
INTRODUCTION
The worldwide escalation of mycobacterial resistance (Dookie et al., 2018; Nguyen et al., 2019) [pulmonary and extrapulmonary tuberculosis (Tb)] to conventional vaccines and antibiotics poses a serious concern to modern medicine (Castan et al., 2014). In 2019, the World Health Organization’s Global Tuberculosis Report estimates the occurrence of 10 million Tb cases globally. Besides this, 484,000 new cases of resistance to rifampin were also reported in a year, from which 78% of cases had multiple drug-resistant (MDR-Tb) (WHO, 2020a). It decreases the effectiveness of current treatments and causes thousands of deaths. Therefore, the need to brainstorm for this disease and its remedies still persist.
Tuberculous meningitis (TbM) is severe form of extrapulmonary Tb which is associated with high mortality of around 13% to 57% even after 12 months of anti-tubercular treatment (Donovan et al., 2019; Rohlwink et al., 2019; Soria et al., 2019; Thwaites et al., 2013). Mycobacterium tuberculosis (MTb) causing TbM in human is creating serious condition globally including in India as the estimated mortality is 627,000 annually (WHO, 2020b). Neuro-inflammation is a key pathological process that eventually forms millary TbM, increasing endovascular pressure, cranial nerve infarction, and obstruction in hydrocephalus that can be observed in computed tomography scan or magnetic resonance imaging (Donovan et al., 2019). Other clinical signs and symptoms of TbM include recurrent periods of chills and fever, headache, abdominal pain, vomiting, altered cautiousness, nausea, hepatomegaly, and hypertension (Rohlwink et al., 2019). Various host genetic factors regulating immunological pattern recognition molecules, such as Toll-like receptors polymorphisms were found to render susceptibility to TbM (Faksri et al., 2018; Gagneux et al., 2006; Thuong et al., 2007).
Vaccine and antibiotics currently used in the treatment are also facing their limitations such as increase in the probability of the emergence of MDR Mycobacterium strains which is due to long treatment duration and improper administration of drugs (Cresswell et al., 2019; Singh et al., 2019). The main drawbacks of current conventional anti-tubercular agents are the hepatotoxicity, various adverse side effects and development of MDR. Drug-resistant bacteria require higher doses of antibiotics that often cause intolerable toxicity (Dookie et al., 2018). Similarly, a vaccine that is currently used to cure Tb is bacillus Calmette–Guérin (BCG) produced from the live, attenuated Mycobacterium bovis. It induces some immune-activating factors and prevents TbM in children, but provide a limited contribution to the cure of patient suffering from pulmonary and latent Tb (Barry et al., 2009). Thus, to overcome the limitations of BCG and to reduce the Tb infection at initial stages, more efficient vaccines are required (Andersen and Doherty, 2005; Darrah et al., 2019; Nguipdop et al., 2016; Nieuwenhuizen and Kaufmann, 2018). The advent of reverse vaccine technology has reduced the time duration and cost of vaccine production over conventional methods. Although many vaccines including whole-cell derived vaccines, recombinant BCGs (Honda et al., 2008; WHO, 2017), recombinant viral vectors, mycobacterial extracts, protein-adjuvant combinations, and reverse vaccine-derived epitope vaccines are produced but they are still in pre-clinical phase or different phases of clinical trials (Sable et al., 2020). Two anti-Tb agent’s bedaquiline (class: diarylquinoline) and delamanid (class: nitroimidazoles) have been introduced to the market (Evans et al., 2016; Grzelak et al., 2019), but soon during the retrospective study on 24 cases of MTb in Iraq, Ghajavand et al. (2019) reported their resistant strain (Polsfuss et al., 2019; Veziris et al., 2017). Therefore, there remains an urgent need to discover new anti-Tb drugs that can shorten the treatment period and overcome the growing problem of drug resistance (Young et al., 2019).
Recently, the epitope-based vaccine designing technique has successfully used in finding the control of infectious diseases like shigellosis (Pahil et al., 2017). In this way, epitope-based vaccine designing is becoming a powerful tool in the stimulation of cellular and humoral immunity against infectious diseases (Majid and Andleeb, 2019).
Outer membrane and secreted proteins of MTb are required for membrane integrity, protection from toxins and are also necessary for pathogenicity and virulence. These proteins help in nutrition uptake as well as guide the bacterial multidrug-efflux pump to extrude the therapeutic drugs thus, enabling resistance in MTb strain when it is inside the host macrophage (Young et al., 2019). Goldberg et al. (2012) in their study discussed that the virulence decreased in peptidoglycan [outer membrane protein (OMP)] mutated strains. In another study by Stamm et al. (2019) they depicted that exoproteome consisting of membrane as well as secreted proteins of MTb that interacts with the eukaryotic membrane to induce host dependent interaction with the Tb bacterium. Therefore, it was hypothesized that OMPs prove to be efficient target for vaccine designing.
In the present study, comparative proteome analysis and reverse-vaccine based techniques have been applied to design a chimeric multi-epitope vaccine against drug-resistant MTb.
MATERIALS AND METHODS
The complete protocol of the study is summarized as a flow chart in Figure 1.
Proteome selection
The genome of drug-sensitive and multidrug-resistant clinical strains of MTb along with reference strains of pathogenic (MTB H37rv), as well as of non-pathogenic (MTB H37ra) and BCG Bovine were retrieved from NCBI. To select a suitable genome sequence, an online database Public database for molecular typing and microbial genome diversity (PubMLST) (Zeng et al., 2017) was used. The proteome of selected strain was retrieved from NCBI and in-silico reverse vaccinology techniques were applied in identifying the potential vaccine targets.
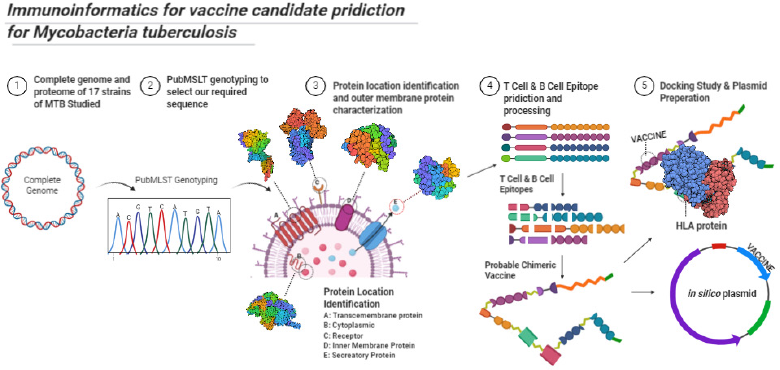 | Figure 1. Protocol of study summarized. [Click here to view] |
Prediction of novel antigenic proteins and their localization
Vaxign server (He et al., 2010) (http://www.violinet.org/vaxign/index.php) [genome and proteome-based vaccine prediction server using filters like transmembrane regions (TM), subcellular localization, adhesion properties] was used for predicting the consensus vaccine candidates (antigenic proteins). The complete proteome of the selected strain in the FASTA format was subjected and the experimental threshold value was assigned as 0.51. All the proteins with values ≥0.51 were considered to possess good adhesion property and selected as consensus antigens. To reduce any cross-reactivity between the developed vaccine and human cell, only non-homologous proteins were considered as vaccine candidates. For this, BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins) analysis was carried out and the sequences having an expectation value (E-value) ≤ 10−4 were considered as homologous sequences and were excluded from the study. Furthermore, the localization of the non-homologous above-identified proteins was sorted as extracellular, periplasmic, OMPs (Laal and Zolla-Pazner, 2010), inner membrane, or cytoplasmic using PSORTb 3.0.2 (database for subcellular location of proteins of bacteria) (Yu et al., 2010), and CELLO (predictive software determining the protein cellular location of bacteria based on support vector machine based on n-peptide composition) (Yu et al., 2006) servers.
Prediction of signal peptide antigens
Signaling nature [classical, non-classical secreted proteins as well as proteins with GPI (Glycosylphosphatidylinositol)-anchor] of the above-selected proteins were predicted by SignalP 4.1, SecretomeP 2.0, and PredGPI (Angala et al., 2014; Pierleoni et al., 2008), respectively. Based on the Sec-dependent pathway, SignalP 4.1 (Henrik, 2017) server was applied for the prediction of the classical group of secretory proteins. The positional limit for prokaryote organisms was set as 70 residues truncation and for remaining parameters, default values were considered. SecretomeP version 2.0 (Bendtsen et al., 2004, 2005) was used for the prediction of non-classical groups of secretory proteins by selecting the default values/options and all the proteins having N–N score ≥ 0.5 were considered as non-classical secreted proteins. Similarly, PredGPI (Angala et al., 2014; Pierleoni et al., 2008) was also used to predict both the presence of the GPI-anchor and the position of the ω-site using default values.
Antigenicity and allergenicity prediction of the screened proteins
After getting insight into the signaling nature of the above-selected proteins, VaxiJen and AntigenPro web servers were used for the screening of consensus antigenic proteins (Doytchinova and Flower, 2007; Magnan et al., 2010) using the default parameter (threshold value > 0.7). Proteins predicted as positive by both the tools were considered as consensus antigens and were subjected to Allergen FP v.1.0 (Dimitrov et al., 2014) tool to investigate the allergic nature of the selected proteins employing the default parameters.
Characterization of physiochemical properties of proteins
TM of the non-allergic proteins were checked using the Transmembrane hidden Markov model (TMHMM) method (Krogh et al., 2001). ABTMpro server (Cheng et al., 2005) (http://scratch.proteomics.ics.uci.edu/) was used to characterize whether a selected protein sequence belongs to transmembrane protein or not. This server also described the probabilities of TM as an alpha-helical or a beta barrel transmembrane protein. As the protein should be soluble in the cytoplasm during over expression in Escherichia coli during large scale vaccine production; therefore, the SOLPro (Magnan et al., 2009) tool was used to depict the solubility of the selected proteins. Finally, the sequence similarity between the selected OMPs of MTb and its intraspecies, i.e., MTb complex (MTbC) was also observed by the Ortho MCL database (Chen, 2006).
T-cell (MHC-I, MHC-II) and B-cell epitopes prediction
For potent epitopes identification, T-cell epitope analysis was performed using four servers (i) Immune Epitope Database (IEDB) Major Histocompatibility Complex (MHC)-I prediction server, (ii) Peptides Naturally Processed by Major Histocompatibility Complex (MHC-NP), (iii) NetCTLpan1.1, and (iv) NetMHCpan 3.0. The IEDB (http://tools.immuneepitope.org/processing/) MHC-I prediction server with default parameters was used to identify the epitopes having the possibility to interact with MHC-I proteins. MHC elution-pattern-based server MHC-NP (http://tools.immuneepitope.org/mhcnp/) was used to predict the probability of a selected peptide can be processed naturally or not. Similarly, the NetCTLpan1.1 server (http://www.cbs.dtu.dk/services/NetCTLpan/) was used to predict the cytotoxic lymphocyte epitopes of proteins. Finally, NetMHCpan3.0 server (http://www.cbs.dtu.dk/services/NetMHCpan/) was used to predict the ability of peptide-MHC class I binding.
Consensus T-cell epitopes having the binding ability to MHC class II molecules were identified by four prediction servers like IEDB, MHC Class-II (http://tools.iedb.org/mhcii/), Propred (https://webs.iiitd.edu.in/raghava/propred/index.html), and NetMHC-II (http://www.cbs.dtu.dk/services/NetMHCII-2.2/) under default parameter conditions.
Similarly, B-cell epitopes prediction was done by ABCPred (https://webs.iiitd.edu.in/raghava/abcpred/ABC_submission.html), the BCPred (https://webs.iiitd.edu.in/raghava/bcepred/bcepred_submission.html), and IEDB server (http://tools.iedb.org/bcell/) with cut-off score value >0.8. Common epitopes in all three servers were considered for further studies.
Epitope characterization
Above predicted epitopes were compared and the common antigenic epitopes were subjected to the IEDB server to identify the epitopes having immunogenic property. The epitopes showing a positive immunogenicity score were shortlisted for antigenic analysis using VaxiJen version 2.0 (Doytchinova and Flower, 2007). According to the criteria described by Khan et al. (2019), peptides showing score value ≥1.0 were selected for the toxicity prediction by the ToxinPred tool.
For the chimeric vaccine, epitopes should be hydrophilic (present on the surface), otherwise they will not be able to initiate the immune reaction in the host cell. The epitope hydropathy was analyzed through the grand average of hydropathy (GRAVY) score analysis through the ProtParam tool. The GRAVY value of epitope was calculated by using the following calculation:
G = grand average of hydropathy value; Ha = hydropathy values of amino acids; N = number of amino acid residues in a given protein
A positive value of GRAVY score indicates the hydrophobic nature and a negative value suggests the hydrophilic nature of proteins.
“MHC restricted allele prediction tool” of the IEDB server was used to identify the MHC class I and II-restricted epitopes. Identified epitopes were further crosschecked by the MHCcluster 2.0 server to confirm the above prediction (Thomsen et al., 2013).
Construction of chimeric vaccine
The chimeric vaccine sequences were designed manually using the results of epitopes analysis. Overlapping sequences of epitopes were merged and three chimeric vaccine candidates (VC1, VC2, and VC3) were constructed by the protocol described by Solanki et al. (2019). Briefly, all the selected epitopes were joined using universal amino acid linker sequences (HEYGAEALERAG and GGGS). Further to enhance the Immunogenicity of constructs distinct adjuvant were added using “EAAAK” linkers at both the termini (N and C). The adjuvant used for VC1, VC2 and VC3 were 50s ribosomal L7/L12 protein (Lee et al., 2014), beta-defensin and HBHA respectively. Further to enhance the vaccine competence, a sequence of 13 amino acid universal epitope (AKVAAWTLKAAAC) also known as non-natural pan-DR (PADRE) (Alexander et al., 2000) was used.
Characterization of vaccine constructs
The above three vaccine constructs were analyzed according to antigenicity, allergenicity, and solubility prediction. For the prediction of allergenic nature, the AlgPred server (Marti et al., 2007) was used, whereas the antigenicity of the constructs was predicted using ANTIGENpro (Magnan et al., 2010) and VaxiJen 2.0 server. Solubility and corresponding probability (≥0.5) of the vaccine constructs were predicted by the SOLpro server (Magnan et al., 2009).
Physiochemical properties [amino acids count, Isoelectric Point (PI) values, their molecular weight, hydropathicity GRAVY score, aliphatic, and instability index] of the vaccine constructs were characterized using the Expasy ProtParam server (Gasteiger et al., 2005). The 2o structure of all three vaccine constructs was predicted by PSIPRED v3.3 program (McGuffin et al., 2000). Furthermore, the tertiary structures of the vaccine constructs (VC1, VC2, and VC3) were predicted by the Phre2 (Kelley et al., 2015) online tool. The structures were saved in .pdb file format.
Molecular docking study
Interaction studies of vaccine constructs (VC1, VC2, and VC3) with 10 different HLA alleles (Axelsson et al., 2015) [(HLA-A*02:01(6EQA), HLA-A*24:02 (4F7M), HLA-B*15:01 (1XR8), HLA-B*35:01 (1A1N), HLA-B*39:01 (4O2E), HLA B*44:02 (1N2R), HLA-B*58:01 (5IM7), HLA-DR2 (DRA*0101, DRB1*1501) (1BX2), HLA-DRA1*0101/DRB5*0101 (1H15) and HLA-DQ2.3 (DQA1*03:01/DQB1*02:01) (4D8P)] was performed using the PatchDock server. 3D structures of all the HLA alleles were obtained from the protein data bank Research Collaboratory for Structural Bioinformatics - Protein Data Bank (RCSB-PDB) and saved in the .pdb file format. The best 10 solutions to the PatchDock were further refined by FireDock.
Codon optimization and in-silico cloning of vaccine construct
Codon optimization was performed by Java Codon Adaptation Tool (JCAT) to enhance the production of heterologous protein (vaccine construct) in E. coli (Chauhan et al., 2019). During optimization, the rho-independent transcription terminators, prokaryotic ribosomal binding sites, and few restriction sites were kept constant. The expression of the vaccine construct was predicted by the Snapgene tool after cloning the gene sequence of a construct in E. coli pET28a vector (Solanki et al., 2019).
RESULTS AND DISCUSSION
Comparative subtractive proteomic approach to screen the MTb strains
The complete genome sequences of seventeen MTb strains were compared by PubMLST and the results are summarized in Table 1. Out of 17 strains, 6 having drug-resistance were found suitable for the study. Furthermore, among the six selected strains, only three clinical strains showing their isolation source from the cerebrospinal fluid sample having a greater possibility to possess MTb virulence were screened. The proteome of possible three clinical strains were further analyzed for similar proteins using multiple alignment tools (data not shown). Finally, to reduce redundancy and based on alignment, MLST values, proteome size the MTb strain LJ319 (NZ_CP026742.1) having 4,025 proteins was selected for the study (Hatolkar et al., 2018).
Prediction of novel antigens
To identify the potential proteins for vaccine construct, all the 4,025 proteins of reference proteome (LJ319) were filtered according to their subcellular localization using vaxign, CELLO and PSORTb tool. Out of 4,025 proteins, 982 different proteins having their localization either in the periplasmic or the extracellular or outer membrane of the bacterial cell were found suitable for the study (data not shown). The rest of the proteins that were present either in the cytoplasm or inner cytoplasmic membrane region were excluded from the study. Cellular localization of bacterial Possibly surface exposed (PSEs) and outer membrane plays an essential role in pathogenesis such as drug efflux pumps, permeability barrier; membrane protein also helps in integrity, active transport, and diffusions of nutrients (Angala et al., 2014). Sajjad et al. (2020) during the study of Acinetobacter nosocomialis also used the above tool for designing multi epitope vaccine which depicts the authenticity of the results obtained through the tools.
Above screened 982 proteins were examined for their adhesion nature through vaxign server, and only 165 OMPs were found to possess the adhesion property. Adhesion and signal properties are the characteristic features of the vaccine candidates described by Chauhan et al. (2019) here vaxign predicts that out of 982 proteins, only 165 OMPs possess the adhesion property. Majid and Andleeb (2019) suggested that the proteins having allergic properties cannot be considered for vaccine candidate prediction. Similarly, for vaccine designing, protein should be soluble in E. coli and host cell for protein production and biological reaction respectively (Magnan et al., 2010); therefore, the screened 165 OMPs were further refined on the above basis and only 35 proteins were found suitable for the analysis.
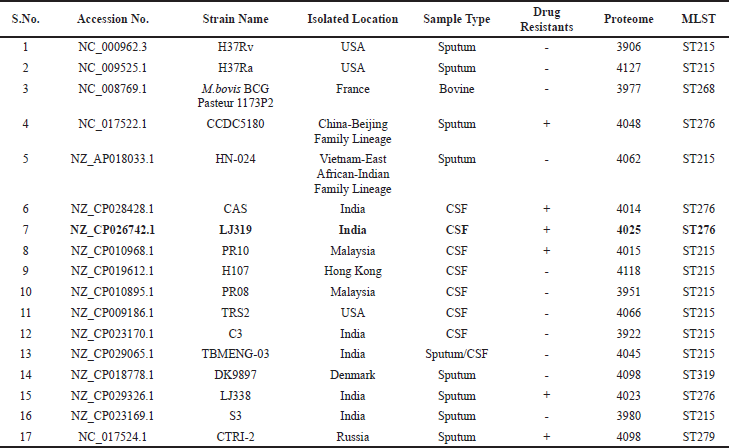 | Table 1. Complete genome sequences of seventeen M. tuberculosis strains compared by PubMLST (Multi Locust Sequence Typing). [Click here to view] |
In the next step, signaling natures of these 165 OMPs were studied through SignalP, SecretomeP 2.0, and PredGPI. SignalP 4.1 based on the Sec-dependent pathway predicts 85 proteins under the “classical secreted proteins.” Similarly, SecretomeP 2.0 predicted 135 proteins as the “non-classical secreted proteins” and PredGPI predicts the presence of 8 GPI-anchor proteins, i.e., the presence of the ω-site.
Alongside, these 165 OMPs were also examined for their antigenicity properties using VaxiJen and ANTIGENpro web server. Results suggest that out of 165, only 36 OMPs found common in both possessing the antigenic property were selected for the study; summarized in Table 2.
Characterization of physiochemical properties of proteins
The identification of TM α-helices by TMHMM method suggests that all the 36 OMPs containing either 0 or 1 helix, confirming their presence in the outer membrane region (Supplementary Table 3). Hence, the results of the TMHMM method validate the finding of vaxign, CELLO and PSORTb tool. Furthermore, when all the 36 OMPs were subjected to the AllergenPro web server to find out if their exist any allergic tendency, then 1 OMP (WP_003401880.1) was found to pose allergic behavior in the host cell, and therefore excluded from the further studies. The solubility of the above OMPs were examined through SOLPro suggesting that out of 35 OMPs, 22 OMPs were soluble whereas, rest 13 OMPs having insoluble nature during overproduction in vitro were excluded from the race of potential vaccine candidates (Supplementary Table 3).
To reduce the pathogenesis of TbM infection, a potential vaccine candidate should have a tendency to also identify the associated intra-pathogenic species. Therefore, the presence of the orthologs sequences of the MTbC in the above selected 22 OMPs were detected by the Ortho MCL server and the results are summarized in Supplementary Table 4. Results suggest that out of 22 OMPs, 6 proteins (WP_031663355.1, WP_031661316.1, WP_016330440.1, WP_009938581.1, WP_003910913.1 and WP_003900236.1) were not common in all the 5-members of MTbC, therefore, were excluded from the potential vaccine candidates list and the filtered 16 OMPs were selected for further studies. Palucci et al. (2016) observed that even a few GGA-GGN repeats of PE/PPEs proteins can play an important role in Tb pathogenesis and provide immunity to host by activating the TLR2-dependent MTb entry into macrophages. Hence, suggests that the family group such as PPE, PE and PE_PGRS proteins influences the antigenic variation and immune system evasion. In another study Ocampo et al. (2014) depicted that antigen Rv1911c (LppC), are lipoproteins representing an important protein present on the cell envelope thereby enhancing MTb pathogen’s virulence. Therefore, considering their important feature these proteins were included among above selected 16 OMPs (Abraham et al., 2018; Kavvas et al., 2018; Phelan et al., 2016).
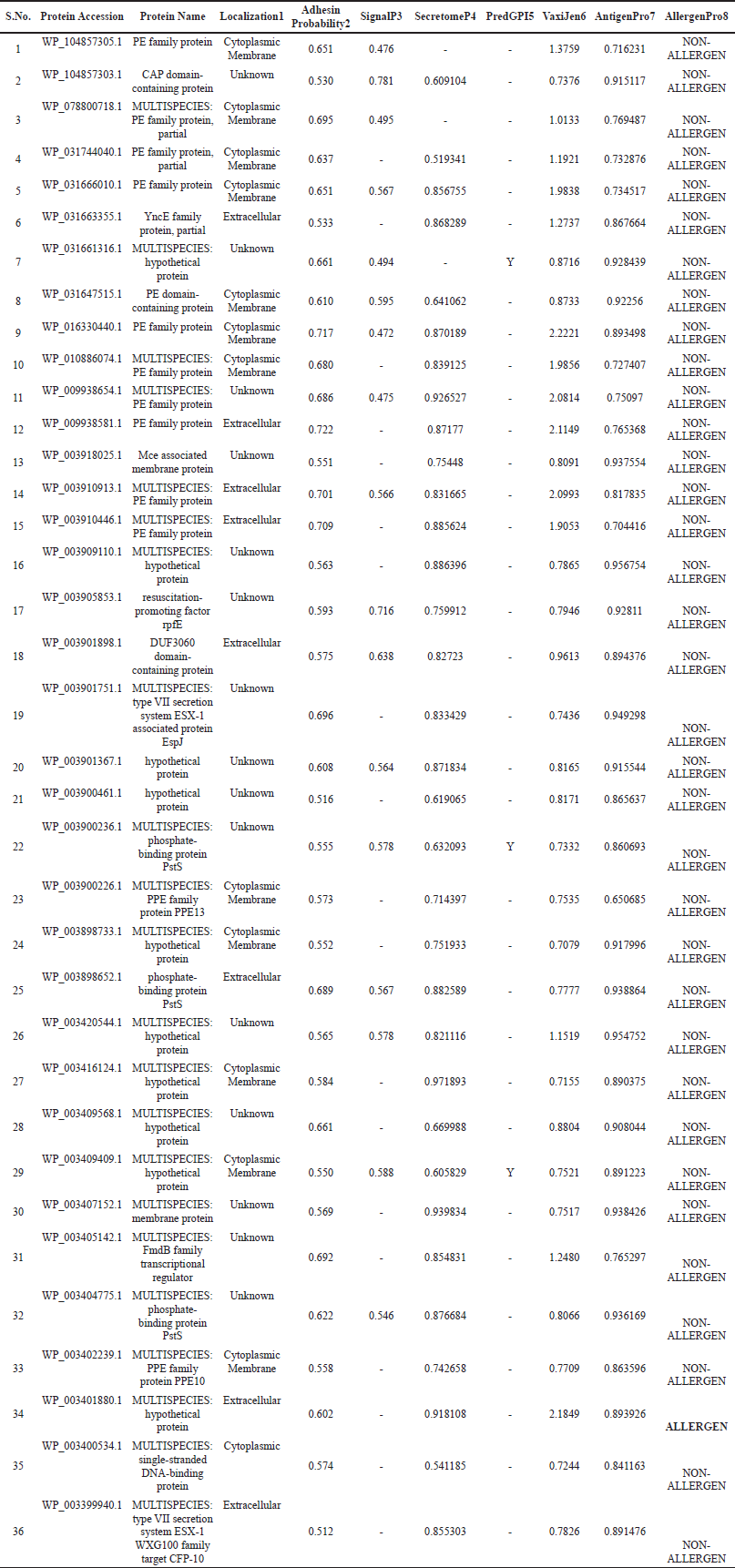 | Table 2: Proteome analysis of LJ319 ( NZ_CP026742.1) strain of M. tuberculosis and outer membrane protein characterization using different servers. 1: Localization using CELLO, PSORBTb; 2: Adhesion property using Vaxign server; 3,4,5: Protein signaling and GPI-anchor by SignalP, SecretomeP and PredGPI; 6,7: Antigenicity prediction using Vaxijen, AntigenPro; 8: Allergenicity by AllergenPro Server. [Click here to view] |
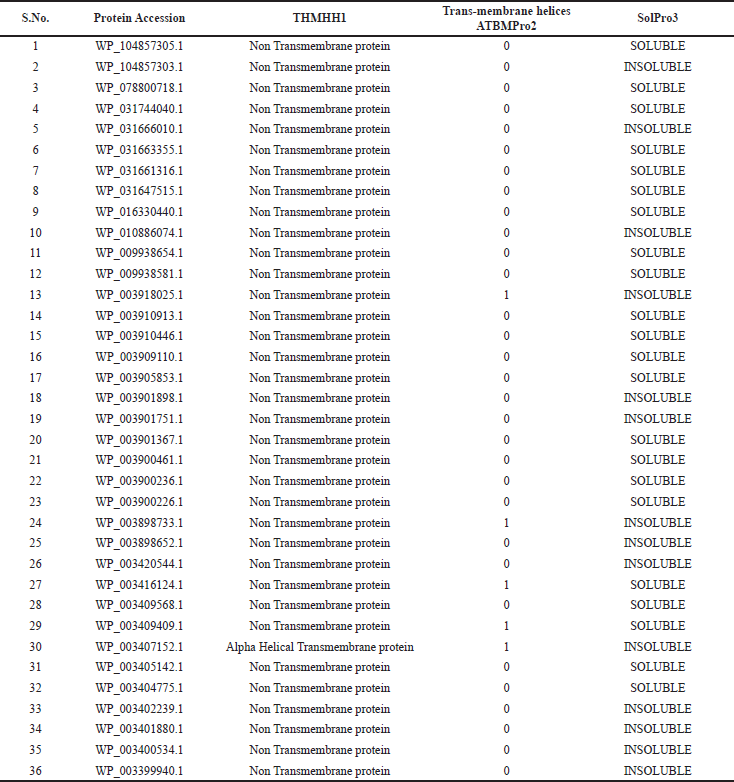 | Table 3. Screening of potential vaccine candidates for transmembrane regions1,2 and solubility property3 during over-expression in plasmid vector in E.coli during vaccine production. [Click here to view] |
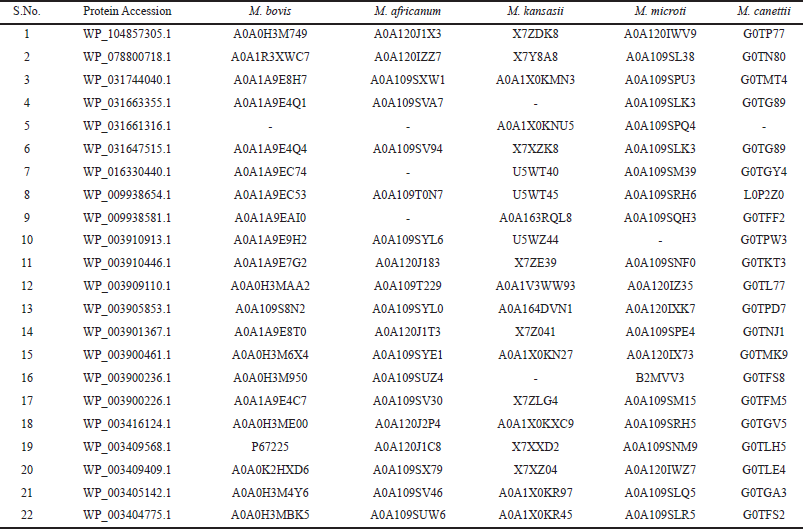 | Table 4: Screening of orthologs associated intra-pathogenic species in the Mycobacterium tuberculosis complex (MTbC) using OrthoMCL. [Click here to view] |
T-cell (MHC-I, MHC-II) and B-cell epitopes prediction
All the 16 OMPs when subjected to IEDB server for epitopes prediction, then based on higher affinity [Inhibitory Concentration (IC) < 50 nM] and good percentile rank (≤0.2), 221 MHC-I, 69 MHC-II, and 81 B-cell epitopes were filtered. To further refine the IEDB prediction for MHC-I and II binding epitopes, MHC-NP, netCTL, netMHC, and Propred tools were used. As a result, 159 MHC-I and 41 MHC-II epitopes, found common in the results of these tools were selected for characterization studies. Similarly, B-cell epitopes were filtered by IEDB, BepiPred linear epitope prediction servers, BCPREDS and ABCPred tools suggesting 31 common B-cell epitopes were suitable for the study.
Epitope characterization
Immunogenicity, antigenicity and toxicity prediction of epitopes
The above selected MHC- I (159), MHC-II (41), and B cell epitopes (31) were subjected to the IEDB immunogenicity prediction tool to check immunological behavior of the epitopes. Using immunogenicity score (>0.038 cutoff value), 98 and 34 MHC-I and MHC II epitopes respectively out of 159 MHC-I and 41 MHC-II epitopes, were picked for further studies that showed higher potency to stimulate naive T cells and also to induce cell-mediated immunity, results are in (Supplementary Table 5). Furthermore, the antigenicity of selected MHC I and II epitopes was evaluated by the VaxiJen web server. A total of 51 (29 MHC- I + 22 MHC-II) epitopes containing antigenicity values more than 0.7 were considered as the potent epitopes (Supplementary Table 5). Similarly, out of 31 B-cell epitopes, only 19 were found immunogenic and antigenic epitopes (Supplementary Table 5). In the next step, cross-reactivity induced by epitopes in the host tissue was figured out through the ToxinPred server and all the 70 epitopes were found to be non-toxic.
Physiochemical analysis of epitopes
The physiochemical properties of epitopes were explored by GRAVY analysis through the ProtParam tool. According to the considered criteria, 40 epitopes (13 from MHC I, 12 from MHC II, and 15 from B cell) having VaxiJen score value >1.0 were subjected to the GRAVY analysis. Among 40 epitopes, only 26 epitopes (MHC I: 4, MHC II: 7, and B cell: 15) having negative score values were predicted as hydrophilic. Above screened hydrophilic epitopes are possibly present in the outer surface, and therefore have a greater tendency to initiate the immunogenicity in the host cell. Hence, chimeric vaccine constructs were designed using all the 26 epitopes.
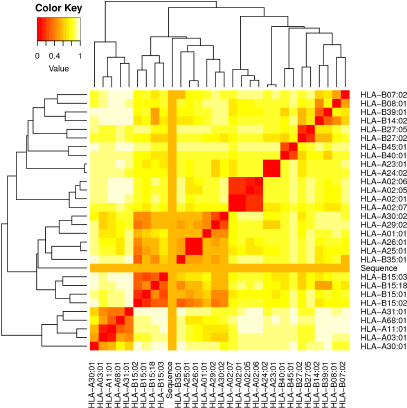 | Figure 2. Heat map analysis of T-cell epitopes using MHC cluster. [Click here to view] |
MHC restriction and cluster analysis of selected epitopes
After physiochemical analysis, the selected epitopes were further validated for the MHC interaction using the MHC cluster and the results are shown as a heat map (Fig. 2) and dynamic tree. The epitopes are clustered according to the interaction with HLA. The red color suggests strong interaction, while the yellow color indicates weak interaction. Selected 4 MHC I and 7 MHC II epitopes showed strong interaction with HLA genes.
Construction of chimeric vaccine
All the shortlisted 26 epitopes {4 MHC I epitopes (107CESGGNWSI115, 398WPIRAPSRL406, 158HYRFTLYHL166 and 104RADRARNTY112), 7 MHC II epitopes (184YGNGGPGGA192, 203WIYGHGGHG211, 205YGHGGHGGA213, 51VEGHTHTIS59, 57IEGDDTDRR65, 90VSPPETTTD98, and 54YRTIDIRNH62) and 15 B-cell epitopes (177AGAIGNGGDGGNGGTS192; 197GSGGDGGNGGNAGLIG212; 231GTGGNGGLLLGFNGTN246; 175GGAGGNGGWLYGNGGP190; 132GLLYGNGGNGGAGDTA147; 355GGAGGAGGRGGWLVGN370; 349GHAGGAGGAGGAGGRG364; 504GGTGGDGGDGGHAGTG519; 467NGGIGGDGAGGGNATS482; 492GGNGGAGGDAGHGGTG507; 495GGAGGNGATGGTGVGN510; 199AGGGGGGTTPTGYLGP214; 169GAGGGDVGGGGAGGTT184; 263GNGNDGNTNFGSGNAG278 and 98GGVGNARADRA RNTYT113)} were used to design the chimeric vaccine. Two linkers HEYGAEALERAG and GGGS were used to join the epitopes. 50S ribosomal protein L7/L12 (rplL) (Accession no. WP_003403353.1), beta-defensin (Accession no. WP_031737436.1), HBHA (Accession no. WP_094028633.1), and PADRE (AKVAAWTLKAAAC) were successfully used as adjuvant (Lee et al., 2014) and linker (Alexander et al., 2000), respectively, for the construction of vaccine candidates by a linker “EAAAK” at both termini (N and C). Satyam et al. (2020) also used same adjuvant during the vaccine construction against Mycobacteroids. VC1, VC2, and VC3 prove their efficacy through their antigenic, allergenic, and toxicity analysis.
Characterization of vaccine constructs
Antigenicity, allergenicity, and solubility prediction
Antigenicity, allergenicity, and solubility of VC1, VC2, and VC3 were predicted by the ANTIGENpro, VaxiJen 2.0, AlgPred, and SOLpro server. The antigenicity score value >0.569 in ANTIGENpro and >1.5596 in VaxiJen 2.0 indicates a satisfactory antigenic property of all the three vaccines constructs. AlgPred server predicted the non-allergenic behavior of VC1, VC2, and VC3. Similarly, SOLpro showed good solubility (>0.9820) of these vaccine constructs during their heterologous expression in the E. coli.
Physicochemical analysis of designed vaccine constructs
ProtParam server suggests the molecular weight of all vaccine constructs ranges between 59 and 72 kDa. All three vaccine constructs are steady in the corresponding pH (Table 6). A negative value (−0.544) of GRAVY (a hydropathic index) analysis suggests the hydrophilic character of the designed constructs which indicates strong interactions with water molecules. Further, the aliphatic index ranges from 49.85 to 59.07 for all vaccines construct suggest protein stability in a defined temperature range. Instability score of all vaccine constructs is <40 showed indicates the good stability of protein to commence an immunogenic reaction. The non-toxic and non-allergenic vaccine may be the good immunotherapy against the pathogenic MTb (Solanki et al., 2019). Based on physiochemical behavior, the shortlisted vaccine constructs (VC1, VC2, and VC3) were subjected for interaction studies.
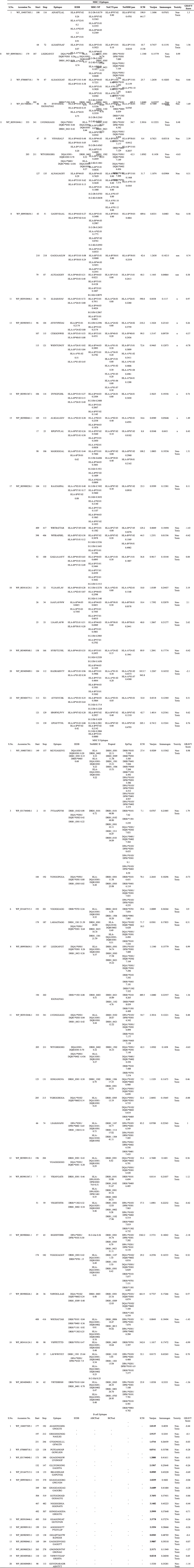 | Table 5. Identification of potent MHC I, MHC II and B cell epitopes and their characterization such as antigenicity, immunogenicity toxicity and hydrophilicity was performed using various servers. [Click here to view] |
 | Table 6. Vaccine construct and their physiochemical characterization. [Click here to view] |
 | Figure 3. Secondary structure prediction of vaccine constructs using PSIPRED. (a) Vaccine construct 1 (VC1) secondary structure shows helix, β-sheets and turns; (b) Vaccine construct 2 (VC2) secondary structures shows helix and β-sheets; (c) Vaccine construct 3 (VC3) secondary structures shows only helix. [Click here to view] |
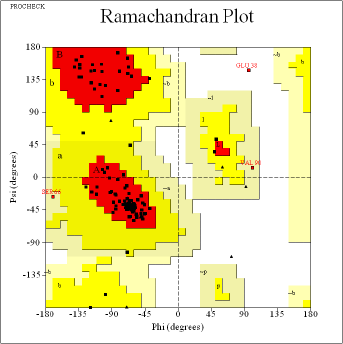
 | Figure 4. (a) Tertiary structure prediction of vaccine constructs VC1 using Phre2. (b) Ramachandran plot analysis of VC1 vaccine construct using RAMPAGE with 90.3% amino acids in most favored region and 8.9% in allowed region. [Click here to view] |
 | Figure 5. In-silico Docking of VC1 vaccine construct (red) with Human HLA alleles (blue and green). (a) HLA-A*02:01 docking with -35.39 binding energy; (b) HLA-A*24:02 with -21.04 binding energy; (c) HLA-B*15:01 with -23.17 binding energy; (d) HLA-DR2 (DRA*01:01-DRB1*15:01) with -32.5 binding energy. [Click here to view] |
 | Figure 6. In-silico cloning of VC1 vaccine construct into pET28a vector for its heterologous expression in E. coli using EcoRI and NdeI restriction enzyme. [Click here to view] |
Structure prediction of selected vaccine constructs
Secondary and tertiary structures of the final vaccine constructs (VC1, VC2, and VC3) were predicted by PSIPRED and Phre2 (Fig. 3a–c, respectively). The predicted secondary structure of VC1, VC2, and VC3 consists of alpha-helix, beta-sheet and beta-turn. The model of VC1, VC2, and VC3 constructs were validated by the Ramachandran plot (Fig. 4).
Interaction of vaccine constructs with HLA allele’s protein
To observe the interaction of vaccine constructs with different HLA alleles of human, vaccine constructs (VC1, VC2, and VC3) were docked with 10 different HLA allele’s retrieved from literature and the results are summarized in Table 7. VC1 have the least global binding energy value with different HLA alleles, i.e., 6EQA (HLA-A*02:01); −35.39, 4F7M (HLA-A*24:02); −21.04, 1XR8 (HLA-B*15:01); −23.17, 1A1N (HLA-B*35:01); −8.29, 4O2E (HLA-B*39:01); −7.69, 1N2R (HLA B*44:02); −11.42, 5IM7 (HLA-B*58:01); −16.4, 1BX2 (HLA-DR2 (DRA*01:01-DRB1*15:01)); −32.5, 1H15 (HLA-DRA1*0101/DRB5*0101); −28.08, and 4D8P (HLA-DQ2.3 (DQA1*03:01/DQB1*02:01)); −1.64.
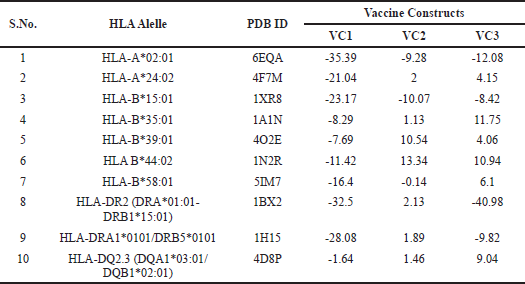 | Table 7. Vaccine constructs [VC1, VC2 and VC3] were docked with 10 different HLA allele’s proteins that correspond to M. tuberculosis susceptibility or pathogenicity. [Click here to view] |
Docking analysis elucidates the efficacy of the designed vaccine in term of binding affinity with HLA alleles. Based on docking analysis, the VC1 was screened as a potential vaccine construct having a tendency to stimulate the immune response as and when required in the host cell. Different adjuvants were also used in the designing process to improve the immune response. Through docking studies, the interaction of VC1 with TLR4/MD2 complex was validated. Satyam et al. (2020) also used TLR4/MD2 complex to predict the efficacy off the vaccine construct. TLR4/MD2 complex has a role in activating Dendritic Cells against a role in Tb.
Best docking was with HLA-A*02:01, HLA-A*24:02, HLA-B*15:01, and HLA-DR2 (DRA*01:01-DRB1*15:01) having binding energies −35.39, −21.04, −23.17, and −32.5 respectively; having interactions of alanine (ALA15) and serine of (SER42 and SER132) amino acids (data not shown); docking results are shown in Supplementary Figure 5a–d (Red depicts vaccine construct VC1 3D structure and blue-green depicts HLA protein 3D structure).
In-silico cloning of VC1 construct for its heterologous expression in E. coli
JCAT was used for cloning and expression prediction of constructed vaccine within the pET28a vector. For in silico cloning experiment the required cDNA sequences were obtained through reverse translation. Codon optimization results suggest 77.50% of constructs was made up of Guanine and Cytosine (GC) content. For the heterologous expression of VC1 in E. coli, its sequences was in-silico cloned into pET28a vector using EcoRI and NdeI restriction enzyme for the addition at 5′ and 3′ ends respectively (Fig. 6). The Codon Adaptation Index (CAI) value (1.0 for VC1) indicates the efficient heterologous expression of VC1 in E. coli cell.
CONCLUSION
The work performed is the stepwise proteomic screening for the identification of a multi-epitope chimeric vaccine targeting the MTb. Filters like subcellular localization, antigenicity, allergenicity, transmembrane α-helices, and solubility were utilized and three vaccine constructs (VC1, VC2, and VC3) were designed. Their secondary and tertiary structures were established through online tools. Based on in silico interaction studies with 10 HLA alleles, the VC-1 construct was found most potential. An in silico cloning studies using pET-28a (+) vector suggests the satisfactory expression and translation efficiency of the VC-1. The proposed anti-tubercular vaccine construct VC1 seems capable to initiate the immune response in the host cell and interact efficiently with HLA alleles. During the designing of VC1, besides, adjuvant (L7/L12 ribosomal protein) linker and PADRE epitopes were also added to enhance the anti-tubercular immune responses. Therefore, vaccine construct VC1, possess all the possible factors which are required to bring about the immunogenicity and feasibility against MTb. Further in vitro and in vivo expression studies in wet lab are needed to validate long-term immunological efficacy of predicted vaccine candidate. Further studies are also needed to detect the vaccine interaction with cell mediated and humoral immunity of the host.
CONFLICT OF INTERESTS
The authors declare no conflict of interests.
FUNDING
None.
REFERENCES
Abraham PR, Devalraju KP, Jha V, Valluri VL, Mukhopadhyay S. PPE17 (Rv1168c) protein of Mycobacterium tuberculosis detects individuals with latent TB infection. PLoS One, 2018; 13:e0207787. CrossRef
Alexander J, del Guercio MF, Maewal A, Qiao L, