
INTRODUCTION
Dipyridamole is an accessory to oral anticoagulation for thromboembolism prophylaxis associated with prosthetic heart valves. It was intended to treat blood clot formation by reducing platelets and endothelial adenosine uptake, which reduces the stimulation of both platelet activating and collagen factors by promoting cyclic adenosine monophosphate (cAMP) accretion (Bult et al., 1991). It is chemically named as 2-[2-[bis(2-hydroxy-ethyl)amino]-4,8-di(piperidin-1yl)pyrimido[5,4-d]pyrimidin-6yl]-(2-hydroxy-ethyl)amino ethanol and the chemical structure of dipyridamole (Yogesh et al., 2012) is shown in Figure 1. The analytical methods reported include high-performance liquid chromatography (HPLC) analysis of dipyridamole (Bridle et al.,1993; Fontani et al., 1983; Hassan et al., 2008; Rao et al., 2016); reverse-phase high-performance liquid chromatography (RP-HPLC) stability, indicating the method for dipyridamole and its impurities (Acharya et al., 2015; Vaghela et sal., 2012); method development and validation for dipyridamole and aspirin by HPLC (Zhang et al., 1997); HPLC methods to identify dipyridamole in human plasma (Barberi et al., 2006); and stability-indicating RP-UPLC in combined capsule formulation (Rajput et al., 2011), revealing the simultaneous determination of dipyridamole and aspirin. Only HPLC methods were reported to determine dipyridamole and there is no UPLC-Q-TOF-MS (ultra-performance liquid chromatography to quadrupole time-of-flight mass spectrometry) analytical approach for the identification of impurities in dipyridamole literature sources. Advanced techniques are needed to recognize and interpret these impurities, when the impurities standards are not available. Therefore, attempts have been made to determine the trace level of Genotoxic impurities (GTIs) accurately; as a result, UPLC-Q-TOF-MS methodology has been developed as a useful approach. The related substance UPLC-MS method has been developed by using volatile MS compatible buffer media which separates and determines the listed impurities (Fig. 1) with 30 minutes runtime. The elution time and resolution were achieved by using the C18 stationary phase column 100 × 3 mm, 3.1 μ, and 1.8 μm. The high-strength silica (HSS) particle is the first and only 100% silica particle designed, tested, and intended for use in applications up to 15,000 psi (1,034 bar). The present method has been validated for its accuracy by spiking the API at 25%, 50%, and 75% for each analyte at the upper limit of quantification (ULOQ) concentration. The retention ranges of the respective analytes were less than 20% of the reported lower limit of quantification (LLOQ) value. International Conference on Harmonization (ICH) and USFDA guidelines were followed to the validate method with reference to specificity, linearity, accuracy, robustness, limit of detection (LOD), limit of quantification (LOQ), precision, and stability.
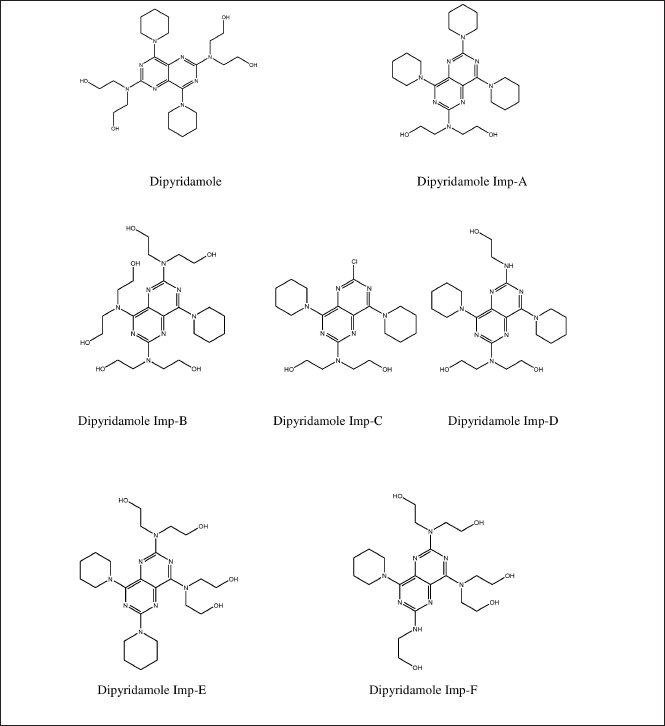 | Figure 1. Structure of dipyridamole and its impurities. [Click here to view] |
The developed and validated UPLC-Q-TOF-MS method is novel and has the advantages of shorter retention time with a volatile buffer system and a lower quantitation limit, which can detect impurities in dipyridamole.
EXPERIMENTAL STUDY
Chemicals and reagents
Dipyridamole-certified analytical reference standard (99.5%) and dipyridamole-related impurities (>95%) were obtained from Subtle Pharmaceuticals Pvt. Ltd, Bangalore. To obtain Milli-Q grade water for UPLC-Q-TOF analysis, distilled water was filtered using the pure lab pulse water purification system (ELGA LabWater, UK). Honeywell Research Chemicals (USA) supplied the LC-MS grade methanol and acetonitrile. Liquid ammonia, ammonium acetate, and glacial acetic acid were purchased from Thermo Fisher Scientific (India). Amber borosilicate glassware was used to handle analyte solutions.
Instrumentation
The Acquity H-class UPLC (Waters Corporation, Milford, MA) was employed, which had an integrated vacuum degasser, automatic sample manager (Serial# C10UPA554M, Waters Corporation, Singapore), high-performance binary solvent manager (Serial# C10UPB081A, Waters Corporation, Singapore), and injection volume range of up to 100 μl with an optional extension loop. An HSS T3 C18 stationary phase (100 × 3 mm, 3.1 μ) was used for chromatographic separation. A photodiode array detector (DAD) was employed in conjunction with a Xevo G2-XS Q-TOF (Serial# YFA1548, Waters Corporation, Wilmslow, UK) for MS detection.
Analytical reference standard solution preparation
Dipyridamole and its related impurities (impurity A, B, C, D, E, and F), 2 mg of each compound, was diluted with methanol up to 10 ml to obtain 200 μg/ml primary stock solutions. By diluting the respective primary stock solutions with methanol at concentrations of 100 μg/ml, 10 μg/ml, 1 μg/ml, 100 ng/ml, 10 ng/ml, 5 ng/ml, and 1 ng/ml, working stock solutions of each analyte were prepared. The standards and quality control samples (QCs) were aliquoted and kept frozen at −20°C in polypropylene tubes. Additional QCs were stored at around −5°C for stability testing.
UPLC-Q-TOF-MS method development and optimization
The working standard solution containing all analytes of interest at a concentration of 500 ng/ml of each analyte was used for method development and optimization.
Method development
The mobile phase is made-up of 10 mM ammonium acetate with 1% acetic acid in water as an aqueous phase (A) (pH = 4.8) and acetonitrile as an organic modifier (B), and it is delivered at a flow rate of 0.6 ml/minute in the following gradient: initially for 5 minutes (10%–30% B), held for 3 minutes, 8–18 minutes (30%–60% B), 18–23 minutes (60%–95% B), held for 2 minutes, 25–26 minutes (95%–10% B), and held at 10% B for 4 minutes. The sample was injected into a volume of 5 μl. The column oven temperature was kept at an optimal level throughout the chromatographic run (22oC). For MS detection, a positive polarity electrospray-combined ionization (ESCi) source was used. The optimal instrument and acquisition parameters are as follows: 50 l/hour cone gas (nitrogen) flow; 900 l/hour desolvation gas (nitrogen) flow; 600oC desolvation temperature; 30 V sampling cone voltage; 150oC source temperature; 80 V source offset voltage; capillary voltage of 3.0 kV; sample infusion flow rate is 5 μl/minute; the collision energy ramp varies from 6 eV (argon, collision gas); and a mass range of 50–1,500 m/z. Positive polarity was used to measure all analytes. All analyses were carried out with the lock spray to assure accuracy and reproducibility; lock mass used was leucine enkephalin. To acquire and process data, Waters Corporation’s MassLynx software (Milford, MA) was used. Table 1 and Figures 2 and 3 show the analytical performance of analytes with the optimized instrument and acquisition parameters.
Method optimization
Effect of pH
By varying the pH of the aqueous phase with ion-pairing reagents and buffers salts, such as acetic acid, ammonium acetate, and ammonia to 4.2, 4.8, and 5.3, respectively, the effect of pH on the chromatographic behavior and ionization efficiency of the analytes was studied.
Effect of stationary phase
Chromatograms were generated to evaluate the chromatographic performance of the analytes of interest using the sub-micron columns syncronis C8 (100 × 4.6 mm, 3 μ), HSS T3 (100 × 3.0 mm, 3.5 μ), and BEH C18 (50 × 2.1 mm, 1.7 μ).
