
INTRODUCTION
Diabetes mellitus (DM) is a chronic hyperglycemic metabolic disorder characterized by impaired fat, carbohydrate, and protein metabolism. There is a high risk of severe complications and comorbidities associated with DM, including cardiovascular diseases and kidney failure (Tabish, 2007). Given the increasing prevalence of type 2 diabetes (T2D) worldwide, strict glycemic control is critical to effectively treat T2D and its life-threatening complications (Schwartz, 2013). Despite the availability of various antidiabetic drugs in clinical use, a successful comprehensive cure of DM remains unachievable. In particular, these drugs are associated with serious adverse effects such as hypoglycemia, gastric irritation, nausea, diarrhea, injection phobia, and other factors that make adherence to therapy so difficult (Chaudhury et al., 2017). It is therefore highly desirable to have a long-acting, stable, and invasive-free drug delivery system to reduce the frequency of repeated administration and increase patient compliance.
Pioglitazone hydrochloride (PGZ; molecular weight = 392.9) is a blood glucose-lowering agent known to increase sensitivity to insulin in the liver and periphery and improve glucose uptake and utilization. PGZ increases insulin-dependent glucose clearance, remediates impaired glucose homeostasis, and lowers plasma glucose concentration, plasma insulin levels, and hemoglobin A1c levels in patients with T2D (Kamel et al., 2019). Following the Biopharmaceutical Classification System, the PGZ is classified as a class II drug, which suffers from poor aqueous solubility (0.3–0.4 μg/ml at pH 6.8), slow dissolution rate, and high permeability through biomembranes after oral administration. The mean serum half-life of PGZ ranges from 3 to 7 hours, making it a candidate for controlled release formulation (Elbary et al., 2008). Accordingly, a variety of drug release systems such as nanolipid formulations (Shaveta et al., 2020), proniosomes/niosomes transgels (Prasad et al., 2016), and self-microemulsifying drug release systems (Hyma and Abbulu, 2013) have been studied to increase the solubility of PGZ and consequently improve its bioavailability and therapeutic efficacy.
Microparticles (MPs) are tiny polymeric spheres with diameters ranging from 10 to 1,000 μm that have recently gained attention as advanced biocompatible materials for tissue engineering and drug delivery (Li et al., 2018). A variety of polymers and copolymers are used for the fabrication of MPs. Ideally, the polymers used to produce MPs for biomedical applications should be inert, stable, safe, biodegradable, and biocompatible. In addition, the type of polymers and copolymers used in the formulation of MPs has a significant impact on the MPs’ physicochemical properties, loading efficiency, and drug release kinetics (Song et al., 2018). In addition, MPs could improve the water solubility of poorly soluble drugs, enhance their bioavailability, ensure controlled and sustained drug release (Khalifa and Abdul Rasool, 2017), and decrease drug toxicity (Oliveira et al., 2017). It was found that these carriers were promising in drugs protection and improving their stability (Boni et al., 2021), targeting the drug to a specific biological site, and reducing the number of doses, thereby improving patient adherence to treatment (Abdul Rasool et al., 2014; Bartos et al., 2021).
Lipids are widely used as an excipient in pharmaceutical formulations for protection against moisture and enzymatic degradation, masking unfavorable taste, controlled release, and improving bioavailability (Haider et al., 2020a). Lipid microparticles (LMPs) are prepared using glyceride, fatty alcohol, fatty acid, and solid wax. It was found that LMPs are an effective carrier for encapsulating poorly water-soluble drugs. They showed superior biocompatibility and better control of drug release than polymeric microparticles, which can be hazardous to the body due to the organic solvents used in the preparation process (Haider et al., 2021). Also, MPs have been applied extensively for antidiabetic therapy to improve their properties and therapeutic efficacy (Rafiee and Abdul Rasool, 2022). Nevertheless, LMPs are complex multiphase systems whose properties are strongly influenced by the type and concentration of lipids and surfactants used. In addition, the production of LMPs has some limitations because of the low reproducibility, expensive materials, and complex manufacturing procedure. Fortunately, formulation optimization techniques can be used to determine the variables that control high-quality product characteristics (Abdul Rasool et al., 2021). The “Design of Experiments (DOE)” and “Quality by Design (QbD)” principles enable efficient investigations of critical process and formulation variables; thereby, experimental runs can be reduced, and consequently, more economically viable products can be developed (Haider et al., 2020b).
This study aims to use the QbD principles to develop and optimize PGZ-loaded LMPs as an oral drug delivery system to improve PGZ solubility in the gastric medium and sustain its release for better bioavailability and patient compliance. The MPs were developed using a modified solvent injection technique. The impact of the lipid amount, surfactant type, and surfactant concentration on particle size (PS), entrapment efficiency (EE%), and PGZ release from the prepared PGZ-LMPs was determined and optimized using a D-optimal factorial design to investigate the relationship between their composition and properties. The prepared PGZ-LMPs were studied for their surface topography, possible drug-lipid interactions, drug release kinetics, and antidiabetic activity in rats.
MATERIALS AND METHODS
Materials
PGZ was obtained from Global Pharma (Dubai, UAE). Cetyl alcohol (CA), poloxamer (Pluronic® F-86), polysorbate (Tween 80®), and sorbitan monooleate (Span 80®) were obtained from Sigma-Aldrich (St. Louis, MO). Potassium dihydrogen phosphate and chloroform were purchased from Fisher Scientific (Pittsburg, PA). Streptozocin (STZ) was acquired from Shanghai Yuanye Biological Technology (Shanghai, China).
QbD approach for optimization of PGZ-LMPs
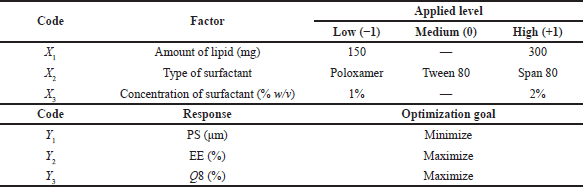
PGZ-LMP characteristics were examined using the Design-Expert® software (Version 12.0, Stat-Ease Inc., Minneapolis, MN) utilizing a D-optimal factorial design. For the experimental design, three distinct, independent factors including amount (mg) of lipid (X1), surfactant type (X2), and concentration of surfactant (X3) were selected as critical material attributes (CMAs) to assess the effect of these formulation parameters on critical quality attributes (CQAs). As a result of the selected levels of the independent variables, the drug-loaded LMPs could be processed in a feasible manner (Table 1).
Next in order, for the optimization of the studied factors and the production of the optimal formula, three responses had been tracked, including PS (Y1), EE% (Y2), and cumulative drug release after 8 hours (Q8) (Y3) (Table 1). The target responses were set at the minimum size of MPs, maximum EE%, and maximum drug release rate in 8 hours. The optimal PGZ-LMP formula was then prepared using the optimal independent variables.
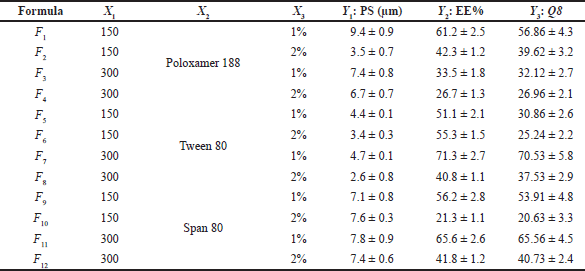
As shown in Table 2, the statistical design software generated 12 different experimental runs (formulations). Experimental runs were conducted randomly to increase predictability and eliminate bias in model variance. The measurements were done in triplicate (n = 3). Polynomial equations were statistically validated by analysis of variance (ANOVA) after the linear, two-factor interaction and quadratic responses were fitted simultaneously.
Preparation of PGZ-LMPs
By adopting the solvent injection technique, PGZ-LMPs were prepared following the experimental design (Schubert and Müller-Goymann, 2003). The procedure was optimized to obtain the fittest stirring speed and time. First, 30 mg of the drug and different amounts of CA were dissolved in ethanol (2 ml), while different amounts of Poloxamer 188, Tween 80, and Span 80 were dissolved in 8 ml of distilled water. The alcoholic solution containing the drug was rapidly injected into the aqueous phase after both solutions were heated to 50°C, followed by continuous stirring at 2,000 rpm for 1 hour. PGZ-LMPs were frozen at −60oC for 4 hours and lyophilized at −50oC and 7 × 10−2 mbar for 48 hours, using Vertical Freeze Dryer BK-FD18S BK-FD18P (China) to yield a dry powder. The powdered samples were then sealed in firmly closed containers, wrapped with parafilm, and stored at 4oC.
 | Table 1. Optimization of PGZ-LMPs based on independent variables (factors) and dependent variables (responses). [Click here to view] |
 | Table 2. DOE and measured responses for PGZ-LMP optimization. [Click here to view] |
Determination of particle size PGZ-loaded microparticles
The average particle size (z-average) of the PGZ-LMP formulations was measured by Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) at a scattering angle of 173o. A sample of 0.2 ml of PGZ-LMP dispersion was diluted with purified water (0.8 ml) and placed in the instrument to measure the PS at room temperature (Abdul Rasool et al., 2020). The PS of PGZ-LMPs was calculated as mean values ± standard deviation (SD) of triplicate measurements (n = 3).
Determination of entrapment efficiency
The loading efficiency of PGZ in the MPs was determined following the preparation of the PGZ-LMP formulae; the MPs were separated from the hydration medium by centrifugation at −10oC and 18 × 103 rpm for 30 minutes. The supernatant was then decanted, and the separated MPs were dissolved in 50 ml of a chloroform/methanol mixture (1:10 v/v) and sonicated for 5 minutes to obtain a clear solution (Bhosale et al., 2016). The amount of the entrapped PGZ was determined by a UV-visible spectrophotometer at λmax 269 nm, and EE% was then computed according to the following formula:
In vitro drug release and kinetics measurements
The in vitro release of PGZ from the prepared LMPs was studied using the dialysis membrane diffusion technique (Al Shuwaili et al., 2016). A sample of 1 ml of each drug-loaded microparticle dispersion, equivalent to 3 mg PGZ, was centrifuged at −9 ± 0.5°C and 15,000 rpm for 30 minutes. The residue was then dispersed in distilled water (1 ml) and placed in the cassette dialysis bags (Slide-A-Lyzer Dialysis Cassette, USA) as a donor compartment fitted with a dialysis membrane of cut-off molecular weight equal to 10,000 da. The dialysis cassettes were then immersed in 150 ml of the dissolution media (0.1 N HCl, pH 1.2) or phosphate-buffered saline (PBS), pH 6.8. The media were stirred continuously at 100 rpm at 37°C ± 0.5. Samples of 3 mL were collected at predetermined time intervals (0.5, 1, 2, 3, 4, 5, 6, 7, and 8 hours) and substituted with the same volume of the freshly prepared phosphate buffer to control the sink condition.
After analyzing the samples spectrophotometrically at 269 nm, the amount of released PGZ at each time interval was measured by converting the absorbance into concentration using the previously prepared PGZ calibration curve in 0.1 N HCl, pH 1.2. The data was normalized against the total amount of PGZ and reported as a percentage of cumulative drug released. The experiments were conducted in triplicate (n =3).
The release kinetics of PGZ from the optimized MPs was studied by fitting the release data to various kinetic models, including the “zero-order kinetics, first-order kinetics, Korsmeyer-Peppas, Hixson–Crowell, Higuchi, and Makoid–Banakar” equations. In addition, the data collected from the in vitro release study were analyzed, and various kinetic parameters were determined using the DDSolver® 2010 add-in program for Microsoft Excel. The model which showed the best fitting to data and could describe the PGZ release mechanism from the MPs was selected based on the correlation coefficient (R2) produced from the linear regression analysis.
Morphological examination
SEM was used to identify the surface topography and morphological features of the optimal PGZ-loaded LMP formula and the blank MPs. The test was performed by placing the dispersion on a clean microscopical glass slide under a vacuum until complete drying. The dried sample was then fixed with conductive tape on a metal stub and coated with a gold-palladium (80%−20%) coat using a Mini Sputter Coater (SC7620, Quorum Technologies, UK). The gold-coated samples were examined at an acceleration voltage of 3 kV, and photomicrographs were taken by a Thermo Scientific Apreo Field Emission SEM (FEI Company, Hillsboro, OR).
FT-IR spectroscopy studies
FT-IR spectroscopy was used to explore the chemical compatibility between the PGZ and the excipients. Samples of the pure drug, the individual formulation constituents (PGZ and CA), their physical mixture, the optimized PGZ-LMPs, and the blank formula were scanned from 4,000 to 400 cm−1 at a resolution of 4 cm−1 using an FT-IR spectrophotometer (JASCO FT-IR 6300, Jasco, Easton, MD). The physical mixture was prepared by mixing an equal molar ratio (1:1) of PGZ and CA with a mortar and pestle for 30 minutes. The stretching and vibrational modes represent the functional groups and the chemical bonding in the tested samples.
DSC analysis
The differential scanning calorimetry (DSC) analysis was conducted to measure possible chemical interactions between PGZ and the excipients in the prepared microspheres. Samples of PGZ, CA, their physical mixture, and the optimized formulation were placed in a flat-bottomed aluminum pan and heated in the DSC-50 (Shimadzu, Kyoto, Japan). To eliminate the oxidative and pyrolytic effects of air, nitrogen gas was pumped into the apparatus. The heating rate was 10°C/min, and the temperature ranged from 0 to 400°C. PGZ-LMPs and physical mixtures were DSC-thermogrammed, and the results were compared with the control groups containing a similar proportion of each component.
Preclinical in vivo study
Animals
The Animal Ethical Committee approved the preclinical study under the Research Unit, Dubai Pharmacy College for Girls (DPCG), Reference No. REC/MPharma/PPD/2019/03. The preclinical study was done at the college’s laboratory. Twenty-five healthy albino Wistar rats weighing between 150 and 250 g were selected for the preclinical in vivo study. The animals were placed separately in cages and cleaned twice daily to ensure their safety and well-being. Three days before the study, the animals were housed in the animal house to adapt to the environment. There was a 12 hour daylight and 12 hour dark cycle in the room, and the temperature was kept between 20°C and 25°C. None of the rats had a history or symptoms of diabetes. Thus, all of them were used to monitor and prove the antidiabetic activity of the prepared PGZ-LMPs. Before the experiment, the rats were fed a regulated fixed diet and fasted for 12 hours. However, the rats had access to only water ad libitum during the test but not to food. The animals were left moving freely throughout the experiment.
