunga TL. Molecular docking of putative compounds in aqueous Muntingia calabura L. Leaf extracts with cytochrome P450 proteins. Asian J Biol Life Sci. 2022;11(1):137.
32. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–61.
33. Marco KPD, Llagas JPB, Barzaga MTA, Heralde III FM. In silico prediction of SARS-CoV-2 epitopes for vaccine development. Philipp J Health Res Dev. 2020;24(4):1–19.
34. Drijvers E, De Roo J, Martins JC, Infante I, Hens Z. Ligand displacement exposes binding site heterogeneity on CdSe nanocrystal surfaces. Chem Mater. 2018;30(3):1178–86.
35. Bawar KM, Cruz LP, Ilao KB, Justiniano JM, Panganiban LM, Fabito DL, et al. In silico identification of novel compounds as quorum-sensing inhibitors in selected waterborne pathogens. Asian J Biol Life Sci. 2021;10(2):367.
36. Stothard P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques. 2008;28:1102–4.
37. Bagaev DV, Vroomans RM, Samir J, Stervbo U, Rius C, Dolton G, et al. VDJdb in 2019: database extension, new analysis infrastructure and a T-cell receptor motif compendium. Nucleic Acids Res. 2020;48(D1):D1057–62.
38. Jensen KK, Rantos V, Jappe EC, Olsen TH, Jespersen MC, Jurtz V, et al. TCRpMHCmodels: structural modelling of pMHC-TCR class I complexes. Sci Rep. 2019;9(1):1–12.
39. Desta IT, Porter KA, Xia B, Kozakov D, Vajda S. Performance and its limits in rigid body protein-protein docking. Structure. 2020;28(9):1071–81.
40. Heo L, Lee H, Seok C. GalaxyRefineComplex: refinement of protein-protein complex model structures driven by interface repacking. Sci Rep. 2016;6(1):1–10.
41. Honorato RV, Koukos PI, Jiménez-García B, Tsaregorodtsev A, Verlato M, Giachetti A, et al. Structural biology in the clouds: the WeNMR-EOSC ecosystem. Front Mol Biosci. 2021;708:729513.
42. Nas JS. Predicting short peptide immunogenic B cell epitopes distinct in RHDV1 and RHDV2 of Oryctolagus cuniculus. Rabbit Genetics. 2020;10(1):1–0.
43. Khan F, Kumar A. An integrative docking and simulation-based approach towards the development of epitope-based vaccine against enterotoxigenic Escherichia coli. Netw Model Anal Health Inform Bioinform. 2021;10(1):1–10.
44. Engelhard VH. Structure of peptides associated with class I and class II MHC molecules. Annu Rev Immunol. 1994;12(1):181–207.
45. Wieczorek M, Abualrous ET, Sticht J, Álvaro-Benito M, Stolzenberg S, Noé F, et al. Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front Immunol. 2017;8:292.
46. Rock KL, Reits E, Neefjes J. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol. 2016;37(11):724–37.
47. Garstka MA, Fish A, Celie PH, Joosten RP, Janssen GM, Berlin I, et al. The first step of peptide selection in antigen presentation by MHC class I molecules. Proc Natl Acad Sci. 2015;112(5):1505–10.
48. Wilson CC, McKinney D, Anders M, MaWhinney S, Forster J, Crimi C, et al. Development of a DNA vaccine designed to induce cytotoxic T lymphocyte responses to multiple conserved epitopes in HIV-1. J Immunol. 2003;171(10):5611–23.
49. Bui HH, Sidney J, Li W, Fusseder N, Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinform. 2007;8(1):1–6.
50. Nas JS. Exploring the binding affinity and non-covalent interactions of anthocyanins with aging-related enzymes through molecular docking. Phil J Health Res Dev. 2020 Sep 28;24(3):9–19.
51. Nas JS, Sanchez A, Bullago JC, Fatalla JK, Gellecanao Jr F. Molecular interactions of cyanidin-3-glucoside with bacterial proteins modulate the virulence of selected pathogens in Caenorhabditis elegans. Asian J Biolo Life Sci. 2021 Jan;10(1):151.
52. Rasmussen M, Harndahl M, Stryhn A, Boucherma R, Nielsen LL, Lemonnier FA, et al. Uncovering the peptide-binding specificities of HLA-C: a general strategy to determine the specificity of any MHC class I molecule. J Immunol. 2014;193(10):4790–802.
53. Di Marco M, Schuster H, Backert L, Ghosh M, Rammensee HG, Stevanovi? S. Unveiling the peptide motifs of HLA-C and HLA-G from naturally presented peptides and generation of binding prediction matrices. J Immunol. 2017;199(8):2639–51.
54. Quiros-Fernandez I, Poorebrahim M, Fakhr E, Cid-Arregui A. Immunogenic T cell epitopes of SARS-CoV-2 are recognized by circulating memory and naïve CD8 T cells of unexposed individuals. EBioMedicine. 2021;72:103610.
55. Kumar N, Admane N, Kumari A, Sood D, Grover S, Prajapati VK, et al. Cytotoxic T-lymphocyte elicited vaccine against SARS-CoV-2 employing immunoinformatics framework. Sci Rep. 2021;11(1):1–14.
56. Jain R, Jain A, Verma SK. Prediction of epitope based peptides for vaccine development from complete proteome of novel corona virus (SARS-COV-2) using immunoinformatics. Int J Pept Res Ther. 2021;27(3):1729–40.
57. Rakib A, Sami SA, Mimi NJ, Chowdhury MM, Eva TA, Nainu F, et al. Immunoinformatics-guided design of an epitope-based vaccine against severe acute respiratory syndrome coronavirus 2 spike glycoprotein. Comput Biol Med. 2020;124:103967.
58. Sarma VR, Olotu FA, Soliman ME. Integrative immunoinformatics paradigm for predicting potential B-cell and T-cell epitopes as viable candidates for subunit vaccine design against COVID-19 virulence. Biomed J. 2021;44(4):447–60.
59. Mock JY, Oh J, Yi J, Daris ME, Hamburger A, Kamb A. Design of TCR structural variants that retain or invert the normal activation signal. ImmunoHorizons. 2021;5(5):349–59.