Effect of solvents
As organic modifiers, different solvents such as methanol and acetonitrile were used, along with 10 mM ammonium acetate and 1% acetic acid in water (pH = 4.8) as the aqueous phase. The flow rate was 0.6 ml/minute.
Effect of mobile phase ratio
Chromatograms were generated to evaluate the chromatographic performance and ionization of the analytes of interest using mobile phases containing 70%, 50%, and 10% acetonitrile in 10 mM ammonium acetate and 1% acetic acid in water (pH = 4.8).
Effect of flow rate
Flow rates of 0.3, 0.5, and 0.6 ml/minute were used, and the chromatograms were recorded.
Effect of ionization source
Following chromatographic separation, analytes were ionized in an ESCi source, where each analyte was simultaneously ionized in ESI and APCI modes for subsequent MS detection and the total ion chromatograms were recorded.
Method validation
The method was validated in accordance with the USFDA’s analytical method validation guidance, which included an assessment of system suitability, specificity, accuracy, linearity, precision (repeatability and intermediate precision), range, quantitation limit, detection limit, and robustness (CDER, 1994; USFDA, 2015; ICH Q2 (R1), 2015).
System suitability
System suitability testing is critical for ensuring the UPLC-Q-TOF-MS system’s quality performance. The capacity factors (k’), injector repeatability [relative standard deviation (RSD)], resolution (Rs), tailing (T), and theoretical plate number (N) of the analyte peaks obtained from the chromatograms of diode array detection (DAD) and Q-TOF-MS (total ion chromatograms) detection were determined as a criterion of system suitability.
Capacity factor (k’)
The capacity factor is a measure of the location of the peak of interest in relation to the void volume, i.e., the nonretained components including the elution time. For the analyte peaks, a k’ value greater than 2 is recommended. For this parameter, a working standard solution containing 500 ng/ml concentrations of all analytes was used. The k’ value is calculated using the following equation:
k’ = (tR − t0)/t0 …….(1)
where tR = retention time of the analyte and t0 = elution time of the void volume or nonretained components.
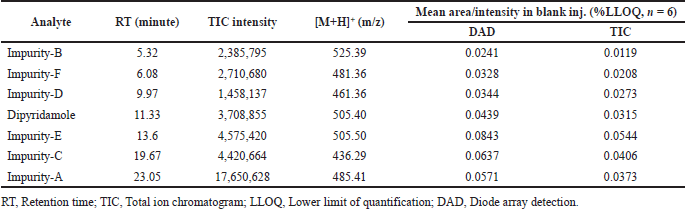 | Table 1. Analytical performance of dipyridamole and its six impurities at 500 ng/ml concentrations per analyte [Click here to view] |
Injection repeatability
Injection precision, expressed as RSD, represents the performance of the DAD and total ion chromatogram (TIC), which includes the plumbing, column, ionization source, and environmental conditions, as well as the time the samples are analyzed without considering the variations in sample preparation and manufacturing.
To calculate the RSD for this parameter, a working standard solution containing 500 ng/ml concentrations of all analytes was injected in six replicates (n = 6). An RSD of 1% is regarded as desirable.
Resolution (Rs)
Metric Rs used for determining how well two peaks are separated. Well-separated peaks are required for quantitation to be reliable. The ratio of the two compounds being measured has little effect on Rs. It is preferable to have Rs more than 2 between the peak of interest and the nearest peaks. A working standard solution with 500 ng/ml concentrations of all analytes was used for this parameter. The following equation is used to calculate Rs value:
where tR1 and tR2 = retention times of respective analytes and tW1 and tW2 = peak width of the extrapolated straight sides to baseline measured at baseline.
Tailing (T)
Quantitation accuracy decreases as peak tailing increases due to difficulties encountered by the integrator in determining where/when the peak ends, thus calculating the area under the peak. The analyst sets the integrator variables for the best calculation of the area for the peak of interest. For this parameter, a working standard solution containing 500 ng/ml concentrations of all analytes was used. It is preferable to have a tailing value of ≤2. The following equation is used to calculate T value:
T = WX / 2 f …….(3)
where Wx = width of peak determined by subtracting 5% (0.05) from the peak height’s baseline and f = distance between peak maximum and peak front at Wx.
Theoretical plate number (N)
N is a measure of column efficiency or the number of peaks that can be located per unit chromatographic runtime. For this parameter, only the analyte peaks in the DAD chromatogram were examined. For each peak on a chromatogram with a fixed set of operating conditions, N is fairly constant. Peak position, particle size in column, column temperature, analyte molecular weight, mobile phase flow rate, and viscosity are all factors that can affect N. A working standard solution containing 500 ng/ml concentrations of all analytes was used for this parameter. The theoretical plate number varies with elution time, but should be >2,000 in general. The following equation is used to calculate N value:
where tR = retention time of analyte and tW = peak width measured at baseline of the extrapolated straight sides to baseline.
Specificity (Selectivity)
Specificity refers to a method’s ability to measure the amount of analyte that is claimed to be measured. The expectation of no detectable interferences is referred as “specificity.” LC-MS is regarded as one of the most preferential techniques available, which allows identity confirmation. The chromatographic resolution (Rs) between the analyte and the closest eluting peak is usually used to determine selectivity in LC-MS. The analysis of a blank sample and spiked blank sample (all analytes at 500 ng/ml) determines specificity. The blank signal (n=6) at analyte retention time must be less than 20% of the analyte’s LLOQ to demonstrate that the analyte is not misidentified, that its identification is not hampered, and that its quantification is not influenced.
Linearity
The determinant of a linear relationship between analyte signals and analyte concentrations in calibration samples is linearity evaluation. Several factors influence signal linearity in an LC-MS method. The ion source’s behavior is said to be linear if the analyte’s ionization efficiency in the ion source is independent of its effluent concentration, i.e., the analyte concentration is proportional to the number of ions generated. A calibration graph is created using seven calibration solutions including a zero sample (blank), viz. 50, 125, 500, 1,000, 2,000, 3,750, and 5,000 ng/ml, derived from independent dilutions of the same stock solution of 200 μg/ml. When 75% of non-zero standards are within 15% of the nominal concentration, the standard curve is considered acceptable (20% for LLOQ).
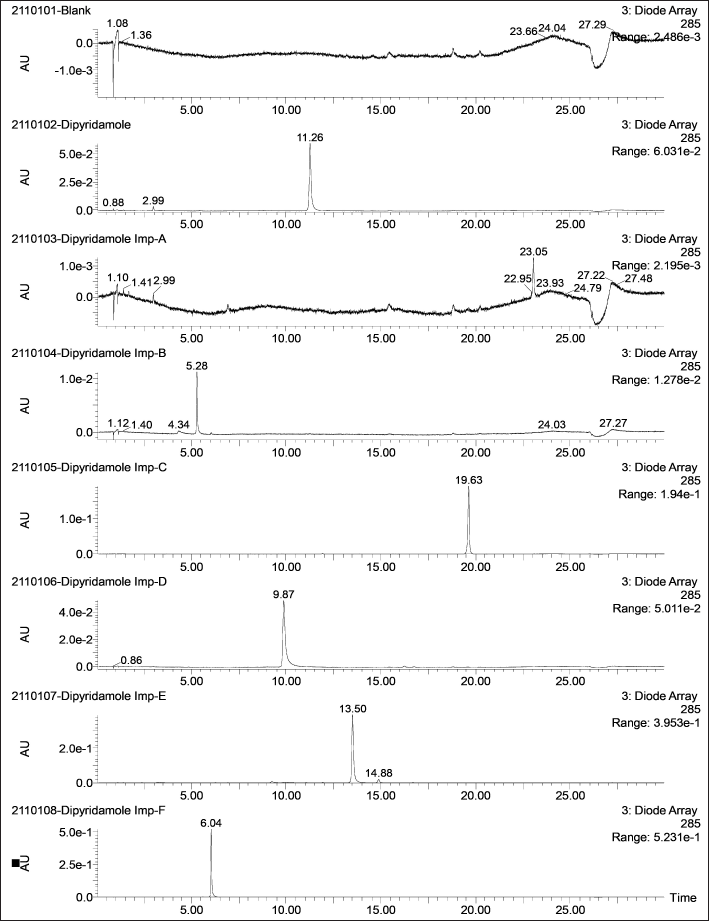 | Figure 2. Representative DAD chromatograms of DP and its six impurities (500 ng/ml) along with a blank. [Click here to view] |
 | Figure 3. Representative total ion chromatograms of DP and its six impurities (500 ng/ml) along with a blank. [Click here to view] |
Linear range
Linear range is defined as the lower and upper concentrations of analyte in the sample for which precision, accuracy, and response function have been established. The concentration range of 0%–100% of the expected analyte concentration (50 ng/ml) is set as the linear range for all analytes.
Sensitivity
Sensitivity is defined as the lowest analyte concentration that can be measured with acceptable accuracy and precision (i.e., LLOQ). It is essential for method optimization and routine instrument monitoring. Sensitivity is also directly related to ionization suppression—in fact, the essence of ionization suppression is a decrease in sensitivity caused by coeluting molecules. The LLOD, ULOD, and LLOQ are sensitivity measures with signal-to-noise ratios of 3:1, 5:1, and 10:1, respectively. To assess the method’s sensitivity, the peak intensities of spiked control samples are compared to the peak intensities of the blank at the retention time range of analytes in a TIC.
Precision
Precision is defined as the degree of agreement (expressed as RSD) between measured values obtained by repeat measurements on the same or similar objects under specified conditions. Precision is a component of measurement uncertainty and is related to the random error of a measurement system.
Repeatability is achieved by repeatedly analyzing (in triplicates) independently prepared quality control (QC) samples of concentrations, viz. 125 ng/ml (lower QC), 1,750 ng/ml (middle QC), and 3,750 ng/ml (higher QC), with each concentration being analyzed in triplicate from a homogeneous sample (200 μg/ml) in the same laboratory by one operator, using one experimental setup and one set of reagents on the same day.
The precision obtained upon the analysis of 125 ng/ml (lower QC), 1,750 ng/ml (middle QC), and 3,750 ng/ml (higher QC) in triplicates within a single laboratory over a period of time is referred to as intermediate precision.
Accuracy
Accuracy refers to the degree of agreement between the measured and true values. The accuracy of a method refers to its ability to provide accurate results. Measurement uncertainty is a quantitative measure of accuracy. Spike/recovery studies are used to assess accuracy. The sample is divided into four aliquots, one of which is analyzed at its original concentration of 50 ng/ml and the remaining three are spiked with 25%, 50%, and 75% of the analyte at ULOQ concentration and expressed as percent recovery [Eq. (5)], as follows:
where Xspike = response of analytes in spiked sample and Xunspike = response of analytes in unspiked sample.
Robustness
The terms robustness and ruggedness describes an analytical method’s ability to remain unaffected by small changes in method parameters (column age, column temperature, mobile phase composition, and so on) and environmental factors (room temperature, air humidity, and so on) and to characterize its reliability during normal usage. It can be regarded as no change in the detected amount of the analyte in a given sample, despite changes in the method parameter. The robustness parameters evaluated in the UPLC separation were pH variation of ±0.5 units, composition of acetonitrile varied at ±2% and aqueous phase additives varied at ±10% of the original values. For Q-TOF-MS detection, the desolvation gas temperature varied at ±10oC and condition of the ion source, i.e., before and after cleaning the ion source. The change in analyte response is expressed as RSD.
RESULTS AND DISCUSSION
UPLC-Q-TOF-MS method development and optimization of dipyridamole and its related impurities A, B, C, D, E, and F
The concentrations of analyzed samples and storage/analysis times, pHs, mobile phases, elution parameters (gradient), column, and ionization source used in the analyses were included in the method development and optimization. Analyte stability is also an important factor in optimizing an analytical method.
Effect of pH
The retention time of dipyridamole and its related impurities decreased as the pH of the mobile phase increased, with impurity B failing to retain. This could be due to the ionized state of impurity B caused by ammonia at higher pH levels. The retention time for pH 3.47 (0.01% acetic acid), pH 4.81 (10 mM ammonium acetate and 1% acetic acid), and pH 5.73 (0.01% ammonia) buffers was 9.54, 5.32, and 0.86 minutes, respectively; thus, pH 4.8 buffer was chosen because it provided a retention time of around 5.32 minutes.
Effect of stationary phase
The syncronis C8 (100 × 4.6 mm, 3 μ) column retained dipyridamole and its related impurities at shorter runtimes, but resolution was lacking. HSS T3 (100 × 3.0 mm, 3.1 μ) generated symmetrical and resolved peaks with analyte retention ranging from 5.32 to 23.05 minutes. BEH C18 (50 × 2.1 mm, 1.7 μ) produced peaks with poor symmetry and resolution.
Effect of solvents
When methanol was used, peak broadening and high back pressure were observed. Peak tailing was observed in the presence of methanol and water. The current study used 10% acetonitrile in 10 mM ammonium acetate and 1% acetic acid in water (pH = 4.8) as the aqueous phase delivered at a flow rate of 0.6 ml/minute because it provided good separation.
Effect of mobile phase ratio
As the mobile phase, 70:30, 50:50, and 10:90% v/v acetonitrile and 10 mM ammonium acetate and 1% acetic acid in water (pH = 4.8) were used as initial composition in a gradient program. Symmetrical peaks eluted at around 5.32 (Imp-B), 6.08 (Imp-F), 9.97 (Imp-D), 11.33 (Dipyridamole), 13.6 (Imp-E), 19.67 (Imp-C), and 23.05 (Imp-A) with a good capacity factor at a 10:90% v/v ratio of acetonitrile and 10 mM ammonium acetate and 1% acetic acid in water (pH = 4.8), and it was chosen for further studies. Ionization of analytes was not affected by the mobile phase ratio.
Effect of flow rate
The flow rate range (0.5 and 0.6 ml/minute), except 0.3 ml/minute, produced symmetrical and resolved peaks with an acceptable capacity factor. For the current study, 0.5 ml/minute was chosen because it has a shorter retention time (25 minutes), better peak shapes, acceptable back pressure, and better separation of impurities from dipyridamole.
Effect of ionization source
The structures of dipyridamole and its related impurities contain abundance of nitrogen atoms that could be used for protonation. However, the full scan ESI mass spectrum contains only one major peak for [M+H]+ at m/z 525.39 (Imp-B), 481.36 (Imp-F), 461.36 (Imp-D), 505.4 (Dipyridamole), 505.63 (Imp-E), 436.29 (Imp-C), and 485.41 (Imp-A), which could be due to the first protonation drastically decreasing the basicity of all nitrogen atoms. Peak intensities were higher in ESI than in APCI, which can be attributed to the greater amount of mobile phase delivered in APCI, which reduces droplet charge leading to ionization suppression.
In conclusion, 0.1% formic acid/10 mM ammonium acetate in water and methanol/acetonitrile as mobile phase were unable to resolve Imp-D and dipyridamole. An aqueous solution of 1% acetic acid in 10 mM ammonium acetate and acetonitrile provided good resolution between Imp-D and dipyridamole. Also tested were BEH C18, 1.0 × 50 mm, 1.7 μ, Accucore C18, 4.6 × 50 mm, 2.6 μ, and HSS T3, 3.0 × 100 mm, 3.5 μ columns. The HSS T3, 3.0 × 100 mm, 3.5μ column provided adequate resolution for all analytes.
Method validation
System suitability
The system suitability parameters (System suitability section) were analyzed upon integration of the analyte peaks in chromatograms of DAD using MassLynx software. Analyte peaks were well resolved with retention times ranging from 5.32 (Imp-B) to 23.05 (Imp-A) minutes. The k’ values of the analytes’ peaks were not less than 2, as required. With 7.5 μl as injection volume, all analytes had better injector repeatability, expressed as %RSD of peak areas (n = 5 for each analyte). The resolution of all analyte peaks was found to be greater than 2, as specified. All peaks were found to be symmetrical, with tailing factors no greater than 2, as specified. Analyte retention on column was confirmed by a theoretical plate count of analyte at a specific retention time for which none of the analytes showed less than 2,000 N. The suitability of analytes for simultaneous analysis using UPLC-Q-TOF-MS method is presented in Table 2.
Specificity (selectivity)
The LC-MS identification revealed no interferences throughout the analyte retention time range. The mean peak areas (DAD) and intensities (TIC) (n = 6; blank) at the retention ranges of the respective analytes were less than 20% of the reported LLOQ value. Table 1 presents the values for specificity of each analyte. Figures 2–4 show DAD and TIC chromatograms, as well as MS spectra for analytes of interest and a blank.
Sensitivity, linearity, and range
The LLOD, ULOD, and LLOQ of the analytes of interest were confirmed using a signal-to-noise (S/N) ratio of the analyte peak intensities (n =3, per analyte) to the intensities observed at the specific analyte retention time in blank (n = 6 per standard injection) of 3:1, 5:1, and 10:1 (Table 3), respectively. In addition, the relative standard deviation (RSD) of the mean peak intensities derived from the TICs of each analyte at LLOQ (n = 6) was determined to be less than 5%. The expected concentration to be quantified was 50 ng/ml for all analytes; so, a 0%–100% range was established. This range was further evaluated for linearity using a simple linear regression equation at seven concentration levels ranging from 50 to 5,000 ng/ml prepared from a single 200 μg/ml stock solution. The chosen linearity was discovered to be linear, with coefficient variance (R2) of analytes of interest greater than 0.999. Furthermore, at ULOQ, the RSD value of analytes of interest was found to be less than 5%.
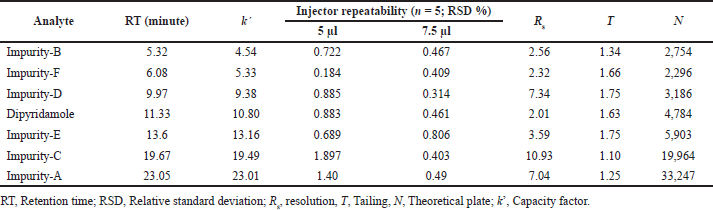 | Table 2. System suitability evaluation of dipyridamole and its six impurities at 500 ng/ml concentrations per analyte. [Click here to view] |
 | Figure 4. Representative mass spectra of DP and its six impurities (500 ng/ml). [Click here to view] |
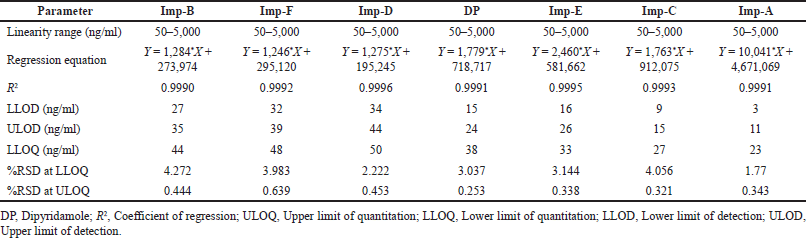 | Table 3. Determination of linearity and sensitivity of dipyridamole and its six impurities using UPLC-Q-TOF-MS. [Click here to view] |
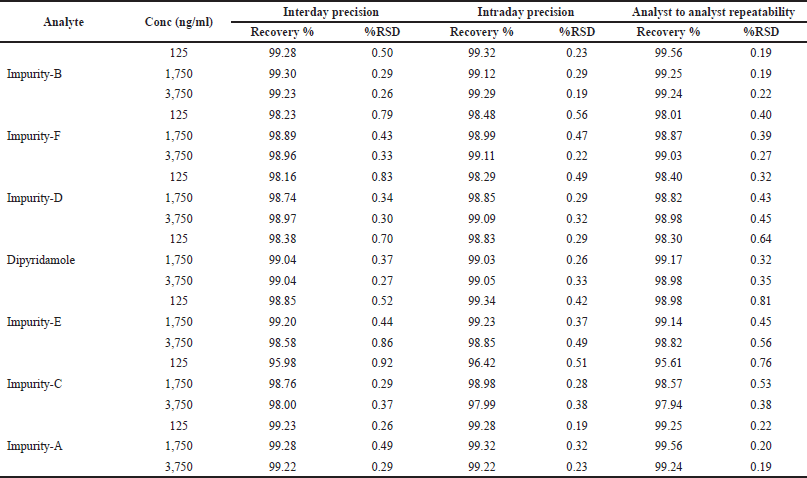 | Table 4. Evaluation of precision and recovery of dipyridamole and its six impurities. [Click here to view] |
Precision
Analytes at the LQC, MOQ, and HQC levels were prepared in sets of two triplicate samples per QC level for repeatability evaluation. The RSD value of analytes was found to be less than 1%. For intermediate precision studies, QC concentrations were prepared fresh before each analysis twice a week for 1 month by two analysts who independently prepared the QCs, integrated the peaks, calculated, and reported the RSD values of the respective analytes (Table 4).
Recovery
The recovery of analytes of interest was evaluated in triplicate (n = 3) using four aliquots at different concentrations. The results were reported as mean percent recoveries with RSD values (Table 4).
Robustness
The obtained RSD values for the evaluated method variation parameters, such as pH variation of ±0.5 units, acetonitrile composition variation of ±2%, and aqueous phase additives variation of ±10% of the original values, were well within the acceptable range. For Q-TOF-MS detection, the desolvation gas temperature was varied at 10oC, and the ion source’s condition, i.e., before and after cleaning, was evaluated, and the obtained RSD values were well within the acceptable range.
