
INTRODUCTION
Diabetes mellitus is a global pertinacious endocrine and metabolic disorder with a significant escalation in the number of new cases leading to increased mortality rates as reported by International Diabetes Federation (2021). It is featured by chronic hyperglycemia due to the insubstantial insulin secretion from the pancreatic beta cells resulting in aberrations in the metabolism of lipids, proteins, and carbohydrates, and is further worsened by organ damage. The prolonged administration of antidiabetic drugs has detrimental effects like gastrointestinal (GI) disorders and obesity, prompting researchers to look for newer safe alternatives. Aldose reductase (AR) (EC 1.1.1.21) is a prototypical rate-limiting monomeric enzyme in the polyol pathway of glucose metabolism. The protein is a triose phosphate isomerase structural motif (315 amino acid residues) that accommodates 10 peripheral α-helical segments enclosing an inner barrel of β-pleated sheet segments and an active site for catalysis. The cofactor nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) is positioned on the apex of the β/α barrel structure, which aids the enzyme in reducing glucose to sorbitol in the polyol pathway (Grewal et al., 2016). AR is not found in all mammalian cell types but resides in insulin-independent tissues such as the kidney, heart, lens, retina, vasculature, Schwann cells of peripheral nerves, placenta, RBC, testis, liver, ovary, cardiac, and brain owing to prolonged diabetic complications (Alexiou et al., 2006; Grewal et al., 2016). Under normal conditions (3.8–6.1 mmol/l), the enzyme hexokinase phosphorylates the glucose in the mammalian cells into glucose 6-phosphate, which further takes part in glycolysis, and this conversion occurs to a larger extent. In contrast, the non-phosphorylated glucose enters the polyol pathway only in trivial amounts (about 3%). Conversely, under hyperglycaemic conditions (>7 mmol/l), exaggerated flux via the polyol pathway constitutes beyond 30% of glucose metabolism (Tang et al., 2012). The hyperglycaemic condition stimulates the activity of AR enormously. It thus promotes glucose metabolism by activating polyol pathway in target tissues (Tang et al., 2012) like nerves, retina (Srivastava et al., 1984), kidney (Ansari et al., 1991; Ohta et al., 1991), placenta (Das and Srivastava, 1985; Vander et al., 1990), RBC (Das and Srivastava, 1985), liver (Petrash and Srivastava, 1982), and heart (Vander Jagt et al., 1990). The resulting sorbitol cannot diffuse rapidly through cell membranes because it is highly hydrophilic and polyhydroxy alcohol. The gradual build-up of sorbitol generates osmotic stress in the retina, kidney, and sciatic nerves, thereby contributing to the progression of diabetic complications like retinopathy, nephropathy, and neuropathy, respectively (Grewal et al., 2016). The cofactor NADPH is critical for the output of antioxidant glutathione (GSH) intracellularly. NADPH is utilized by AR, which brings forth abatement in the levels of NADPH, eventually leading to the decline in GSH. Consequently, the depleted levels of NADPH/NADP+ ratio and reduced NAD+ potentially produce excessive reactive oxygen species (ROS) and evokes oxidative stress (Brownlee, 2001). Usually, the ROS-generated toxic aldehydes are reduced to inactive alcohols by AR. However, during hyperglycemia, AR transforms the excess glucose to sorbitol which is further oxidized to fructose by succinate dehydrogenase (Brownlee, 2001; Grewal et al., 2016).
The aberrant activation of the polyol pathway seems to be the plausible mechanism for tissue-based pathologies and chronic disorders in diabetic patients. AR plays an influential role in these serious predicaments, considering that innumerable AR inhibitors (ARIs) have emerged as potential therapeutic drug candidates. Moreover, adapting ARIs has been proven to be an assured strategy to impede significant complications like atherosclerosis, sepsis, asthma, uveitis, ovarian cancer, and colon cancer as these inhibitors recede the sorbitol flux in polyol pathway of glucose metabolism (Grewal et al., 2016; Lorenzi, 2007; Sever, 2021). A significant effort has been made over the years to report a plethora of ARIs that neutralize the associated pathologies and these ARIs were further evaluated in preclinical trials and quite a few have been progressed to the late phase of clinical trials. Although certain ARIs proved effective in the in vitro studies, there are growing concerns about the undesirable effects owing to low in vivo efficacy, skin allergic reactions, and hepatic toxicities (Meyler, 2016). So, there is an actual need to develop risk-free and effective medications.
Presently, epalrestat (a carboxylic acid derivative) is the only commercially available ARI in Japan since 1992 to manage diabetic neuropathy and it was recently authorized for marketing in China and India (Grewal et al., 2016). Quercetin is an effective antioxidant but has low oral bioavailability (BA) due to its high hydrophilic nature (Durán-Iturbide et al., 2020). Many ARIs were discontinued in the development phase because of low clinical efficacy issues and poor pharmacokinetic profiles. For instance, tolrestat exhibited poor efficacy in clinical trials and was subsequently withdrawn as this compound brought forth fatal hepatic necrosis (Lagorce et al., 2017). Though sorbinil was reported to be a safe ARI for human use after comprehensive in vitro and in vivo tests, it was later taken off the market for augmenting hypersensitivity reactions. These challenges have prompted us to search for a wide diversity of new ARIs devoid of severe adverse impacts. In the present study, we summarized the explored ARIs cited in the literature (Grewal et al., 2016) and generated their respective pharmacophores. Through in silico analysis, we simultaneously validated their absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties and the best binding affinity of those compounds to target AR. In the final analysis, two compounds viz., ZINC89259516 (Butein pharmacophore) and ZINC13349982 (Benzylisoquinoline pharmacophore) have emerged from this study which could be quite promising to combat diabetes and associated complications.
