
INTRODUCTION
Staphylococcus aureus (S. aureus) is a Gram-positive bacterium found on human skin and nasal cavity and causes pyogenic disease, sepsis, osteomyelitis, and endocarditis (Lakhundi and Zhang, 2018). Methicillin-resistant S. aureus (MRSA) and vancomycin-resistant S. aureus (VRSA) cause nosocomial infections (McGuinness et al., 2017). These resistant strains are not confined only to hospitals but have also spread in the community (community-acquired MRSA: caMRSA) (Khan et al., 2018). The caMRSA infection causes serious diseases such as necrotizing pneumonia and can be life-threatening. An outbreak of this species affected several people in the early 2000s, and the disease spread worldwide. The spread of caMRSA infections is predicted to become a major problem in the future (Khan et al., 2018).
The de novo synthesis pathway, a nucleic acid synthesis pathway in S. aureus, produces the purine nucleotides ATP and GTP (Li et al., 2011). Inhibition of this pathway leads to decreased nucleic acid synthesis, which ultimately kills bacteria (Kobayashi et al., 2014). Dihydrofolate reductase (DHFR), which is involved in the de novo synthesis pathway, is a known target for developing antibacterial drugs and is gathering attention as an attractive drug target against MRSA and VRSA (He et al., 2020; Kobayashi et al., 2014). Trimethoprim (TMP) is a classical DHFR-targeting antimicrobial agent used to treat infections caused by S. aureus. TMP is prescribed together with sulfamethoxazole (SMX) as co-trimoxazole (Bactrim) to prevent the emergence of resistant mutations in bacteria (Wróbel et al., 2020). SMX is a specific inhibitor of the dihydropteroate synthesis (DHPS) enzyme. It has been reported that the Phe98 group in DHFR is mutated to Tyr (F98Y) in many pathogenic S. aureus variants (Wang et al., 2022). Staphylococcus aureus with mutated DHFR developed resistance to the diaminopyrimidine (DAP) ring of TMP. Therefore, identifying compounds different from TMP and those that are devoid of the DAP ring would help develop new therapeutic agents for patients infected with TMP-resistant S. aureus strains, as well as MRSA, VRSA, and caMRSA strains (Holland et al., 2014). Drug discovery targeting TMP-resistant S. aureus DHFR (TMP-resistant saDHFR) has attracted a great deal of attention (Huang et al., 2019; Keshipeddy et al., 2015; Reeve et al., 2019; Wang et al., 2022).
Structure-based drug screening (SBDS) has remained one of the most effective computational methods for developing new drugs (Pinzi and Rastelli, 2019). Based on the three-dimensional structure of the target protein, chemical compounds that bind to the pocket structure of the target were searched using protein-compound docking tools. In silico SBDS was performed using docking simulation tools such as GOLD (Scarpino et al., 2018), DOCK (Kinjo et al., 2013), GLIDE (Reddy et al., 2020), FRED (Gentile et al., 2020), AutoDock Vina (Trott and Olson, 2010), and Hex (Uciechowska-Kaczmarzyk et al., 2019). Multistep in silico SBDS, which consists of two or more docking simulation tools, has been used as a more effective way to identify active compounds (Taira et al., 2017). In particular, a high hit rate can be achieved by utilizing a compound screening method in which multiple docking simulations are linked hierarchically (Kobayashi et al., 2014; Kuriki et al., 2021).
The emergence of drug-resistant strains threatens the pool of first-line drugs, and this issue has necessitated the rapid and continuous development of novel antimicrobial agents. Computer-aided drug discovery provides a solution because, in comparison with screening based on biological experiments, the technique enables a more rapid identification of lead compounds. However, there is still room to improve efficiency in distinguishing true or false positives. In this study, the structure of TMP-resistant saDHFR was used as a target protein to identify novel antimicrobial compounds that are effective against multidrug-resistant S. aureus strains. Many attempts at hierarchical in silico drug screening have been made (Pinzi and Rastelli, 2019). This study reports the parallel use of two flexible docking simulation tools based on genetic algorithms. We established an AutoDock Vina (ADV)-GOLD parallel compound screening (PCS) method and identified novel antimicrobial chemical compounds. The dose-dependent inhibitory effects of the active compounds were confirmed and IC50 values were determined. The molecular dynamics (MD) simulation results indicated that the compounds directly interacted with TMP-resistant saDHFR.
MATERIALS AND METHODS
Compound structure library
The three-dimensional structural compound library (154,118 compounds from ChemBridge) was obtained from the Ressource Parisienne en Bioinformatique Structurale (RPBS) web-based database [available at http://bioserv.rpbs.jussieu.fr/RPBS/cgi-bin/Ressource.cgi?chzn_lg=an&chzn_rsrc=Collections (accessed Jun 10, 2012)]. The compound library was filtered using ADME/Tox filters to exclude compounds that were inappropriate as drugs. Compound structure libraries with multiple conformations were created, with a maximum of 10 conformations per compound. Compound multicoordinates were generated using the LowModeMD method (Labute, 2010).