Study protocol
The dose of PGZ (10 mg/kg) was decided based on different literature reports (Silva-Abreu et al., 2019; Suke et al., 2013). Furthermore, we conducted a pilot study on a group of two rats before initiating our study to evaluate the acute toxicity of the PGZ-LMP dose for 48 hours after oral administration. It showed no lethal effect on any rat. We concluded that the oral dose of 10 mg/kg is safe to use.
The diabetic model was induced in healthy rats using STZ intraperitoneal injection (45 mg/kg) (Kapoor et al., 2019). Following this, the rats were casually divided into five groups (control group, PGZ solution, marketed PGZ tablets, PGZ-LMPs, and blank LMPs) comprised of five animals each. The animals in the control group were injected with saline to ensure that the rats went through similar conditions. Then, fasting blood glucose level (BGL) was measured by a digital glucometer (GLUCO DR, Korea) after 72 hours of the streptozotocin administration. Fasting BGL value ≥200 mg/dl was regarded as a diabetic condition and was admitted to the experimental study (Shaveta et al., 2020). A dose equivalent to 10 mg/kg body weight was dispersed in saline and administered orally using a feeding cannula. Rats’ lateral tail veins were sampled at 0, 0.5, 1, 2, 3, 4, 6, 8, 10, and 24 hours following treatment administration. The BGL was recorded on the digital glucometer by taking one drop of blood from each rat’s tail and placing it in the glucometer strip. After 24 hours, the % reduction in BGL was calculated using
Different groups’ BGL scores were compared statistically and expressed as mean ± standard deviation (SD)
Statistical analysis
The in vitro experiments were carried out in triplicate, and the results are expressed as mean ±SD. The two-tailed unpaired Student’s t-test and one-way ANOVA were employed to compare two groups and test the equality of several means, respectively. The results were statistically considered significant at p < 0.05. The Design-Expert® software (Version 12.0, Stat-Ease Inc., Minneapolis, MN) or SPSS (SPSS® Statistics software program, Version 17.0, International Business Machines Corp., Armonk, NY) were used for statistical analyses.
RESULTS AND DISCUSSION
Design and preparation of PGZ-LMP formulations
To maximize the therapeutic effect of PGZ, the principle of the quality target product profiles (QTPPs) was constructed based on the characteristics of PGZ-LMPs designed for oral administration (Scalia et al., 2015). Also, depending on our prior experience with the development and optimization of LMP formulations, we achieved the critical process parameters (CPPs), CMAs, and CQAs.
A solvent injection method was used to prepare PGZ-LMPs, which is known for its ease of production and low cost (Schubert and Müller-Goymann, 2003). The stirring speed and stirring time were fixed at 2,000 rpm and 1 hour, respectively, to generate the smallest particle size. The choice of the lipid and stabilizers is very important as they affect the ability of the formulation to improve the drug solubility and control its release. The lipid that we chose, CA, is an FDA-approved solid lipid with suitable pharmaceutical and biological properties. It is biodegradable, biocompatible, and nonimmunogenic and has no taste or smell. Accordingly, it is suitable for incorporation in formulations for oral administration to control drug release (Gupta et al., 2013). Stabilizer addition is essential to facilitate the formation of uniform microparticles, enhance drug entrapment, and prevent aggregation during storage. Surface active agents (SAA) such as Poloxamer 188, Tween 80, and Span 80 were used as stabilizers because of their satisfactory solubilizing and surfactant properties. Therefore, their use is promising in various pharmaceutical formulations (Haider et al., 2020b).
The PGZ-LMPs were fabricated using process parameters selected based on preliminary trials and previous studies. Using the previously described experimental design (Table 2), the response data for each of the 12 experimental runs were obtained and multiple linear regression analysis and ANOVA were used to investigate the individual effects of the independent factors on the selected responses; in addition, a second-order polynomial equation was derived as follows:
Y = βo + β1X1 + β2X2 β3X3 + β11X12 β22X22 β33X32 + β12X1X2 + β13X1X2 + β23X2X3 (3)
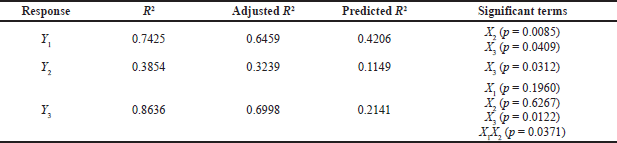
where Y is the response, β1, β2, β3, …, and β23 are the regression coefficients, and X1, X2, and X3 are the studied factors at the specified levels. The equation in terms of coded factors can be used to estimate the response for the given levels of each factor. Moreover, they can be used to identify the relative impact of the factors by comparing the factor regression coefficients. Three and higher-order interactions were not considered. The predicted R2 values were in agreement with the adjusted R2 for all responses, indicating the model’s reliability in estimating all response values (Table 3). Adequate precision was used to measure the signal-to-noise ratio to ensure that the model could navigate the design space. The resultant adequate precision was significant and more than four (the desirable value) for all responses.
Determination of particle size
There is a critical relationship between particle size and the stability of the LMPs, EE%, drug release profile, dissolution rate, drug absorption, cellular uptake, and bioavailability. Smaller particles with a narrow distribution of sizes tend to be more stable and less prone to aggregation and physical instability (Wang et al., 2016). Furthermore, particle size reduction can efficiently enhance the wettability and solubility of drugs, as particle size reduction can increase drugs’ dissolution and bioavailability to a certain extent (Zhang et al., 2020). The graphical analysis of the effects of the formulation variables on particle size displayed that the average size of the prepared PGZ-LMPs ranged from 2.57 ± 0.8 μm to 9.38 ± 0.9 μm (Table 2 and Fig. 1).
This wide range of particle sizes suggests that formulation variables, especially the type of SAA used and its concentration, are critical and significantly affect the analyzed response. Particles formed by formulations containing Poloxamer 188 were the largest, whereas those prepared using Tween 80 were the smallest. According to the statistical analysis, the type of the SAA (X2) and its concentration (X3) had a significant effect on particle size (p < 0.05) while the amount of CA (X1) did not. The particle size equation obtained from the analysis was
PS = 5.99 + 0.7383 X2 – 2.22 X22 – 0.815 X3. (4)
Previous studies showed that using various SAA with different hydrophilic-lipophilic balance (HLB) resulted in size variation of O/W emulsion particles (Housaindokht et al., 2012). On the other hand, the concentration of the SAA has an inverse effect on the size of PGZ-LMPs, where increasing the SAA concentration from 1% w/v to 2% w/v resulted in a significant reduction in the particle size. These results are in good correlation with previous work studying the effect of various concentrations of poloxamer on the size of the particles in nanostructured lipid carriers and observing that MPs prepared with a high ratio of surfactant to lipid had the smaller particle size (Zirak and Pezeshki, 2015). This trend might be expected at a relatively low quantity of SAA (<2% w/v) as increasing SAA concentration means more SAA particles are present to cover any newly formed LMP surfaces at a convenient rate during preparation (Qian and McClements, 2011).