CONCLUSION
UPLC-Q-TOF-MS methodology for dipyridamole and its impurities has found to be highly efficient compared to other techniques in terms of mobile phase, runtime, and in lower detection limit. A selective, precise, sensitive, accurate, and robust approach for simultaneous quantification of dipyridamole and its related genotoxic impurities was developed and proved to be suitable for UPLC-Q-TOF-MS analysis. The approach was validated and could be used in laboratories for routine analyte analysis.
AUTHORS’ CONTRIBUTIONS
Concept and design, acquisition of data, interpretation of data and drafting the article were carried out by Menaka. Statistical analysis, critical revision, supervision, and final approval were carried out by Ramya Kuber.
ACKNOWLEDGMENTS
The authors would like to thank Subtle Pharmaceuticals Pvt Ltd for their support in providing standard and impurities. They also acknowledge the Institute of Excellence, Vijnana Bhavan, University of Mysore, for providing facilities to carry out the research work.
CONFLICT OF INTERESTS
The authors declare no conflict of interests.
ETHICAL APPROVAL
This study does not involve experiments on animals or human subjects.
FUNDING
There is no funding to report.
DATA AVAILABILITY
All data generated and analyzed are included within this research article.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Acharya, Daniel A, Gyadangi B, Ramsamy S. Isolation, characterization of a potential degradation product of aspirin and an HPLC method for quantitative estimation of its impurities. J Chrom Sci, 2015; 53(9):1491–7. CrossRef
Barberi-Heyob M, Merlin JL, Pons L, Calco M, Weber B. A sensitive isocratic liquid chromatography assay for the determination of dipyridamole in plasma with electrochemical detection. J LiqChrom, 2006; 17:1837–48.ß CrossRef
Bridle JH, Brimble MT. A stability indicating method for dipyridamole. Informa Heal Drug Dev Ind Pharm, 2008; 19:371–81. CrossRef
Bult H, Fret HR, Jordaens FH, Herman AG. Dipyridamole potentiates platelet inhibition by nitric oxide. Thromb Haemost, 1991; 66(3):343–9. PMID: 1746006. CrossRef
Center for Drug Evaluation and Research. Validation of chromatographic methods. Docket ID: FDA-2013-S-0610, 1994. Available via https://www.fda.gov/media/75643/download
Fontani F, Finardi GP, Targa G, Besana GP, Ligorati M. Purity evaluation of dipyridamole by high-performance liquid chromatography. J Chrom A, 1983; 280:181–7. CrossRef
Hassan H, Hammud, Fawzy A. El Yazbib, Mohamad E. Mahrous, Ghassan M. Sonji, Nada M. Sonji. Stability-indicating spectrofluorimetric and RP-HPLC methods for the determination of aspirin and dipyridamole in their combination. OpeSpect J, 2008; 2:19–28. CrossRef
ICH Q2 (R1).Validation of analytical procedures: text and methodology. In International conference on harmonization of technical requirements for registration of pharmaceuticals for human use. Switzerland: Geneva. 2005.
Rajput AP, Sonanis MC. Development and validation of a rapid RP-UPLC method for the determination of aspirin and dipyridamole in combined capsule formulation. Int J Pharm Pharma Sci, 2011; 3:156–60.
Rao AS, Rao K, Dadichand AS, Punna Rao AML, Balaswami B. Development and validation of RP-HPLC method for assay of dipyridamole in formulations. J Pharm Sci Res, 2016; 8(5):256–9.
USFDA. Analytical procedures and methods validation for drugs and biologics. Docket ID: FDA-2015-N-0007, 2015. Available via https://www.fda.gov/media/87801/download
Vaghela B, Rao SS, Reddy SP. Development and validation of a stability indicating RP-LC method for the estimation of process related impurities and degradation products of dipyridamole retard capsules. Int J Pharm Pharm Sci, 2012; 4:615–22.
Yogesh D Bommegowda, Dinesh B Shenoy, Menaka T, Chethan BP, Nagaraja G, Fazil Baig, Sandeep K N, Venkanna Babu. Synthesis of novel pyrimido-[5, 4-d] pyrimidines by sequential nucleophillic substitution reaction. J Chem Pharm Res, 2012; 11:4888–93.
Zhang J, Miller RB, Jacobus R. Development and validation of a stability-indicating HPLC method for the determination of degradation products in dipyridamole injection. Chromatographia, 1997; 44:247–52. CrossRef