MATERIALS
Computational tools and database sources
The crystal structure of the AR enzyme was retrieved from the RCSB PDB database (https://www.rcsb.org/structure/1t41). The protein structure was pre-processed, and a consequent molecular docking study was performed using AutoDock4.2 (MGL tools version 1.5.6) (https://autodock.scripps.edu/download-autodock4/). The binding affinity of the ligand with the target binding site was predicted using this ADT4.2 software suite. The ligands were obtained from the ZINCPharmer database (http://zincpharmer.csb.pitt.edu/pharmer.html), and all the desirable ligands were saved in 3D conformer from the web-based server Pubchem (https://pubchem.ncbi.nlm.nih.gov). Open Babel (https://github.com/openbabel/openbabel/releases/tag/openbabel-3-1-1) was used to interconvert the chemical file formats of ligands from .sdf to .pdb format. Publicly accessible online tools like SwissADME (http://www.swissadme.ch/), ADMETlab2.0 (https://admetmesh.scbdd.com/), PreADMET (https://preadmet.webservice.bmdrc.org/) and ProTox- II (https://tox-new.charite.de/protox_II/) were used for ADMET profiling by incorporating the canonical SMILES (Simplified Molecular Input Line Entry System) procured from the PubChem database. The 2D and 3D visualization of the docked protein-ligand complexes and their intermolecular interactions were screened using BIOVIA Discovery Studio visualizer v21.1.0.20298 (https://discover.3ds.com/discovery-studio-visualizer-download) and AutoDock softwares.
EXPERIMENTAL
Generation of pharmacophore models
ZINCPharmer database works on the similarity search and produces pharmacophore models to analyze the accurate 3D structure of the preferred ligand-protein interaction pattern. This uses an array of ligand descriptors such as aromatic rings, hydrogen bond donors (HBD), hydrogen bond acceptors (HBA), hydrophobic areas, and charge transfer. Coordinates define ligands, and the interaction of these coordinates with the protein of interest signals the best fit for biological activity.
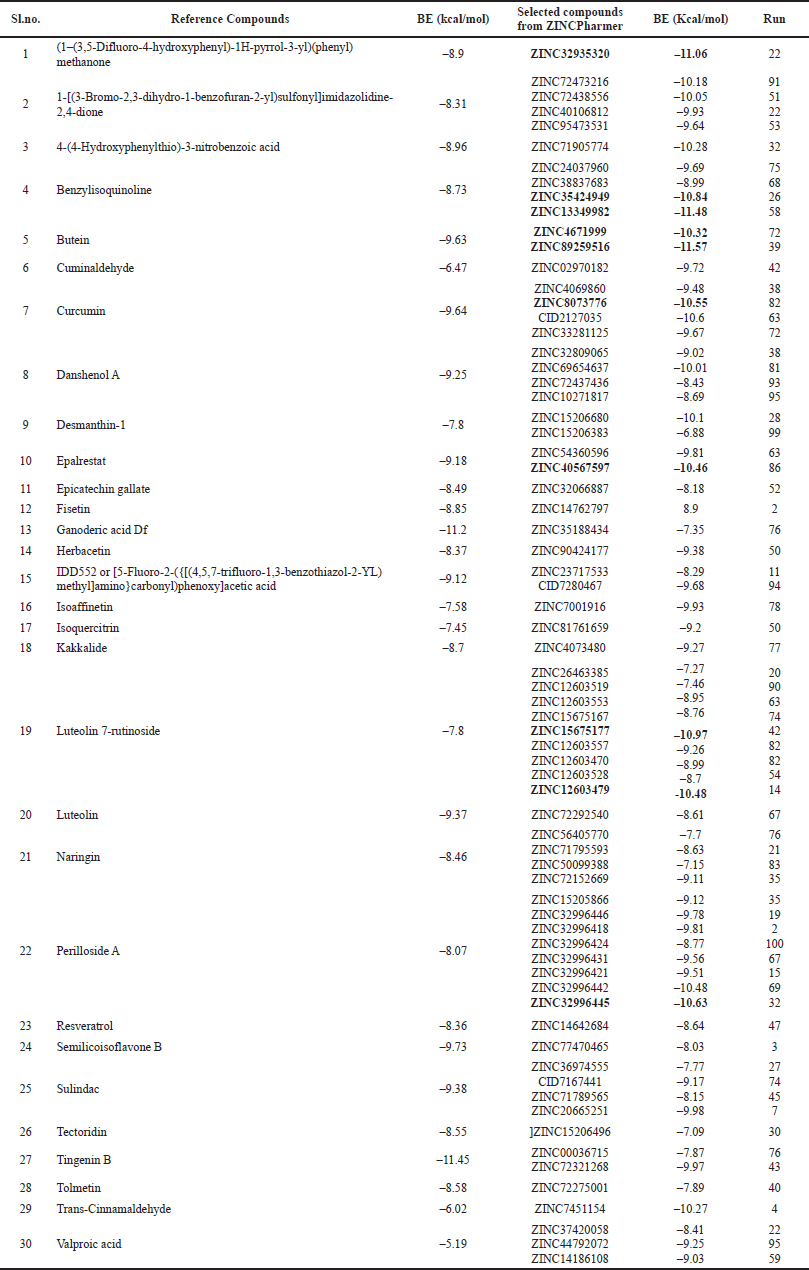 | Table 1. Comparative binding energies (kcal/mol) of the reference compounds and their corresponding ZINC IDs. [Click here to view] |
Dataset of ARIs
Thirty diverse ARIs were retrieved from the literature (Grewal et al., 2016) (Table 1). We explored 418 hit compounds with the extremity of likeness to the query pharmacophores by utilizing the ZINCPharmer database. Out of the in silico ADMET study, 70 potential hit compounds were selected and further considered for docking analysis (Fig. 1).
Prediction of physicochemical, pharmacokinetics, and druglikeness features of compounds
ADME profiling
The SMILES notation of 418 desired compounds were derived from PubChem and these strings were used to predetermine a pool of ADMET parameters cited below. The quantitative estimate of druglikeness (QED) and other physicochemical descriptors such as the molecular weight (MW), topological polar surface area (TPSA), number of hydrogen bond donors (nHBD), number of hydrogen bond acceptors (nHBA), solubility (LogS), distribution coefficient (LogD), partition coefficient (LogP), number of rotatable bonds (nRB), Caco2 permeability, plasma protein binding (PPB), clearance (CL), half-life (T1/2) were predicted via ADMETlab2.0 web server.
The pharmacokinetic parameters were also predicted for the absorption and distribution of drugs inside the body using the SwissADME (Swiss Institute of Bioinformatics, Switzerland) web tool. These properties include blood–brain barrier (BBB) permeation, GI absorption, cytochrome P450 (CYP) isoforms inhibition, plasma glycoprotein (P-gp) substrate, molar refractivity (MR), skin permeability (Log Kp), and BA score. Other criteria, such as plasma glycoprotein (P-gp) inhibitor and human ether-a-go-go-related gene (hERG) blockers, were estimated using the openly accessible PreADMET tool. The selected compounds were also evaluated for their compliance with the basic rules like Lipinski, Pfizer, Veber, Egan, GSK, and Golden Triangle as a general rule of thumb for druglikeness properties.
Toxicity screening by ProTox-II
Toxicity check dominates ADME analysis and turns out to be a crucial determining element since toxic drugs detrimentally affect the advancement of drugs (Lagorce et al., 2017; Maliehe et al., 2020). A few significant parameters like hepatotoxicity, carcinogenicity, immunotoxicity, Ame’s mutagenicity, cytotoxicity, median lethal dose (LD50), and acute toxicity class (class 1 to 6) were predicted using an instantly available ProTox-II server to evaluate the toxicity profile of the selected ligands. The molecular properties and toxicological endpoints assessed from all the aforementioned web tools were deemed to screen the best drug candidate among the selected compounds.
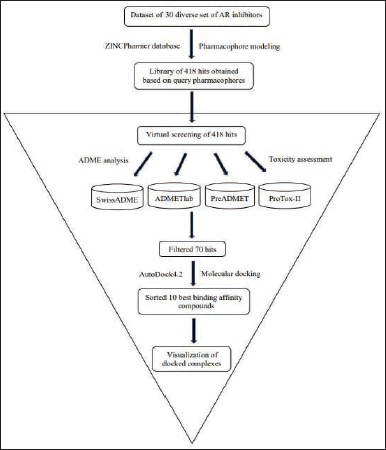 | Figure 1. Schematic workflow summarizing the virtual screening process implemented in the identification of the best leading compound. [Click here to view] |
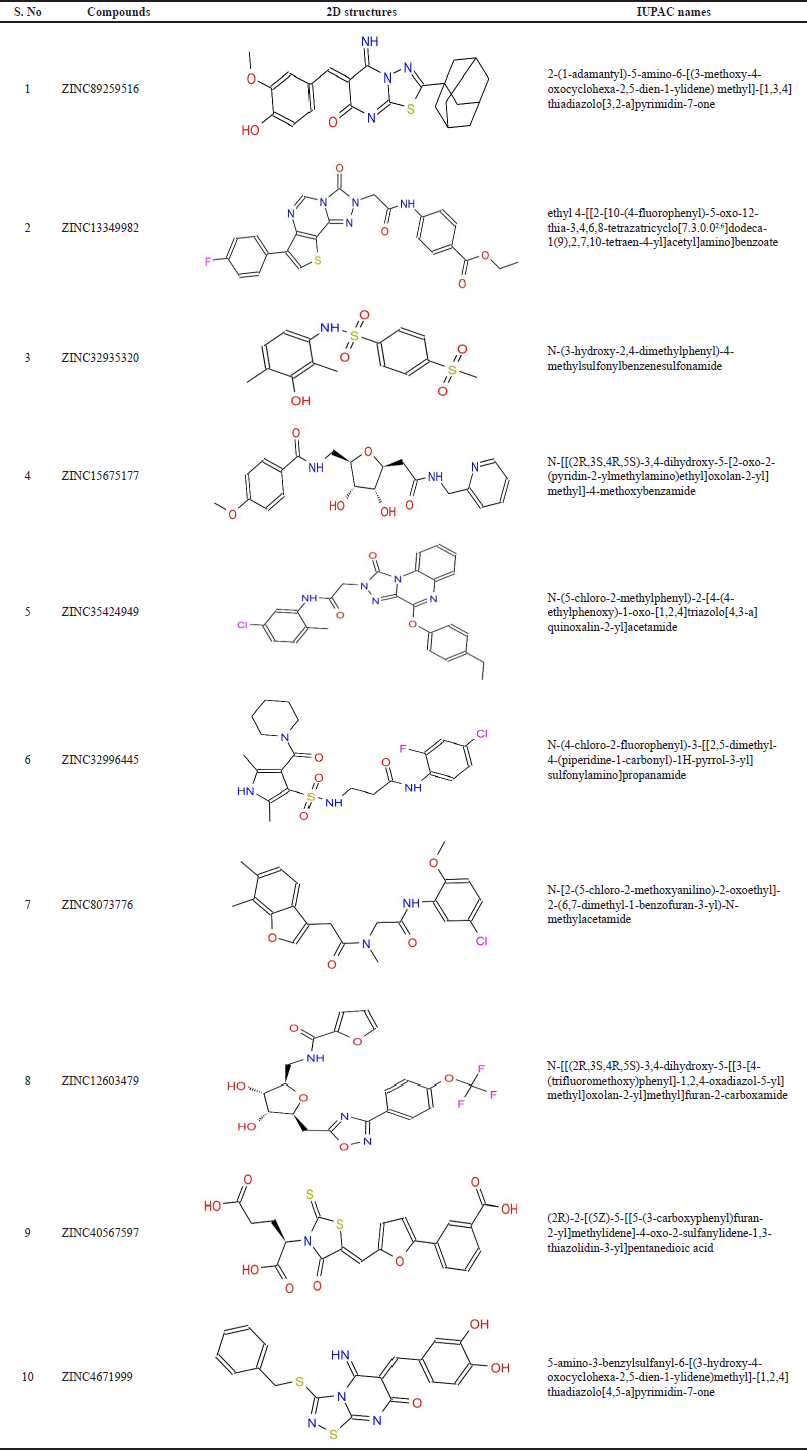 | Table 2. Chemical structures and IUPAC names of the top-scored compounds. [Click here to view] |
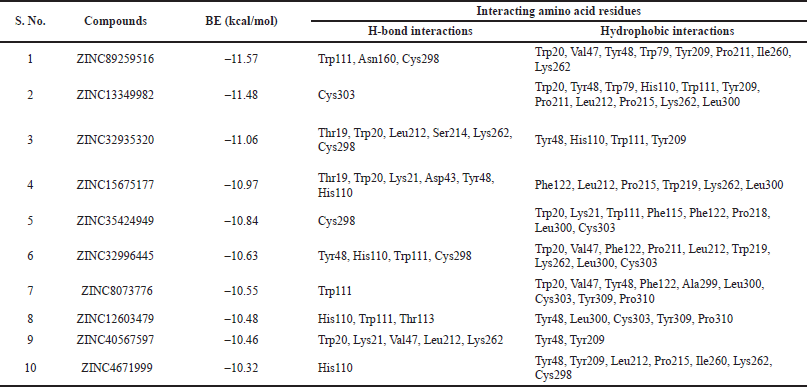 | Table 3. Docking results of the best ten compounds ranked based on their lowest to highest binding energies. [Click here to view] |
Molecular docking
Protein preparation
The X-ray crystallographic structure of human AR complexed with NADP and IDD552 (PDB ID: 1T41) was retrieved in PDB format from the RCSB PDB database. This template was additionally formatted using AutoDock and optimized for docking studies. The preferred resolution for docking was less than 2 Å. The protein structure was refined by deleting water molecules and heteroatoms, as retaining them could prove problematic in further analysis. Polar hydrogen atoms were added because there might be higher chances that few H-atoms are missed during protein structure determination. This is essential for the accurate computation of partial atomic charges and finding the ligand’s binding affinity against the protein. Furthermore, Kollman charges were added to the protein, missing atoms were repaired, and the protein was assigned AD4 type. A target.pdbqt file competent enough for docking was generated from the conventional PDB file.
Ligand preparation
A total of 418 hits were selected based on all the query pharmacophores by virtual screening via ZINCPharmer. The 70 compounds filtered in conformity with ADMET study were edited in ADT software. Open Babel program was used to convert the .sdf format of the 3D conformer ligands (downloaded from PubChem) into .pdb format. Gasteiger charges were computed, and the number of active torsions was set for each ligand, generating a ligand.pdbqt file.
Protein-ligand docking
The existing target and ligand .pdbqt files were chosen. In the best interest of performing blind docking, a grid box was set up to enclose the target enzyme’s entire surface to scan for the available binding sites. The docking parameters were configured by applying the Lamarckian Genetic Algorithm (LGA) to generate rigid conformations of the compound. Docking runs were fixed to 100 with a population size of 300 individuals and a maximum number of 2,500,000 energy evaluations to achieve an ideal crystallographic pose of the ligand and accurate outcomes. After running the AutoDock program, a docked log file was generated comprising root mean square deviation (RMSD) scores and all the binding poses inclusive of their binding affinities. The docking pose exhibiting the least binding energy (BE) was considered the best pose as it represents a stronger binding affinity to the target enzyme. The 2D and 3D visualization and analysis of the docked protein-ligand complexes were carried out using the Discovery studio visualizer.
RESULTS AND DISCUSSIONS
In silico analysis of physicochemical, pharmacokinetic and druglikeness properties
In silico ADMET evaluation is a streamlined approach to ensure the potentiality of the selected compounds for further exploration of therapeutic drug candidates. A compound is advised to be an ideal drug candidate for metabolism if it complies with the ADME profile.
Absorption
Analytical drug design is essentially based on Lipinski’s rule of five, which estimates the drug-like nature of the molecules. All the tested compounds listed in Table 4 were found to hold MW < 500, indicative of ameliorated absorption from the GI tract, diffused and transported (Daina et al., 2017; Srimai and Ramesh, 2013). Other molecular descriptors, including nRB (vital filter to measure molecular flexibility and good BA), nHBA, nHBD, and LogP, have no deviations from the acceptable norms. Compounds with TPSA > 140 Å were presumed to be poorly absorbed, while those with TPSA < 140 Å suggested good absorption (Table 5). The obtained results favor Lipinski’s rule and are assumed to be agreeable oral bioavailable agents. All compounds also adhered to Pfizer’s and GSK’s rules (Supplementary Table 1). Solubility influences the absorption and distribution of active drugs (Lagorce et al., 2017; Stegemann et al., 2007). Compounds with high aqueous solubility can easily permeate across cell membranes and exhibit good absorption (Daina and Zoete, 2016; Srimai and Ramesh, 2013). The computed prediction of LogS showed that compound 2 (–1.697) and compound 4 (–1.551) have high solubility. Compounds 1, 3, 9, and 10 have moderate solubility with the recognized values of −3.666, −3.337, −3.052 and −3.662, respectively, whereas compounds 5, 6, 7, and 8 were found to be poorly soluble (Table 4). We predicted Caco-2 Permeability, P-gp substrate, P-gp inhibitor, 20% BA (F20%), and 30% BA (F30%) to evaluate the extent of absorption of the compounds. The calculated data of Caco-2 permeability implied low to moderate permeability, ranging from –4.758 to −6.202 cm/s.
Distribution
Egan BOILED-Egg (Brain Or IntestinaL EstimateD permeation method) is a distinct 2D graphical view displayed using SwissADME, which concomitantly prefigure the inhibitors permeation in both the GI tract and brain. In an Egan’s egg graph (Fig. 5), the yellow (yolk) and white ellipses symbolize the BBB permeant molecules and GI permeant molecules, respectively (Lynch and Price, 2007). The results signified that all compounds, exclusive of compounds 7 and 8, predicted a high potential for GI absorption. Only compound 10 exhibited BBB penetration, and the rest of the compounds were incapable of passing across the BBB, inferring little detrimental impact on central nervous system (CNS) (Table 6). Hence, BBB permeant drugs may not be recommended for peripheral targets to avert the CNS side effects. P-glycoprotein (P-gp) is a membrane-spanning transport protein that governs the influx and efflux of miscellaneous drugs. Compounds 1, 2, 5, 7, and 10, highlighted in red circles, were estimated to be non-substrates for P-gp (Table 6), meaning there is no effluence of compounds from the cells, while compounds 3, 4, 6, 8, and 9, visible in blue dots, act as P-gp substrates (Fig. 5).
Metabolism
Drug metabolism and elimination in the human body is a top priority mechanism catalyzed by CYP enzymes predominantly sited in the liver and intestine. CYP450 monooxygenase isoforms, namely; CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4, are fundamental determinants in the optimization of curative effectiveness and lethality of numerous drug candidates. Compounds estimated as non-inhibitors of CYP isoforms have a strong probability of getting metabolized and serve as exceptionally good oral bioavailable agents. In contrast, CYP isomer inhibitors cause metabolic failure and show undesirable adverse effects being the cause of poor BA (Srimai and Ramesh, 2013; Lynch and Price, 2007). The induction and inhibition of these enzymes by multifarious compounds were predicted and tabulated in Table 6. All compounds were revealed to be non-inhibitors of CYP1A2 except compound 1. About 50% of the compounds were non-inhibitors of CYP2C9, and CYP3A4, and 40% of the compounds were inhibitors of CYP2C19, and likewise, CYP2D6 was inhibited only by compound 6. The skin permeability (LogKp) parameter evaluates the compounds that might necessitate transdermal administration (Srimai and Ramesh, 2013). The LogKp values of the selected compounds (Table 6) were in the range of –9.01 to –6.2 cm/s, which slightly varied from the recommended range (−0.7 to −5 cm/s), assumed to be impermeable.
Excretion
Total CL of the drug is a hybrid parameter that influences half-life (T1/2) (along with VD) and BA (along with oral absorption), thereby determining the frequency of clinical drug dosage to sustain a plateau drug concentration (Berellini et al., 2012; Lagorce et al., 2017). Compounds 1, 5, 8, and 10 showed moderate CL displaying values within the optimum range of 5–15 ml/minute/kg compared to other compounds that showed poor CL of <5 ml/minute/kg (Table 7).
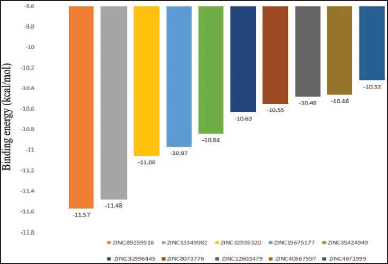 | Figure 2. Bar plot of binding energies of the top-scored 10 compounds. [Click here to view] |
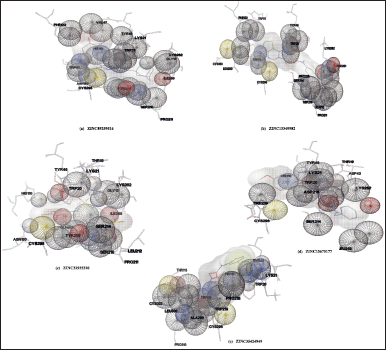 | Figure 3. Wire mesh surface view of the docking interactions of compounds generated by Autodock. Portions of AR that are in contact with the ligand are shown with space-filling spheres. [Click here to view] |
CL = Rate of drug elimination from plasma / Plasma concentration of the drug.
BA of the compounds
It is proposed that the compounds displaying BA score of 0.55 signify 55% probability of being bioavailable and adhere to Lipinski’s RO5 (Lipinski et al., 2001; Srimai and Ramesh, 2013). This study screened all compounds as good indicators of active drugs with adequate oral absorption (Table 7). The computed QED scores of the compounds are provided in Table 7. Amongst the test compounds, compounds 1, 3, 10 were interpreted as attractive drug candidates (QED score > 0.67), compounds 2, 6, 9 as moderate drugs (score 0.54–0.65), and compounds 4, 5, 7, 8 as unattractive drugs (score < 0.54).
Toxicological predictions
The toxicity of the compounds is an integral step in the drug discovery process as this attribute scrutinizes the organ damage (hepatotoxicity) and toxicological endpoints viz; immunotoxicity, carcinogenicity, mutagenicity (Ames test), and cytotoxicity (Mishra et al., 2016; Srimai and Ramesh, 2013). The median LD50 values were computed in mg/kg, and they were interpreted as six acute toxicity classes viz., Class I and II—fatal if consumed, Class III—toxic if consumed, Class IV—harmful if consumed, Class V—perhaps harmful if consumed, Class VI—non-toxic. In this present study, we computed these descriptors for all the selected compounds using ProTox-II. We tested for compliance with being non-toxic which could serve as the basis for appropriate utilization as therapeutic drug candidates. Table 8 summarizes the findings that remarkably revealed all the test compounds to be non-mutagenic, non-cytotoxic, non-carcinogenic, non-immunotoxic, and non-hepatotoxic. LD50 values were noted to be within 611–5,000 mg/kg limits. Compounds 1, 2, 3, 7, and 9 were categorized as Class V, whereas other compounds 4, 5, 6, 8, and 10 belong to class IV (Table 8). The hERG is used to assess the pro-arrhythmic risk of novel drug candidates (Srimai and Ramesh, 2013; Yu et al., 2016). The compounds were revised for another descriptor, hERG using an online accessible server PreADMET. All of them were presumed safe as they exhibited low to medium risks of blocking the hERG channel. This assessed data is tabularized in Table 9.