Crystal structures of target proteins
The X-ray crystal structure of DHFR (PDB ID: 3M09) of the TMP-resistant S. aureus used in this study was obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (PDB) [available at https://www.rcsb.org/ (accessed May 15, 2017)].
Molecular surface extraction and pocket search
The protein structure of 3M09 was downloaded and calculations were performed to add hydrogen atoms and charges to the 3D structure of DHFR, as described in a previous study (Kuriki et al., 2021). Before performing the in silico SBDS, molecular surface extraction and pocket searches were performed. We used the DMS program to extract the molecular surface and the sphgen program to search for pockets [(available at https://dock.compbio.ucsf.edu/ (accessed April 2, 2017)]. The protein–ligand interaction (LI) region was restricted to the region close to the active site of the TMP-resistant saDHFR. NADP+, a coenzyme of DHFR, was prebound to the protein and other ligands were excluded.
Three-step hierarchical in silico SBDS with PCS method
In silico SBDS was performed using DOCK version 6.4, GOLD suite version 5.2.2, and ADV 1.1.2. DOCK, a grid docking tool that was used for the first screening. The scoring function of DOCK was calculated by predicting the protein–ligand binding affinity using van der Waals and electrostatic interaction energies. The binding affinities between the protein and the compounds selected in the first screening were evaluated using the genetic algorithm (GA) flexible docking simulation tools GOLD and ADV in the second screening effort. In the third screening, although the screening system was the same as in the second screening, docking simulation was performed for a 3D compound library with multiple configurations (compounds selected from the second screening). Finally, the compounds were filtered based on the following criteria: the score of the postscoring function RF-ScoreVS (Wang and Zhang, 2017), the presence of important interactions, and Lipinski’s rule of five (Lipinski et al., 2012).
Compounds
All candidate compounds (JP1–9) used in this study were purchased from ChemBridge Corporation (San Diego, CA) and dissolved in dimethyl sulfoxide (DMSO, Sigma). Table 1 lists the compound numbers, names, and ChemBridge IDs.
Bacterial species and growth inhibition assay
Staphylococcus epidermidis was purchased from the Microbial Materials Development Laboratory, RIKEN BioResource Center (Saitama, Japan). S. epidermidis was cultured overnight in 2 ml of the culture medium [composition: 1% peptone (BD), 1% beef extract (BD), and 0.5% NaCl (Wako, Japan), adjusted to pH 6.9] at 37°C and 240 rpm. The cultured S. epidermidis cells were diluted 52-fold and seeded in 96-well plates under 3 conditions: 0.3% DMSO (negative control), ampicillin (positive control), and the candidate compound. After 6 h of incubation at 37°C and 240 rpm, the turbidity (OD595) of the culture medium was measured using a microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA).
 | Table 1. Details of the compounds used in this study. [Click here to view] |
Molecular dynamics simulation
MD simulations were performed using the ligand–protein complex structures predicted by GOLD docking simulations. The GROMACS package with the CHARMM36m force field was used as the MD tool [available at: https://www.gromacs.org// (accessed Jan 15, 2020)]. A simulation system consisting of proteins, compounds, water molecules, and ions was constructed using the CHARMM-GUI web server [available at https://www.charmm-gui.org/ (accessed Feb 15, 2021)]. TIP3P was used as a water molecule. The cut-off distance for the van der Waals force and electrostatic interaction between the atoms was 1.2 nm. The particle mesh Ewald method was used to calculate long-range electrostatic interactions. The LINCS constraint algorithm was used for the energy minimization, equilibration, and production MD calculations. Energy minimization calculations were carried out in up to 5,000 steps using the steepest descent algorithm. Equilibration calculations were then carried out in one step under NVT conditions (310.15 K), followed by two steps under NPT conditions (310.15 K, 1 bar). Finally, 30 ns production MD calculations were performed with a time step of 2 fs. MD trajectories were analyzed using g_rms in the GROMACS package.
Statistical analysis
All statistical analyses were performed using R (The R Foundation for Statistical Computing, Vienna, Austria) and GraphPad Prism (GraphPad Prism Software, Inc., San Diego, CA).
RESULTS
The parallel compound screening method
We established the PCS method using the GA docking tools GOLD and ADV. This method selects a common population of compounds ranked in the top scores of the two docking tools. The datasets in the useful (docking) decoys-enhanced (DUD-E) database [available at http://dude.docking.org/ (accessed Jun 24, 2019)] were used for accuracy validation (Mysinger et al., 2012). The DUD-E database contained 102 target protein structures and datasets of their active and inactive ligand structures. To evaluate the prediction performance of docking simulation tools, the docking score values were analyzed using the receiver operating characteristic (ROC) curve and the area under the curve values. PCS is a method of binarization (classification) based on whether the compound is in the common population, and it does not provide specific parameters. Thus, the ROC curve analysis cannot be used to validate the accuracy of the PCS method. Therefore, the PCS method was evaluated using enrichment factor (EF) and success rate (SR) values (Wang and Zhang, 2017). The EF is a measure of the number of active compounds included in candidate compounds. The SR is a measure that calculates the proportion of proteins in a group of target proteins for which at least one active ligand is obtained. The accuracy of the PCS method was verified for all 102 target proteins in the DUD-E. We used a 200-compound dataset consisting of 20 true-active ligands and 180 pseudo-active compounds (decoys) that are similar to the active ligands but did not bind to the protein for each target protein. The results are presented in Table S1. The percentage of the EF2% (PCS) ≥ EF2% (GOLD) group was 74.5%, and the percentage of EF2% (PCS) ≥ EF2% (ADV) was 82.4%. The SR values for the GOLD, ADV, and PCS methods were 81.4, 83.3, and 85.3%, respectively. In terms of both EF2% and SR, the PCS method performed better than GOLD and ADV. Among the two indicators, the PCS method showed superior performance compared to the conventional method GOLD alone and ADV alone.
Three-step hierarchical in silico SBDS including PCS
A 3-step hierarchical in silico SBDS approach (Fig. 1) was used to screen 154,118 compounds in the structural library [available at http://bioserv.rpbs.jussieu.fr/RPBS/cgi-bin/Ressource.cgi?chzn_lg=an&chzn_rsrc=Collections (accessed Jun 10, 2012)] for TMP-resistant saDHFR (PDB ID: 3M09). In the first screening, rigid grid-docking simulations were performed using DOCK. In the second screening, we used the GA-based docking tools, GOLD and ADV in parallel, to screen compounds in singular coordination using the PCS method. In the third screening, the same PCS method as that in the second screening was applied to compounds with multiple conformers. The interactions between the protein and compound were then checked using the LI tool as a filter to confirm the binding mode near the pocket (Ahmed et al., 2021). The RF score was then calculated using RF-Score-VS (Wang and Zhang, 2017) using GOLD scores, a postscoring function that uses machine learning. Finally, the compounds were filtered by applying Lipinski’s rule of five. The final selected nine compounds are listed in Table 1. Table 2 presents the GOLD and ADV scores for each compound.
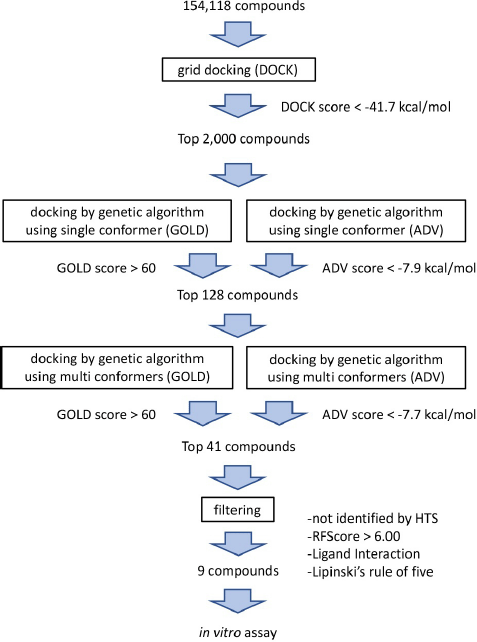 | Figure 1. Flowchart of the three-step hierarchical in silico SBDS with PCS method. [Click here to view] |
Bacterial growth inhibition assay for the candidate compounds
Nine candidate compounds (named JP1–9) predicted via the three-step hierarchical in silico SBDS were analyzed for their growth inhibitory effects on bacteria. S. epidermidis (biosafety level 1) was used as a model S. aureus bacterium in this growth inhibition assay (Kobayashi et al., 2014) because our laboratory is not equipped to perform experiments using S. aureus (biosafety level 2). The amino acid sequences of TMP-resistant saDHFR and S. epidermidis DHFR were analyzed using BLAST [available at https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed Aug 20, 2017)] and UniProt [available at https://www.uniprot.org/ (accessed Aug 20, 2017)]. The residues near the active site of TMP-resistant saDHFR were completely conserved in S. epidermidis, and the RMSD value between both protein structures was 0.543 Å (Kobayashi et al., 2014). JP5 and JP9 showed a strong inhibitory effect among the candidate compounds, while JP7 showed a weak inhibitory effect (Fig. 2). Figure 3A–C shows the structures of the three compounds that had an inhibitory effect on bacterial growth, and these three compounds were not structurally similar to each other. Both GOLD and ADV predicted that all three compounds would bind close to the active site of the DHFR (Fig. 3D–F, data of ADV not shown). The dose-dependent effects of two compounds (JP5 and JP9) on the growth of S. epidermidis were investigated. The IC50 value of JP9 was 6.34 ± 0.47 µM, while the effect was weaker for JP5, with an IC50 value of 56.94 ± 3.16 µM (Fig. 4).
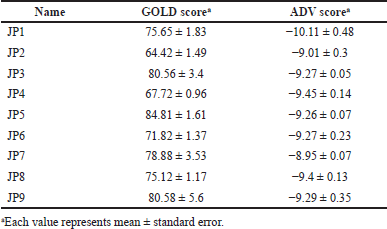 | Table 2. Score values for the nine candidate compounds identified by SBDS with the PCS method. [Click here to view] |
Binding mode prediction of the hit compounds
The interactions between TMP-resistant saDHFR and hit compounds (JP5 and JP9) were evaluated using the LI and protein–LI fingerprint (PLIF; data not shown) tools in Molecular Operating Environment (MOE) version 2011. 10 (Ahmed et al., 2021). Figure 5 shows the binding mode predictions by LI using the ligand–protein complex structures with the highest score calculated by GOLD in the third screening step. All hit compounds were located near the active site of DHFR (Fig. 3D–F). The results of the combined LI and PLIF analyses using multiple conformers are shown in Table 3. The analyses suggested that both the active compounds interacted with Phe92 (Fig. 5 and Table 3). JP5 appeared to form a hydrogen bond with Phe92, and JP9 binds to Phe92 via arene–arene interaction. The benzyl group of JP5 formed a hydrogen bond with Leu28, and the carboxyl and amide groups of JP9 formed a hydrogen bond with His30 and Leu20, respectively. In addition, PLIF analysis with multiple conformers revealed the potential interactions of JP9 with Val6, His23, Asp27, and Thr11 (Table 3).
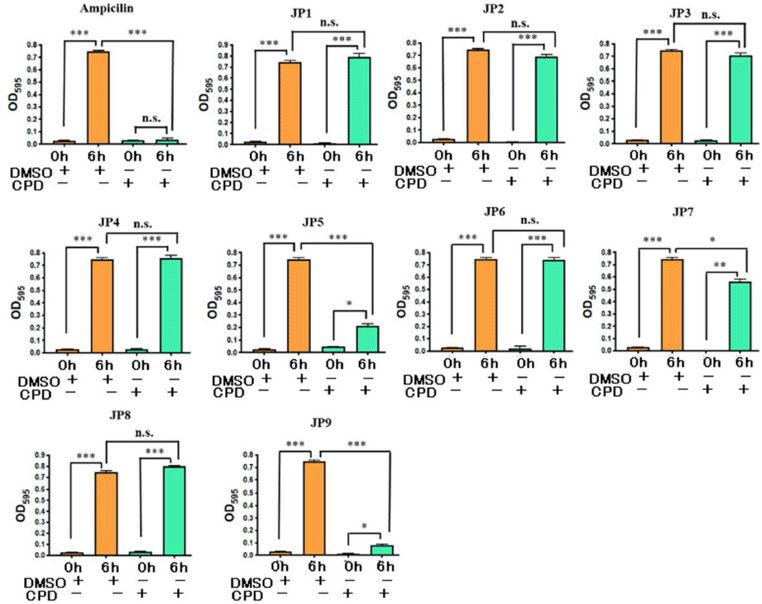 | Figure 2. Results of growth inhibition assay of S. epidermidis by candidate compounds (JP1–9). The concentration of all compounds was 100 µM. DMSO (0.3%) and ampicillin (100 µg/mL) were used as the negative and positive controls, respectively. All values represent mean ± SEM of four independent experiments. Bonferroni’s all-pairs comparison test was performed (n.s.: not significant; ***: p < 0.001; **: p < 0.002; *: p < 0.033). CPD: compound. [Click here to view] |
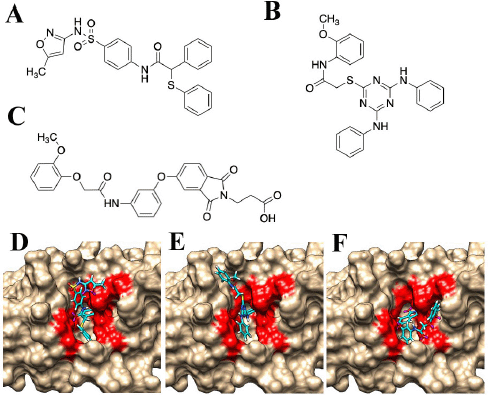 | Figure 3. Structures of the compounds with antibacterial activity. (A) JP5, (B) JP7, and (C) JP9. The complex structures of compounds and the TMP-resistant saDHFR are shown in (D–F). (D) JP5–TMP-resistant saDHFR. (E) JP7–TMP-resistant saDHFR. (F) JP9–TMP-resistant saDHFR. The amino acid residues of the active site pockets are shown in red color. [Click here to view] |
Analysis of compound-protein interaction by MD simulation
MD simulations of 30 ns were performed using the ligand–protein complex structures, JP5–TMP-resistant saDHFR, and JP9–TMP-resistant saDHFR, predicted by GOLD in the third screening. The 30 ns simulations with the complex structure of the TMP–TMP-resistant saDHFR protein were also performed. The complex structure was generated by docking simulation using GOLD (GOLD score = 67.70). A previous study showed that ligand binding ability could be assessed with high accuracy by analyzing ligand RMSD values in MD simulations with 56 proteins and 569 ligands (Guterres and Im, 2020). The time-dependent change of ligand RMSD value was analyzed for 30 ns production MD simulations, and the ligand RMSD value of TMP exceeded 0.65 nm by 2 ns and then exceeded 1.0 nm after 16 ns. At 2 ns, TMP dissociated from the active site pocket and never returned to the pocket. The mean RMSD value of TMP was 0.66 nm, indicating that TMP does not bind to the TMP-resistant saDHFR protein stably. On the contrary, the ligand RMSD values of JP5 and JP9 were stable throughout the simulation, with mean ligand RMSD values of 0.25 nm and 0.27 nm, respectively, indicating that JP5 and JP9 may stably and directly bind to the TMP-resistant saDHFR protein.
Toxicity assessment using the toxicity prediction tools
The toxicities of the candidate compounds were assessed by calculating the predicted LD50 values for oral administration in rats using TEST, a toxicity prediction tool [available at http://www.epa.gov/nrmrl/std/qsar/qsar.html (accessed Aug 20, 2020)]. TEST predicted LD50 values of 1902.8 and 3147.1 mg/kg body weight for JP5 and JP9, respectively. JP5 and JP9 were predicted to have sufficiently low toxicity compared with TMP (LD50 = 1648.1 mg/kg body weight). ToxiM, a machine learning-based toxicity prediction tool, estimated the toxicity classification scores of JP5, JP9, and TMP (Sharma et al., 2017). The toxicity scores of JP5, JP9, and TMP were 0.978, 0.860, and 0.926, respectively, suggesting that JP9 was less toxic than TMP.
DISCUSSION
We constructed and validated a novel screening method, the PCS method, and found that it performed better than the conventional methods. PCS was incorporated into the screening pathway targeting TMP-resistant saDHFR, and three compounds with growth inhibitory effects on S. epidermidis were identified. The interactions of the compounds (JP5 and JP9) identified in this study were predicted by LI and PLIF analyses. JP5 was predicted to form a hydrogen bond with Leu28, which is the same as that of TMP (Kobayashi et al., 2014). The significance of the Leu20 interaction in wild-type saDHFR has also been reported in previous studies investigating other inhibitory compounds (Kobayashi et al., 2014). LI analysis also predicted an interaction between JP9 and His30. These are likely to explain the difference in the growth inhibitory effects of JP5 and JP9. Leu20 is considered an important residue not only in wild-type saDHFR but also in TMP-resistant saDHFR. Furthermore, compounds with growth inhibitory effects commonly interact with Phe92. Therefore, interactions with Leu20, His30, and Phe92 are likely to be important for inhibiting the enzyme activity of DHFR.
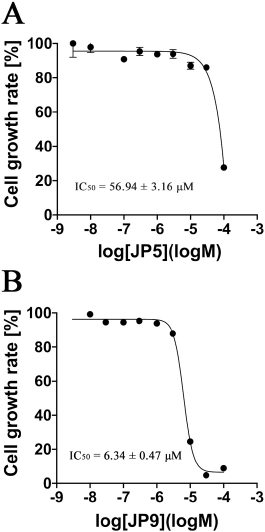 | Figure 4. Results of the verification of the dose-dependent effect of JP5 and JP9 on the growth of S. epidermidis. The vertical axis represents the bacterial growth rate (%) of S. epidermidis after 6 hours of incubation, and the horizontal axis represents the concentration of each compound on a logarithmic scale (log M). Each plot represents mean ± SEM of four independent experiments. The IC50 values were determined using nonlinear regression analysis. Each IC50 value was determined using four independent experiments and nonlinear regression analysis. (A) JP5 and (B) JP9. [Click here to view] |
There exists compatibility between docking simulation tools and target proteins (Pereira et al., 2016). Hence, selecting an appropriate tool for each target protein is important to identify hit compounds with a high probability. However, in the absence of a sufficient dataset, the ROC curve analysis cannot be used to assess the validity of a target protein/tool combination. Even in the absence of a sufficient ligand dataset, the PCS method can provide information on the compatibility between the docking tool and target protein. If the percentage of common compounds in the total compounds selected by each of the two tools is zero or very low, then it indicates that one or both tools may not be suitable for evaluating the target protein. Such information on the compatibility between target proteins and tools is extremely important for planning strategies for in silico drug discovery.
MD simulations using the complex structures of JP5 and JP9 with TMP-resistant saDHFR showed that the complexes remained stable for 30 ns with low RMSD values, suggesting that JP5 and JP9 can bind directly to TMP-resistant saDHFR. On the contrary, TMP deviated from the active site pocket of the TMP-resistant saDHFR early during the 30 ns MD simulation and did not return to the pocket. JP9 inhibited the growth of the bacteria at low concentrations (IC50 value = 6.34 µM) and was also predicted to be less toxic than TMP by the two independent tools for toxicity prediction. Considered together, multiple lines of evidence from in silico simulations and in vitro experiments suggest that JP9 is a promising molecule for future antimicrobial drug development. The hit rate of the three-step in silico SBDS method with the newly proposed screening system with PCS was 22% (33%, including the one that showed weak growth inhibition). This hit rate is extremely high compared to experimental high-throughput screening. Not only the active compounds but also the compound that showed only a weak growth inhibition effect is likely useful as lead compounds for the next step of drug discovery, which is to search for analogues with modified functional groups. The three-step in silico SBDS method used in this study is thought to be effective in terms of both time and cost for identifying new active lead compounds.
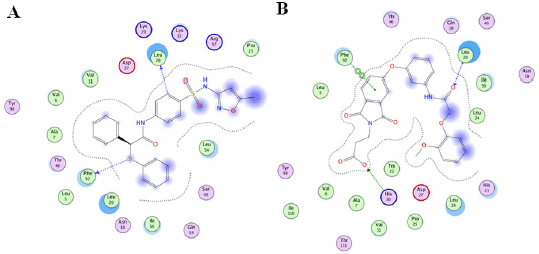 | Figure 5. Binding mode prediction of TMP-resistant saDHFR and hit compounds using LI. [Click here to view] |
 | Table 3. Predicted interaction residues of the compounds by PLIF or LI analyses. [Click here to view] |
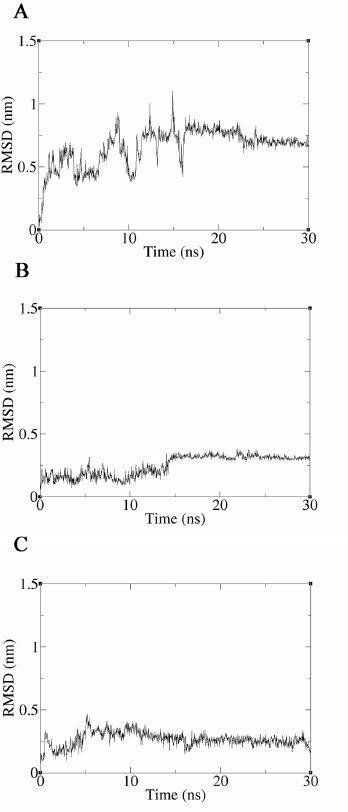 | Figure 6. Results of the interaction analysis between TMP-resistant saDHFR and compounds using MD simulations. Time-dependent changes of ligand RMSD values for TMP (A), JP5 (B), and JP9 (C) during 30 ns production MD simulations. The vertical and horizontal axes represent the ligand RMSD (nm) and time (ns), respectively. [Click here to view] |
CONCLUSION
In this study, compared with the conventional strategy, the PCS method exhibited advantages in the two metrics, EF2% and SR, and out of nine experimentally validated candidates, JP5 and JP9 were identified as active inhibitors. Furthermore, the MD simulation strongly suggested that JP5 and JP9 can form stable complexes with the active sites in TMP-resistant saDHFR. Considering that JP5 and JP9 have unique structures, these compounds could be novel leads for anti-infectives against S. aureus with TMP resistance. In the context of effectively identifying lead compounds, we believe that integrating the PCS method into an in silico SBDS platform could improve the computer-assisted strategy of identifying competitive inhibitors.
ACKNOWLEDGMENTS
This work was supported in part by a Grant-in-Aid for Scientific Research (26460145) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. The authors thank Kohei Kuriki for his descriptive support.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included within this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Ahmed MF, Santali EY, El-Haggar R. Novel piperazine-chalcone hybrids and related pyrazoline analogues targeting VEGFR-2 kinase; design, synthesis, molecular docking studies, and anticancer evaluation. J Enzyme Inhib Med Chem, 2021; 36:307–18. CrossRef
Gentile F, Agrawal V, Hsing M, Ton A-T, Ban F, Norinder U, Gleave ME, Cherkasov A. Deep docking: a deep learning platform for augmentation of structure based drug discovery. ACS Cent Sci, 2020; 6:939–49. CrossRef
Guterres H, Im W. Improving protein-ligand docking results with high-throughput molecular dynamics simulations. J Chem Inf Model, 2020; 60:2189–98. CrossRef
He J, Qiao W, An Q, Yang T, Luo Y. Dihydrofolate reductase inhibitors for use as antimicrobial agents. Eur J Med Chem, 2020; 195. CrossRef
Holland TL, Arnold C, Fowler VG. Clinical management of Staphylococcus aureus bacteremia: a review. JAMA, 2014; 312:1330–41. CrossRef
Huang DB, Noviello S, Balser B, Scaramucci A, Corey GR. A Pooled analysis of the safety and efficacy of iclaprim versus vancomycin for the treatment of acute bacterial skin and skin structure infections in patients with intravenous drug use: phase 3 REVIVE studies. Clin Ther, 2019; 41:1090–6. CrossRef
Keshipeddy S, Reeve SM, Anderson AC, Wright DL. Nonracemic antifolates stereoselectively recruit alternate cofactors and overcome resistance in S. aureus. J Am Chem Soc, 2015; 137:8983–90. CrossRef
Khan A, Wilson B, Gould IM. Current and future treatment options for community-associated MRSA infection. Expert Opin Pharmacother, 2018; 19:457–70. CrossRef
Kinjo T, Koseki Y, Kobayashi M, Yamada A, Morita K, Yamaguchi K, Tsurusawa R, Gulten G, Komatsu H, Sakamoto H, Sacchettini JC, Kitamura M, Aoki S. Identification of compounds with potential antibacterial activity against Mycobacterium through structure-based drug screening. J Chem Inf Model, 2013; 53:1200–12. CrossRef
Kobayashi M, Kinjo T, Koseki Y, Bourne CR, Barrow WW, Aoki S. Identification of novel potential antibiotics against Staphylococcus using structure-based drug screening targeting dihydrofolate reductase. J Chem Inf Model, 2014; 54:1242–53. CrossRef
Kuriki K, Taira J, Kuroki M, Sakamoto H, Aoki S. Computer-assisted screening of mycobacterial growth inhibitors: Exclusion of frequent hitters with the assistance of the multiple target screening method. Int J Mycobacteriol, 2021; 10:307–11.
Labute P. LowModeMD–implicit low-mode velocity filtering applied to conformational search of macrocycles and protein loops. J Chem Inf Model, 2010; 50:792–800. CrossRef
Lakhundi S, Zhang K. Methicillin-resistant Staphylococcus aureus: molecular characterization, evolution, and epidemiology. Clin Microbiol Rev, 2018; 31. CrossRef
Li X, Hilgers M, Cunningham M, Chen Z, Trzoss M, Zhang J, Kohnen L, Lam T, Creighton C, G C K, Nelson K, Kwan B, Stidham M, Brown-Driver V, Shaw KJ, Finn J. Structure-based design of new DHFR-based antibacterial agents: 7-aryl-2,4-diaminoquinazolines. Bioorg Med Chem Lett, 2011; 21:5171–6. CrossRef
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev, 2012; 64:4–17. CrossRef
McGuinness WA, Malachowa N, DeLeo FR. Vancomycin resistance in Staphylococcus aureus. Yale J Biol Med, 2017; 90:269–81.
Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J Med Chem, 2012;55:6582–94. CrossRef
Pereira JC, Caffarena ER, dos Santos CN. Boosting docking-based virtual screening with deep learning. J Chem Inf Model, 2016; 56:2495–506. CrossRef
Pinzi L, Rastelli G. Molecular docking: Shifting paradigms in drug discovery. Int J Mol Sci, 2019; 20. CrossRef
Reddy KK, Rathore RS, Srujana P, Burri RR, Reddy CR, Sumakanth M, Reddanna P, Reddy MR. Performance evaluation of docking programs- glide, gold, autodock & surflexdock, using free energy perturbation reference data: a case study of fructose-1, 6-bisphosphatase-AMP analogs. Mini Rev Med Chem, 2020; 20:1179–87. CrossRef
Reeve SM, Si D, Krucinska J, Yan Y, Viswanathan K, Wang S, Holt GT, Holt GT, Ojewole AA, Estrada A, Agabiti SS, Alverson JB, Gibson ND, Priestley ND, Wiemer AJ, Donald BR, Wright DL. Toward broad spectrum dihydrofolate reductase inhibitors targeting trimethoprim resistant enzymes identified in clinical isolates of methicillin resistant Staphylococcus aureus. ACS Infect Dis, 2019; 5:1896–906. CrossRef
Scarpino A, Ferenczy GG, Keser? GM. Comparative evaluation of covalent docking tools. J Chem Inf Model, 2018; 58:1441–58. CrossRef
Sharma AK, Srivastava GN, Roy A, Sharma VK. ToxiM: a toxicity prediction tool for small molecules developed using machine learning and chemoinformatics approaches. Front Pharmacol, 2017; 8. CrossRef
Taira J, Morita K, Kawashima S, Umei T, Baba H, Maruoka T, Komatsu H, Sakamoto H, Sacchettini JC, Aoki S. Identification of a novel class of small compounds with anti-tuberculosis activity by in silico structure-based drug screening. J Antibiot (Tokyo), 2017; 70:1057–64. CrossRef
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem, 2010; 31:455–61. CrossRef
Uciechowska-Kaczmarzyk U, Chauvot de Beauchene I, Samsonov SA. Docking software performance in protein-glycosaminoglycan systems. J Mol Graph Model 2019; 90:42–50. CrossRef
Wang C, Zhang Y. Improving scoring-docking-screening powers of protein-ligand scoring functions using random forest. J Comput Chem, 2017; 38:169–77. CrossRef
Wang S, Reeve SM, Holt GT, Ojewole AA, Frenkel MS, Gainza P, Keshipeddy S, Fowler VG, Wright DL, Donald BR. Chiral evasion and stereospecific antifolate resistance in Staphylococcus aureus. PLoS Comput Biol, 2022; 18. CrossRef
Wróbel A, Arciszewska K, Maliszewski D, Drozdowska D. Trimethoprim and other nonclassical antifolates an excellent template for searching modifications of dihydrofolate reductase enzyme inhibitors. J Antibiot (Tokyo), 2020; 73:5–27. CrossRef
SUPPLEMENTARY MATERIAL
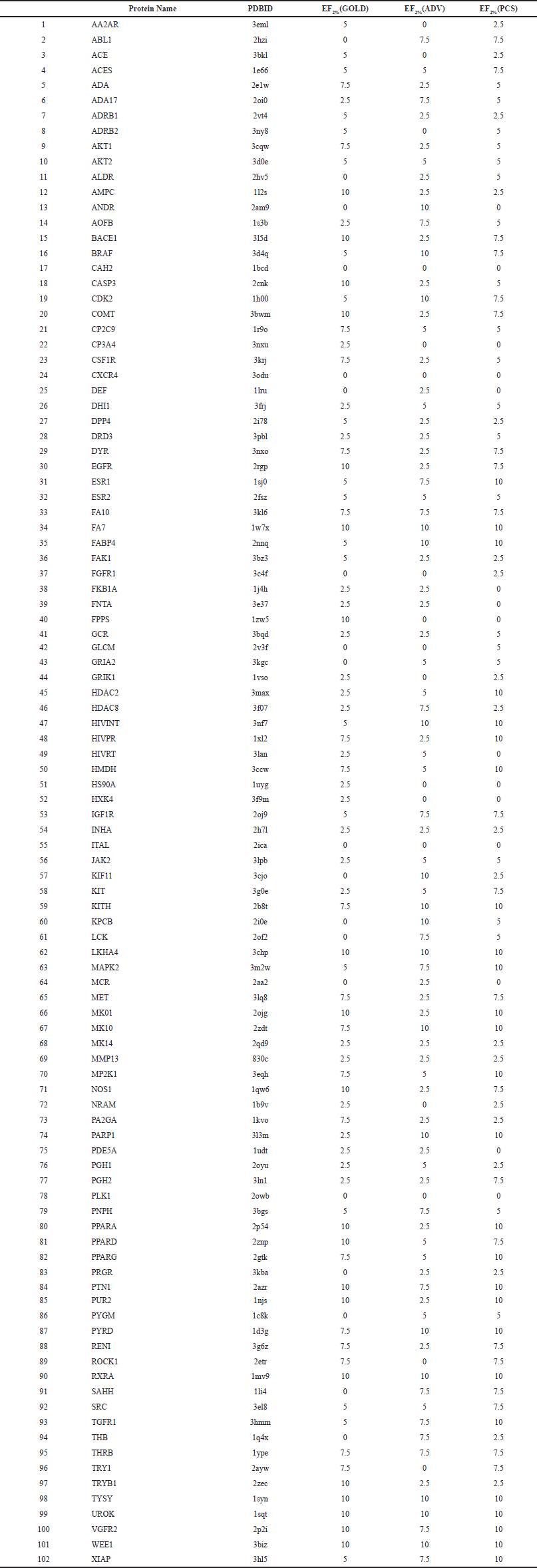 | Table S1. Accuracy validation results of DUD-E No. 1-102 proteins. [Click here to view] |