 | Table 3. Results of the experimental design. [Click here to view] |
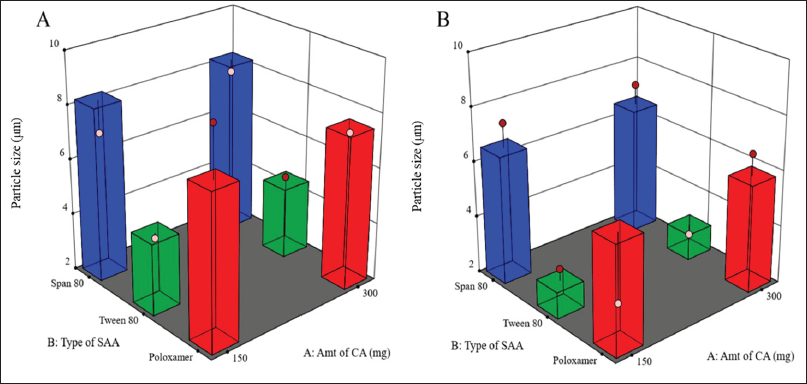 | Figure 1. Surface response chart representing the effect of type of SAA and amount of CA on particle size using 1% (A) and 2% (B) concentration of SAA. [Click here to view] |
Determination of EE%
The EE% of the drug is a critical factor that helps assess the drug loading capacity of the microparticles. Moreover, EE% significantly affects drug release from LMPs, where higher drug loading increases the concentration gradient and enhances the rate of drug release.
In addition, the sufficient concentration of the drug loading may also reduce the size of the administered dose, the cost of manufacturing, and the amount of excipients needed for the manufacturing of the microparticles, specifically SAA, which has been associated with in vivo toxicity when used in large amounts (Haider et al., 2020b). The EE% is indeed highly affected by different factors such as the method of preparation, physicochemical properties of the drug, and type and concentration of lipids and SAA. Our results showed that the PGZ-LMP formulae exhibited an EE% ranging from 26.7% to 71.3%, where the maximum drug entrapment was obtained using 300 mg CA in 1% Tween 80 (Table 2). The statistical analysis proved that only the surfactant concentration had a significant effect on the drug EE% (Table 3). The polynomial predictive equation produced for the EE% was
EE% = +47.26 – 9.23X3. (5)
As shown in Figure 2, the use of a higher concentration of SAA resulted in a significant decrease in the percentage of drugs entrapped within the LMPs (p = 0.0312).
These results were consistent with those obtained by other scientists (Rahman et al., 2010; Thakkar et al., 2005), who reported a decline in EE% in association with increasing SAA concentration due to the drug’s solubility improvement in the external medium and the associated increase in its partition from the lipid matrix of the MPs to the external medium. As mentioned earlier, LMPs prepared using Tween 80 (less than 2% w/v) generated the smallest particle size, suggesting the capability of this combination to improve the drug loading to a great extent without compromising the particle size. Also, as a result of the low crystallinity of the excipients, the lipid matrix was able to accommodate more drugs at higher drug loadings.
It was also noticed that larger MPs prepared using 1% w/v Tween 80 entrapped a bigger amount of the drug than LMPs with the larger particle size prepared using 2% w/v Tween 80 and the same amount of CA.
On the other hand, Chen et al. (2017) observed the same results when studying gefitinib-loaded PLGA MPs and suggested that this may be due to drug loss at the surface upon partitioning from the internal matrix-forming material into the external aqueous phase during the solidification of the MPs. These processes usually occur mainly at the surface of the MPs when they have not solidified yet. Once the particles solidify, the drug is entrapped within the matrix. The relatively higher surface: volume ratio of smaller particles results in relatively more drug loss and hence less EE% of smaller MPs.
In vitro drug release
The solubility of PGZ is pH dependent, where PGZ, a weak base (pKa = 6.8), is more soluble at lower pH. In the beginning, the release of PGZ from the prepared formulations was studied in both 0.1 N HCl and PBS pH 6.8 for 8 hours. In PBS, no PGZ was detected in the release media from the prepared PGZ-LMP formulations after 8 hours of the in vitro study due to the low PGZ solubility at higher pH. On the other hand, the PGZ release from drug-loaded LMPs exhibited a distinctive biphasic release pattern in 0.1N HCl (Fig. 3).
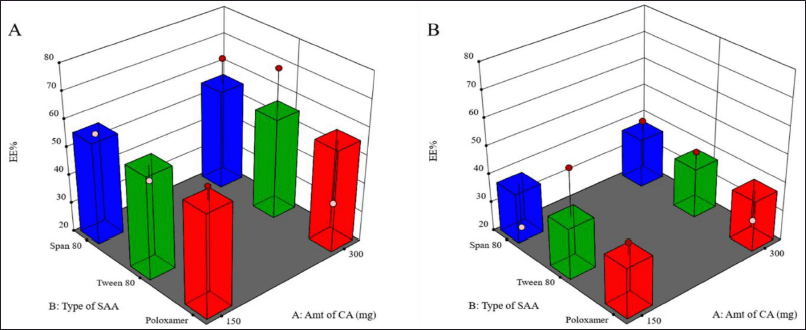 | Figure 2. Surface response chart representing the effect of type of SAA and amount of CA on EE% using 1% (A) and 2% (B) concentration of SAA. [Click here to view] |
The first phase was distinguished by a faster drug release from the MPs for up to 3 hours, especially in the formulation containing 1% SAA, where more than 50% of the drug was released in a burst release pattern followed by a slower constant drug release over 5 hours of the run. A burst in drug release may result from the accumulation of PGZ in the outer surface of LMPs as a result of phase separation during the crystallization of CA and the solidification of the particles. As a result, those drug particles at the lipid/water interface dissolved faster, leading to accelerated release of the drug to the dissolution medium. However, the sustained release pattern occurred when the drug molecules embedded in the matrix of the microparticles were slowly released because of the partition between the oil and water phases, drug diffusion, or matrix erosion (Khamanga and Walker, 2012).
Calculating the Q8 values from the release profiles of different PGZ-LMPs was used to assess their ability to sustain drug release. The prepared PGZ-LMPs could prolong and control the release of the drug to a varying degree (average Q8 values between 20% and 71%), as shown in Table 2. The polynomial equation describing the correlation between the tracked independent variables and Q8 was as follows:
Q8 = 41.71 + 3.86 X1 – 2.82 X21 – 0.6725 X22 – 9.93 X3 − 13.21 X1X21 + 9.13 X1X22. (6)
As shown in Table 3 and Figure 3, SAA concentration (X3) exerted a significant impact on the Q8 response (p = 0.0122). Increasing SAA concentration resulted in a reduction in Q8 values. LMPs prepared using 1% Tween 80 and 300 mg of CA were the highest Q8 values. This may be due to a decrease in hydrophobic interaction between SAA with high HLB when added at a low concentration and the lipid phase resulting in rapid degradation of the lipid matrix and an increment of the amount of drug released (Haider et al., 2020b). The amount of lipid incorporated in the LMP formulae did not affect the drug release. The results in Table 3 showed that increasing the amount of lipid from 150 to 300 mg had no significant effect on the drug release (p > 0.05). Similarly, no significant difference in drug release was reported when changing the type of SAA. Nevertheless, an interaction between the amount of lipid incorporated into the formulation and the type of surfactant was statistically significant (p < 0.05). According to these results, the hydrophobic interactions between lipid chains and SAA molecules affect both the rigidity and permeability of LMPs.
Selection of the optimal PGZ-LMPs
Based on the response surface analysis of the D-optimal surface design, the optimum levels of the studied factors were estimated for PGZ-LMP preparation (Fig. 4). We aimed to minimize particle size (<9 μm) to improve drug solubility and maximize EE% and Q8 to ensure optimal drug release after oral administration. Thus, simultaneous optimization of all three responses was performed, and the desirability value was used to select the optimal formulation for Y1, Y2, and Y3.
The highest desirability value (0.77) was represented in the formulation containing 300 mg CA and 1% w/v Tween 80 (Fig. 4). The optimal PGZ-LMPs selected by the statistical design were prepared and characterized for their PS, EE%, and Q8. The optimal selected formulation produced an observed PS, EE%, and Q8 of 4.64 μm, 69.55%, and 68.72%, respectively (Table 4). As a result of these findings, the optimal PGZ-LMP formulation, proposed by QbD, offered a promising formulation of suitable particle size, maximum drug-loaded LMPs, and highest drug release Q8; hence, it was selected for further investigation. For some of the experiments, PGZ-free LMPs (blank LMPs) were prepared using the optimal formulation.
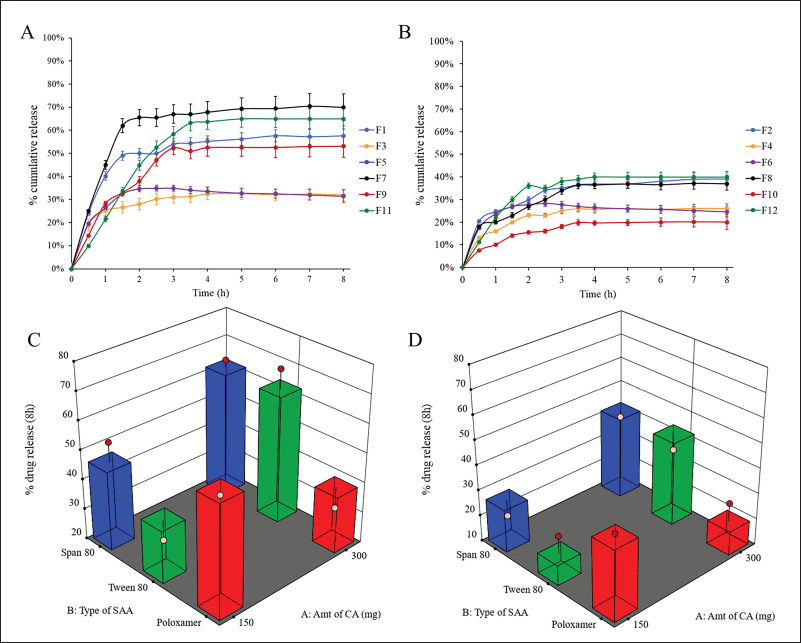 | Figure 3. In vitro release profiles of PGZ from different drug-loaded LMPs containing 1% (A) and 2% (B) (w/v) SAA and surface response chart representing the effect of type of SAA and amount of CA on Q8 using 1% (C) and 2% (D) (w/v) SAA. [Click here to view] |
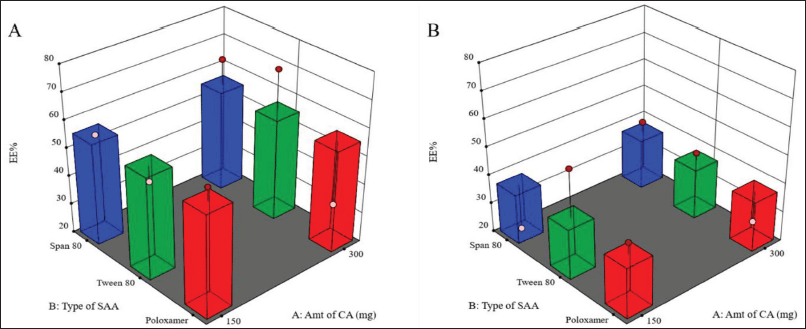 | Figure 4. Selection of optimal PGZ-LMP formula (flagged) among different formulations using 1% (A) and 2% (B) concentration of SAA. [Click here to view] |
 | Table 4. Parameters optimized along with predicted and observed responses. [Click here to view] |
Morphological examination
The morphology, surface topography, and structure of the optimal PGZ-LMP formula were investigated using an optical microscope and SEM. Optical micrographs showed that the particles were spherical and existed as discrete entities in the field (Fig. 5). SEM examination of the blank microparticles and the optimized PGZ-LMPs (F7) revealed the formation of the uniform, monodisperse, and typical spherical shape of LMPs. The existence of clusters of microparticles was also observed, which may be attributed to the incomplete solidification of the microparticles during the time frame of the preparation process. Further magnification showed that both blank microparticles and PGZ-LMPs had a rough and coarse surface which may be due to the crystallization of the lipids on the surface of the microparticles (Wu et al., 2019). In addition, the drug-loaded LMPs were significantly larger than that of the blank microparticles indicating the loading of the drug.
FT-IR spectroscopy analysis
The pure PGZ, CA, their physical mixture, and the optimal blank LMPs and PGZ-LMPs were analyzed by FT-IR spectroscopy, and the peaks relevant to the chemical functional groups were revealed in the spectra (Fig. 6). As a whole, the constituents displayed the typical bands associated with their functional groups. The major IR peaks assigned to PGZ, namely, 1,148, 1,509, 1,683, and 658 cm−1, corresponded to the stretching vibration of C-O, C=N, C=O, and C-S, respectively. On the other hand, CA had two characteristic peaks at 2,847 and 2,914 cm−1 representing C-H and O-H stretching vibration, respectively.
PGZ and CA peaks in the physical mixture indicate that there is no chemical interaction between PGZ and the lipid used in the preparation of the LMPs. Furthermore, in the spectrum of the optimized formula, stretching vibration bands from the lipid matrix appeared at the same wavelength, but none of the pure PGZ peaks were present, confirming the encapsulation of the pure PGZ into the LMPs.
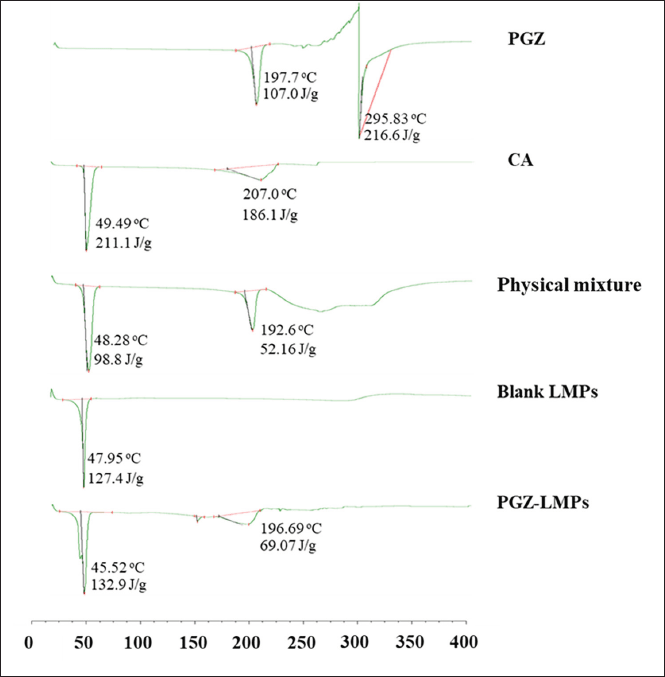
Thermal analysis
The DSC thermograms of the pure PGZ, lipid matrix, PGZ-lipid physical mixture, blank formula, and PGZ-LMP are shown in Figure 7. The pure PGZ and CA thermograms showed a sharp endothermic peak associated with their melting at 197.7°C and 49.49°C, respectively. The PGZ-lipid physical mixture showed both endothermic peaks of PGZ and lipid at 48.28°C and 192.6°C, respectively. PGZ-LMPs showed a broad endothermic peak at 45.52°C, whereas blank LMPs showed a broad endothermic peak at 47.95°C. There was a small shift in the peak mostly due to the drug incorporation into the lipid matrix. Also, in the drug-loaded microparticles, the PGZ endothermic peak was reduced, indicating an amorphous conformation, resulting in homogeneous solid mixtures with the lipids. This could mean the improved dissolution of the drug when PGZ-LMPs were placed in aqueous media. Similarly, the encapsulation of curcumin in stearic acid and tripalmitin lipid particles showed complete drug solubilization within the lipid matrix (Behbahani et al., 2017).
Drug release kinetics modeling
PGZ in vitro release data were implemented in different kinetic models (Table 5). The optimum model would be the one with the highest correlation coefficient (R2). A model was selected based on its R2 value with the closest value to 1.
According to the Makoid–Banakar model, the correlation of the observed PGZ released from the microparticles versus the predicted amount was found visually acceptable for the optimal PGZ-LMP formulation with an R2 value of 0.9915 (Table 5 and Fig. 8).
DDSolver was employed to perform further analysis to confirm the kinetic model fitting to the in vitro release data by generating the “Model Selection Criterion (MSC) and Akaike Information Criterion (AIC).” The release of PGZ from the optimal LMP formula showed the best fitting to the Makoid–Banakar kinetic model with a high value of R2 adjusted and MSC and a low AIC. Additionally, because of the low release exponent n, the release behavior was consistent with the Fickian diffusion mechanism (Wu et al., 2021).
Preclinical in vivo study
The in vivo pharmacological effect of the optimal PGZ-LMP formula was assessed and compared to that obtained from marketed PGZ tablets following oral administration. The low-dose STZ-induced diabetic albino Wistar rats were used for comparing the antidiabetic effect (Fig. 9a). The mean baseline BGL in the normal control group was 91.7 mg/dl, while the diabetic group showed a significant increase in BGL of up to 314.7 mg/dl. The animals treated with the marketed product and PGZ-LMPs presented mean BGL of 243.1 and 218.7 mg/dl, respectively. The PGZ-LMPs-treated group exhibited a significant reduction in BGL, 72.82% (p < 0.05*), compared to the diabetic group and the group the received the marketed PGZ. This was superior to the marketed PGZ oral tablets, which showed no significant difference between both the diabetic and PGZ solution groups. Animals treated with marketed PGZ tablets showed a 60.8% reduction in BGL over 24 hours, while treatment with blank LMPs showed a minimal decrease in BGL. The results confirmed that optimization of drug-loaded LMP formulation resulted in higher therapeutic effectiveness and a longer duration of action for the encapsulated PGZ than the marketed product.
 | Figure 5. PGZ-LMP morphological investigation showing a light microscopic image of PGZ-LMPs at 40× (A), SEM micrographs of blank LMPs (B and C), and PGZ-LMPs (D and E) at 2.5k× and 50k×, respectively. [Click here to view] |
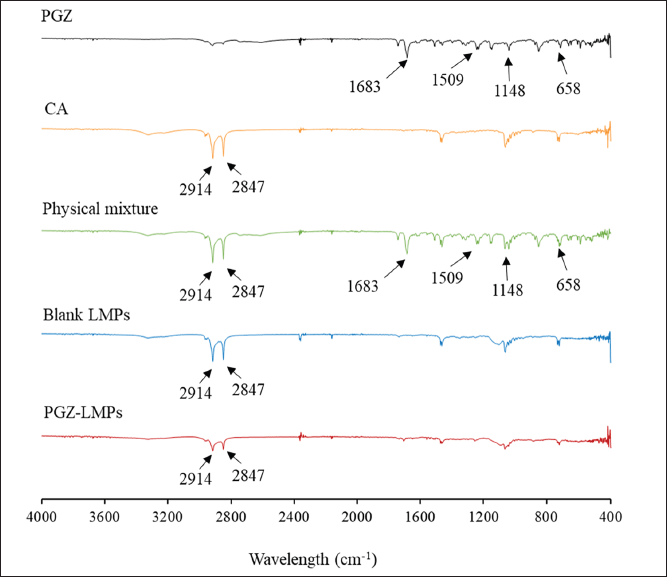 | Figure 6. Characterization of optimal PGZ-LMPs showing FT-IR spectra of PGZ, CA, their physical mixture, and the optimal blank LMPs and PGZ-LMPs. [Click here to view] |
 | Figure 7. Physicochemical characterization of optimal LMP formulation showing thermal analysis of the PGZ, CA, their physical mixture, and the optimal blank LMPs and PGZ-LMPs. [Click here to view] |
 | Table 5. PGZ release mechanism from the optimal formulation. [Click here to view] |
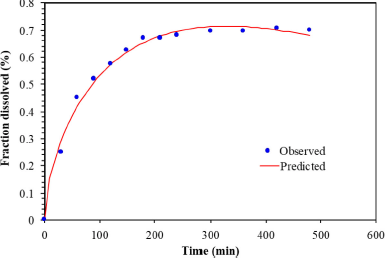 | Figure 8. The correlation of PGZ released versus the predicted amount of PGZ released from PGZ-LMP of F1–F12 by the Makoid–Banakar model. [Click here to view] |
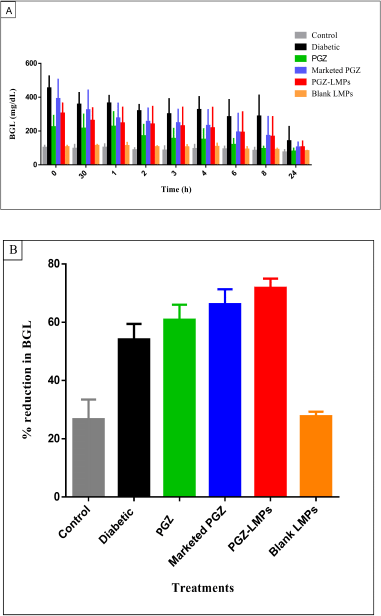 | Figure 9. BGL measurements in control and treatment groups (A) and % reduction in BGL (B) after each treatment for 24 hours. Data presented as a mean ± SD (n = 5). [Click here to view] |
CONCLUSION
PGZ was efficiently loaded into LMPs composed of cetyl alcohol and different concentrations of surfactants (Tween 80, Span 80, or Poloxamer 188). The LMPs were prepared by the solvent injection method. The procedure of preparation was optimized for the stirring speed and time of stirring to achieve the maximum size and drug loading into the LMPs. The selected optimized formula (F7) showed a high EE% (71.3% ± 1.293) and a mean particle size of 4.73 ± 0.06 μm with a PDI of 0.27 ± 0.06. The formation of the microparticles was confirmed by SEM examination. Moreover, DSC and FT-IR spectroscopy analysis confirmed the successful loading of PGZ into the MPs.