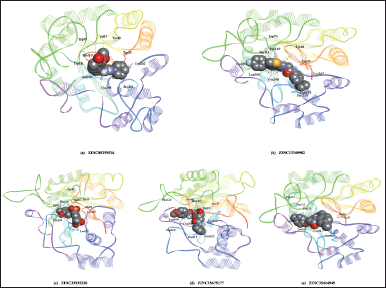 | Figure 4. 3D visualization of the best five docking poses of different compounds in the binding pocket of AR. Protein residues are depicted in line ribbon form whereas the docked ligands are presented in CPK model. [Click here to view] |
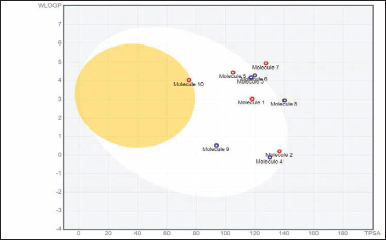 | Figure 5. Evaluation of analysed compounds by BOILED-Egg approach. [Click here to view] |
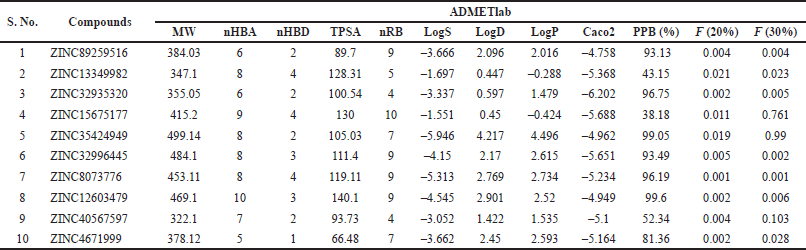 | Table 4. Parameters evaluated for absorption of the selected compounds. [Click here to view] |
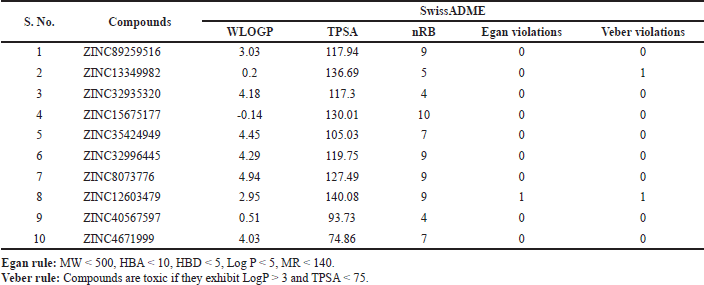 | Table 5. Predicted druglikeness of the compounds based on Egan and Veber rules. [Click here to view] |
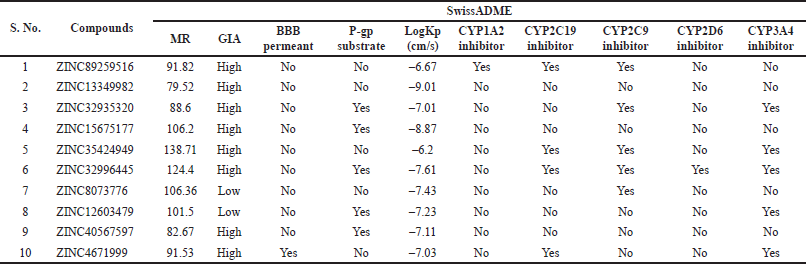 | Table 6. Parameters evaluated for distribution and metabolism of the compounds. [Click here to view] |
Molecular docking studies
Molecular docking results in the generation of BE and is considered a primary parameter to delineate the strength and affinity of the ligand-protein interactions. BE and ligand’s affinity for the target protein are inversely correlated. Therefore, if the BE is low, it implies a high ligand’s affinity for the target protein and vice versa. In our present docking study, we intended to quest for the ligand that reveals the least BE thus demonstrating the greatest affinity among the test compounds. After successful docking of all the test compounds, the generated ligand-protein complex models were analyzed with substantive parameters such as BE, hydrogen bond interactions, hydrophobic interactions (π-π interactions/alkyl/π-sigma/π-alkyl interactions), RMSD of binding site residues and orientation of the docked compound within the binding site.
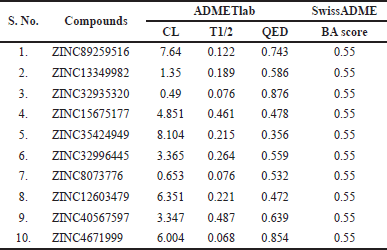 | Table 7. Predicted BA and excretion parameters of the filtered compounds. [Click here to view] |
As summarized in Table 1, the reference compounds and their Zinc IDs represented BE with the target AR ranging between −11.57 and −6.88 kcal/ mol. Out of 70 ZINC IDs, the 10 best binding affinity compounds (Table 2) were ranked in consonance with the docking scores. The intermolecular interactions between AR and the top 10 compounds were analyzed (Table 3) and their binding energies are comprehensively illustrated in a bar graph (Fig. 2). The docking interactions of the top five scored compounds with the target AR are shown in Figure 3. The in silico docking analysis evidenced that the top-scored compound 1 (ZINC89259516) exhibited the least BE score of –11.57 kcal/mol, which was confirmed to have a strong binding affinity for the target AR. This lead compound (ZINC89259516) evinced significant hydrogen bond interactions in the binding pocket of AR with key amino acid residues viz., Trp111, Asn160, Cys298 and hydrophobic bonds with Trp20, Val47, Tyr48, Trp79, Tyr209, Pro211, Ile260, and Lys262 (Fig. 4a). Compound 2 formed only one hydrogen bond with Cys303 and hydrophobic interactions with Trp20, Tyr48, Trp79, His110, Trp111, Tyr209, Pro211, Leu212, Pro215, Lys262, and Leu300 with docking score –11.48 kcal/mol (Fig. 4b) whereas compound 3 displayed hydrogen bonds with Thr19, Trp20, Leu212, Ser214, Lys262, Cys298 and hydrophobic bonds with Tyr48, His110, Trp111, Tyr209 with docking score −11.06 kcal/mol (Fig. 4c).
As we aimed at identifying the potent pharmacophores of the reference compounds targeting AR enzyme, we proposed a computational workflow involving the collection of pharmacophores, virtual screening, and molecular docking study which successfully resulted in the lead molecules; ZINC89259516 (Butein pharmacophore) and ZINC13349982 (Benzylisoquinoline pharmacophore). Finally, based on the resulting in silico data, we confirm that these two compounds impressively attributed to high binding affinity endowed with agreeable ADMET profile as compared to the other compounds. In this regard, these molecules may be employed as potent anti-diabetic compounds to inhibit AR, eventually abating diabetic complications. These computational findings could justifiably serve as a great benefit for further exploration of these lead compounds in pre-clinical and clinical trials of the drug discovery process.
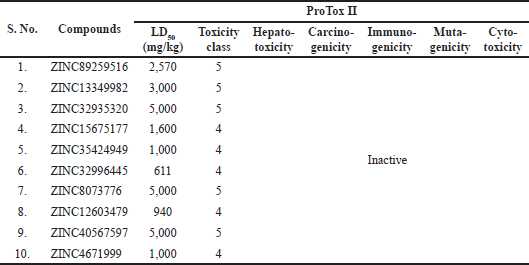 | Table 8. Toxicity prediction of the selected compounds by ProTox II. [Click here to view] |
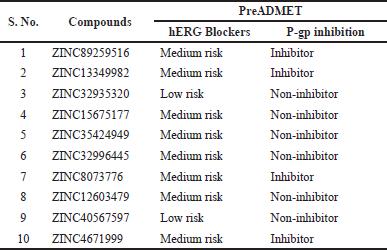 | Table 9. Toxicity prediction of the selected compounds by PreADMET. [Click here to view] |
CONCLUSION
In the current study, we analyzed molecular docking results for the selected compounds which do not deviate from Lipinski and Pfizer rules. Taking into account the outcome, we hereby report two hit molecules that were successfully docked with excellent binding affinity to AR i.e., Butein pharmacophore ZINC89259516 (–11.57 kcal/mol) followed by Benzylisoquinoline pharmacophore ZINC13349982 (–11.48 kcal/mol). Precise insight into these two compounds ADMET assessment profiles and interaction patterns suggested good oral BA, druglikeness, and no toxicity premonitions on the tested criteria. On another note, these potential compounds may get authorized as ARIs only after subsequent approval by follow-up experiments (in vitro and in vivo) on the appropriateness of safe therapeutic antidiabetic drug candidates. As discussed earlier, the activated AR pathway upregulates numerous glucose toxicity pathways such as PKC, HBP, ROS, and AGEs. In this regard, diabetic complications may not be prevented by ARIs alone, but they may serve as potential adjuvant therapy to annul the furtherance of diabetic complications.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
KG is grateful to University Grants Commission for providing UGC-BSR start-up grant No.F.30-546/2021(BSR)- 29/07/2021.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
AVAILABILITY OF DATA AND MATERIALS
All data produced or analyzed during this study are included in this published article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Alexiou P, Pegklidou K, Chatzopoulou M, Nicolaou I, Demopoulos VJ. Aldose reductase enzyme and its implication to major health problems of the 21(st) century. Curr Med Chem, 2009; 16:734–52; doi: https://doi.org/10.2174/092986709787458362
Ansari NH, Bhatnagar AS, Liu Q, Srivastava SK. Purification and characterization of aldose reductase and aldehyde reductase from human kidney. Biochem Int, 1991; 4(25):755–65.
Berellini G, Waters NJ, Lombardo F. In silico prediction of total human plasma clearance. J Chem Inf Model, 2012; 52(8):2069–78; doi: https://doi.org/10.1021/ci300155y
Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature, 2001; 414(6865):813–20; doi: https://doi.org/ 10.1038/414813a
Chung SS, Chung SK. Aldose reductase in diabetic microvascular complications. Curr Drug Targets, 2005; 6(4):475–86; doi: https://doi.org/ 10.2174/1389450054021891
Das B, Srivastava SK. Purification and properties of aldehyde reductases from human placenta. Biochim Biophys Acta, 1985a; 840(3):324–33; doi: https://doi.org/10.1016/0304-4165(85)90212-0
Das B, Srivastava SK. Purification and properties of aldose reductase and aldehyde reductase II from human erythrocyte. Arch Biochem Biophys, 1985b; 2(238):670–9; doi: https://doi.org/10.1016/0003-9861(85)90213-9
Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep, 2017; 7:42717; doi: https://doi.org/10.1038/srep42717
Daina A, Zoete V. A BOILED-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem, 2016; 11(11):1117–21; doi: https://doi.org/10.1002/cmdc.201600182
Durán-Iturbide NA, Díaz-Eufracio BI, Medina-Franco JL. In Silico ADME/Tox profiling of natural products: a focus on BIOFACQUIM. ACS Omega, 2020; 5(2)6:16076–84; doi: https://doi.org/10.1021/acsomega.0c01581
Dunlop M. Aldose reductase and the role of the polyol pathway in diabetic nephropathy. Kidney Int Suppl, 2000; 77:S3–12; doi: https://doi.org/10.1046/j.1523-1755.2000.07702.x
Grewal AS, Bhardwaj S, Pandita D, Lather V, Sekhon BS. Updates on aldose reductase inhibitors for management of diabetic complications and non-diabetic diseases. Mini Rev Med Chem, 2016; 2(16):120–62; doi: https://doi.org/10.2174/1389557515666150909143737
International Diabetes Federation. IDF Diabetes atlas. 10th edition, International Diabetes Federation, Brussels, Belgium, 2021.
Li Q, Hwang YC, Ananthakrishnan R, Oates PJ, Guberski D, Ramasamy R. Polyol pathway and modulation of ischemia-reperfusion injury in type 2 diabetic BBZ rat hearts. Cardiovasc Diabetol, 2008; 7:33; doi: https://doi.org/10.1186/1475-2840-7-33
Lagorce D, Douguet D, Miteva MA, Villoutreix BO. Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci Rep, 2017; 7:46277; doi: https://doi.org/10.1038/srep46277
Lorenzi M. The polyol pathway as a mechanism for diabetic retinopathy: attractive. elusive, and resilient. Exp Diabetes Res, 2007; 61038; doi: https://doi.org/10.1155/2007/61038
Lynch T, Price A, The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Phys, 2007; 76(3):391–6.
Lipinski CA, Lombardo FB, Dominy W, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev, 2001; 46(1–3):3–26, doi: https://doi.org/10.1016/s0169-409x(00)00129-0
Maliehe TS, Tsilo PH, Shandu JS. Computational evaluation of ADMET properties and bioactive score of compounds from encephalartos ferox. Pharmacognosy J, 2020; 12(6):1357–62: doi:10.5530/pj.2020.12.187.
Meyler DC. Side effects of drugs: the international encyclopedia of adverse drug reactions and interactions. Indian J Pharmacol, 2016; 48(2):224.
Mishra S, Sharma CS, Singh HP, Pandiya H, Kumar N. In silico ADME, bioactivity and toxicity parameters calculation of some selected anti-tubercular drugs. Int J Pharm Phytopharmacol Res, 2016; 6(77).
Ohta M, Tanimoto T, Tanaka A. Localization, isolation and properties of three NADPH-dependent aldehyde reducing enzymes from dog kidney. Biochim Biophys Acta, 1991; 3(1078):395–403; doi: https://doi.org/10.1016/0167-4838(91)90162-s
Ohta M, Tanimoto T, Tanaka A. Localization, isolation and properties of three NADPH-dependent aldehyde reducing enzymes from dog kidney. Biochim Biophys Acta, 1991; 3(1078):395–403; doi: https://doi.org/10.1016/0167-4838(91)90162-s
Oates PJ. Aldose reductase, still a compelling target for diabetic neuropathy. Curr Drug Targets, 2008; 9(1):4–36; doi: https://doi.org/10.2174/138945008783431781
Obrosova IG, Chung SS, Kador PF. Diabetic cataracts: mechanisms and management. Diabetes Metab Res Rev, 2010; 26(3):172–80; doi: https://doi.org/10.1002/dmrr.1075
Petrash JM, Srivastava SK. Purification and properties of human liver aldehyde reductases. Biochim Biophys Acta, 1982; 707(1):105–14; doi: https://doi.org/10.1016/0167-4838(82)90402-2
Ramana KV. ALDOSE REDUCTASE: new insights for an old enzyme. Biomol Concepts, 2011; 2(1–2):103–14; doi: https://doi.org/10.1515/bmc.2011.002
Sever B. An extensive research on aldose reductase inhibitory effects of new 4H-1,2,4-triazole derivatives. J Mol Struct, 2021; 1224:129446; doi: https://doi.org/10.1016/j.molstruc.2020.129446
Srivastava SK, Ansari NH, Hair GA, Das B. Aldose and aldehyde reductases in human tissues. Biochim Biophys Acta, 1984; 800(3):220–7; doi; 10.1016/0304-4165(84)90399-4.
Srimai V, Ramesh M, Satya Parameshwar K, Parthasarathy T. Computer-aided design of selective cytochrome P450 inhibitors and docking studies of alkyl resorcinol derivatives. Med Chem Res, 2013; 22:5314–23.
Stegemann S, Leveiller F, Franchi D, De Jong H, Lindén H. When poor solubility becomes an issue: from early stage to proof of concept. Eur J Pharm Sci, 2007; 31(5):249–61; doi: https://doi.org/10.1016/j.ejps.2007.05.110
Tang WH, Martin KA, Hwa J. Aldose reductase, oxidative stress, and diabetic mellitus. Front Pharmacol, 2012; (3):87; doi: https://doi.org/10.3389/fphar.2012.00087
Vander DL, Jagt LA, Hunsaker B, Robinson LA, Stangebye LM, Deck LM. Aldehyde and aldose reductases from human placenta. Heterogeneous expression of multiple enzyme forms. J Biol Chem, 1990a; 19(265):10912–8.
Vander Jagt DL, Robinson B, Taylor KK, Hunsaker LA. Aldose reductase from human skeletal and heart muscle. Interconvertible forms related by thiol-disulfide exchange. J Biol Chem, 1990b; 265(34):20982–7.
Yu HB, Zou BY, Wang XL, Li M. Investigation of miscellaneous hERG inhibition in large diverse compound collection using automated patch-clamp assay. Acta Pharmacol Sin, 2016; 37(1):111–23; doi: https://doi.org/10.1038/aps.2015.143
SUPPLEMENTARY MATERIAL
Supplementary data can be downloaded from the journal’s website