The F7 batch demonstrated a sustained in vitro release pattern at 8 hours of the run. Also, the Makoid–Banakar equation was the best model to fit and describe the drug release (R2 = 0.9666) significantly (p < 0.05) compared to other kinetic models. In addition, F7 showed valuable enhancement of the antidiabetic activity of PGZ and prolonged its effect in the albino Wistar rats for up to 24 hours. The results concluded that, in comparison to the marketed formulation and control groups, PGZ-LMPs could be suggested as an efficient carrier to maintain a sustained, controlled release of PGZ and to improve its oral bioavailability with a significant reduction in the blood glucose level. These promising results open the door for further investigations.
ACKNOWLEDGMENTS
The authors acknowledge Dubai Pharmacy College for Girls, Dubai, United Arab Emirates, for supporting this study with funds and facilities. They are also grateful to the College of Pharmacy, University of Sharjah, Sharjah, United Arab Emirates, for their assistance with the SEM, DSC, and FT-IR tests.
CONFLICTS OF INTEREST
No conflicts of interest have been declared by the authors.
FUNDING
This research was funded by the Dubai Pharmacy College for Girls, Dubai, United Arab Emirates.
ETHICAL APPROVAL
The Animals Ethical Committee, Research Unit, Dubai Pharmacy College for Girls, approved this study (Reference No. REC/MPharm/PPD/2019/03). The study follows the Guide for the Care and Use of Laboratory Animals.
DATA AVAILABILITY
This research article includes all generated and analyzed data.
AUTHORS’ CONTRIBUTIONS
Mona Hasan Rafiee performed the laboratory work, animal study, data analysis, and writing and original draft preparation. Bazigha K. Abdul Rasool (corresponding author) planned and supervised the work and reviewed/edited the manuscript. Mohammed Haider helped with data analysis and draft writing. Hanan S. Anbar helped in conducting the animal study and its statistical data analysis.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Abdul Rasool BK, Azeez O S, Lootah HA, Abusharbain IM, Abu-Alhaj HA, Nessa, F. Extended-release niosomal hydrogel for ocular targeting of piroxicam: in vitro and ex vivo evaluation. J Pharm Res Int, 2014; 4(21):2494–510; doi.org/10.9734/BJPR/2014/13723 CrossRef
Abdul Rasool BK, Khalifa A, Abu-Gharbieh E, Khan R. Employment of alginate gastroretentive floating in situ gel for controlled delivery of celecoxib: solubilization and formulation studies. Biomed Res Int, 2020; 2020. doi: 10.1155/2020/1879125 CrossRef
Abdul Rasool BK, Mohammed AA, Salem YY. The optimization of a dimenhydrinate transdermal patch formulation is based on the quantitative analysis of in vitro release data by DDSolver through skin penetration studies. Sci Pharm, 2021; 89:33; doi.org/10.3390/scipharm89030033 CrossRef
Al Shuwaili AH, Abdul Rasool BK, Abdulrasool AA. Optimization of elastic transfersomes formulations for transdermal delivery of pentoxifylline. Eur J Pharm Biopharm, 2016; 102:101–14. doi: 10.1016/j.ejpb.2016.02.013 CrossRef
Bartos C, Varga P, Szabó-Révész P, Ambrus R. Physico-chemical and in vitro characterization of chitosan-based microspheres intended for nasal administration. Pharmaceutics. 2021;13(5):608; doi: 10.3390/pharmaceutics13050608 CrossRef
Behbahani ES, Ghaedi M, Abbaspour M, Rostamizadeh K. Optimization and characterization of ultrasound assisted preparation of curcumin-loaded solid lipid nanoparticles: application of central composite design, thermal analysis and X-ray diffraction techniques. Ultrason Sonochem, 2017; 38:271–80; doi: 10.1016/j.ultsonch.2017.03.013 CrossRef
Bhosale UM, Galgatte UC, Chaudhari PD. Development of pioglitazone hydrochloride lipospheres by melt dispersion technique: optimization and evaluation. J App Pharm Sci, 2016; 6(1):107–17; doi: 10.7324/JAPS.2016.600118 CrossRef
Boni FI, Cury BSF, Ferreira NN, Gremião MPD. Ionic cross-linking as a strategy to modulate the properties of oral mucoadhesive microparticles based on polysaccharide blends. Pharmaceutics, 2021; 13(3):407; doi: 10.3390/pharmaceutics13030407 CrossRef
Chaudhury A, Duvoor C, Reddy Dendi VS, Kraleti S, Chada A, Ravilla R, Marco A, Shekhawat NS, Montales MT, Kuriakose K, Sasapu A, Beebe A, Patil N, Musham CK, Lohani GP, Mirza W. Clinical review of antidiabetic drugs: implications for type 2 diabetes mellitus management. Front Endocrinol (Lausanne), 2017; 8:6; doi: 10.3389/fendo.2017.00006 CrossRef
Chen W, Palazzo A, Hennink WE, Kok RJ. Effect of particle size on drug loading and release kinetics of gefitinib-loaded PLGA microspheres. Mol Pharm, 2017; 14:459–67; doi.org/10.1021/acs.molpharmaceut.6b00896 CrossRef
Elbary AA, Kassem MA, Abou Samra MM, Khalil RM. Formulation and hypoglycemic activity of pioglitazone-cyclodextrin inclusion complexes. Drug Discov Ther, 2008; 2(2):94–107.
Gupta NV, Gowda DV, Balamuralidhara V, Khan MS. Preparation and comparative bioavailability studies of indomethacin-loaded cetyl alcohol microspheres. J Pharm (Cairo), 2013; 2013:109837; doi: 10.1155/2013/109837 CrossRef
Haider M, Abdin SM, Kamal L, Orive G. Nanostructured lipid carriers for delivery of chemotherapeutics: a review. Pharmaceutics, 2020a; 12(3):288; doi: 10.3390/pharmaceutics12030288 CrossRef
Haider M, Elsayed I, Ahmed IS, Fares AR. In situ-forming microparticles for controlled release of rivastigmine: in vitro optimization and in vivo evaluation. Pharmaceuticals (Basel), 2021; 14(1):66; doi: 10.3390/ph14010066 CrossRef
Haider M, Elsherbeny A, Jagal J, Hubatová-Vacková A, Saad Ahmed I. Optimization and evaluation of poly(lactide-co-glycolide) nanoparticles for enhanced cellular uptake and efficacy of paclitaxel in the treatment of head and neck cancer. Pharmaceutics, 2020b; 12(9):828; doi: 10.3390/pharmaceutics12090828 CrossRef
Housaindokht MR, Nakhaei PA. Study the effect of HLB of surfactant on the particle size distribution of hematite nanoparticles prepared via the reverse microemulsion. Solid State Sci, 2012; 14(5):622–5; doi: 10.1016/j.solidstatesciences.2012.01.016 CrossRef
Hyma P, Abbulu K. Formulation and characterization of self-microemulsifying drug delivery system of pioglitazone. Biomed Prev Nutr, 2013; 3(4):345–50; doi: 10.1016/j.bionut.2013.09.005 CrossRef
Kamel R, El-batanony R, Salama A. Pioglitazone-loaded three-dimensional composite polymeric scaffolds: a proof of concept study in wounded diabetic rats. Int J Pharm, 2019; 570:118667; doi: 10.1016/j.ijpharm.2019.118667 CrossRef
Kapoor G, Pathak DP, Bhutani R, Husain A, Jain S, Iqbal MA. Synthesis, ADME, docking studies and in vivo anti-hyperglycaemic potential estimation of novel Schiff base derivatives from octadec-9-enoic acid. Bioorg Chem, 2019; 84:478–92. doi: 10.1016/j.bioorg.2018.12.004 CrossRef
Khalifa AM, Abdul Rasool BK. Optimized mucoadhesive coated niosomes as a sustained oral delivery system of famotidine. AAPS PharmSciTech, 2017; 18(8):3064–75; doi: 10.1208/s12249-017-0780-7 CrossRef
Khamanga SM, Walker RB. In vitro dissolution kinetics of captopril from microspheres manufactured by solvent evaporation. Dissolution Technol, 2012; 19:42–51.
Li W, Zhang L, Ge X, Xu B, Zhang W, Qu L, Choi CH, Xu J, Zhang A, Lee H, Weitz DA. Microfluidic fabrication of microparticles for biomedical applications. Chem Soc Rev, 2018; 47(15):5646–83; doi: 10.1039/c7cs00263g CrossRef
Oliveira PM, Matos BN, Pereira PAT, Gratieri T, Faccioli LH, Cunha-Filho MSS, Gelfuso GM. Microparticles prepared with 50-190kDa chitosan as promising non-toxic carriers for pulmonary delivery of isoniazid. Carbohydr Polym, 2017; 174:421–31; doi: 10.1016/j.carbpol.2017.06.090 CrossRef
Prasad PS, Imam SS, Aqil M, Sultana Y, Ali A. QbD-based carbopol transgel formulation: characterization, pharmacokinetic assessment and therapeutic efficacy in diabetes. Drug Deliv, 2016; 23(3):1057–66; doi: 10.3109/10717544.2014.936536 CrossRef
Qian C, McClements DJ. Formation of nanoemulsions stabilized by model food-grade emulsifiers using high-pressure homogenization: factors affecting particle size. Food Hydrocoll, 2011; 25:1000–8; doi: 10.1016/j.foodhyd.2010.09.017 CrossRef
Rafiee MH, Abdul Rasool BK. An overview of microparticulate drug delivery system and its extensive therapeutic applications in diabetes. Adv Pharm Bull, 2022 12(4):730–46; doi: 10.34172/apb.2022.075 CrossRef
Rahman Z, Zidan AS, Habib MJ, Khan MA. Understanding the quality of protein-loaded PLGA nanoparticles variability by Plackett-Burman design. Int J Pharm, 2010; 389(1–2):186–94; doi: 10.1016/j.ijpharm.2009.12.040 CrossRef
Scalia S, Young PM, Traini D. Solid lipid microparticles as an approach to drug delivery. Expert Opin Drug Deliv, 2015; 12(4):583–99; doi: 10.1517/17425247.2015.980812 CrossRef
Schubert MA, Müller-Goymann CC. Solvent injection as a new approach for manufacturing lipid nanoparticles--evaluation of the method and process parameters. Eur J Pharm Biopharm, 2003; 55(1):125–31; doi: 10.1016/s0939-6411(02)00130-3 CrossRef
Schwartz SS. Optimizing glycemic control and minimizing the risk of hypoglycemia in patients with type 2 diabetes. Drugs Context. 2013; 2013:212255; doi: 10.7573/dic.212255 CrossRef
Shaveta S, Singh J, Afzal M, Kaur R, Imam SS, Alruwaili NK, Alharbi KS, Alotaibi NH, Alshammari MS, Kazmi I, Yasir M, Goyel A, Ameeduzzafar. Development of solid lipid nanoparticle as carrier of pioglitazone for amplification of oral efficacy: formulation design optimization, in-vitro characterization and in-vivo biological evaluation. J Drug Deliv Sci Technol, 2020; 57:101674; doi: 10.1016/j.jddst.2020.101674 CrossRef
Silva-Abreu M, Gonzalez-Pizarro R, Espinoza LC, Rodríguez-Lagunas MJ, Espina M, García ML, Calpena AC. Thiazolidinedione as an alternative to facilitate oral administration in geriatric patients with alzheimer’s disease. Eur J Pharm Sci, 2019; 129:173–80; doi: 10.1016/j.ejps.2019.01.008 CrossRef
Song R, Murphy M, Li C, Ting K, Soo C, Zheng Z. Current development of biodegradable polymeric materials for biomedical applications. Drug Des Devel Ther, 2018 Sep 24; 12:3117-3145; doi: 10.2147/DDDT.S165440 CrossRef
Suke SG, Negi H, Mediratta PK, Banerjee BD, Sharma KK. Anti-arthritic and anti-inflammatory activity of combined pioglitazone and prednisolone on adjuvant-induced arthritis. Eur J Pharmacol, 2013; 718 (1–3):57–62; doi: 10.1016/j.ejphar.2013.09.019 CrossRef
Tabish SA. Is diabetes becoming the biggest epidemic of the twenty-first century? Int J Health Sci (Qassim), 2007; 1(2):V–VIII.
Thakkar H, Sharma RK, Mishra AK, Chuttani K, Murthy RR. Albumin microspheres as carriers for the antiarthritic drug celecoxib. AAPS PharmSciTech, 2005; 6(1):E65–73. doi: 10.1208/pt060112 CrossRef
Wang Y, Sun T, Zhang Y, Chaurasiya B, Huang L, Liu X, Tu J, Xiong Y, Sun C. Exenatide loaded PLGA microspheres for long-acting antidiabetic therapy: Preparation, characterization, pharmacokinetics and pharmacodynamics. RSC Adv, 2016; 6:37452–62; doi: 10.1039/c6ra02994a CrossRef
Wu C, Luo X, Baldursdottir SG, Yang M, Sun X, Mu H. In vivo evaluation of solid lipid microparticles and hybrid polymer-lipid microparticles for sustained delivery of leuprolide. Eur J Pharm Biopharm. 2019; 142:315–21; doi: 10.1016/j.ejpb.2019.07.010 CrossRef
Wu S, Gong Y, Liu S, Pei Y, Luo X. Functionalized phosphorylated cellulose microspheres: design, characterization and ciprofloxacin loading and releasing properties. Carbohydr Polym, 2021; 254:117421; doi: 10.1016/j.carbpol.2020.117421 CrossRef
Zhang W, Liu C, Chen S, Liu M, Zhang L, Lin S, Shu G, Yuan Z, Lin J, Peng G, Zhong Z, Yin L, Zhao L, Fu H. Poloxamer modified florfenicol instant microparticles for improved oral bioavailability. Colloids Surfaces B Biointerfaces, 2020; 193:111078; doi: 10.1016/j.colsurfb.2020.111078 CrossRef
Zirak MB, Pezeshki A. Effect of surfactant concentration on the particle size, stability and potential zeta of beta carotene nano lipid carrier. Int J Curr Microbiol App Sci, 2015; 4:924–32.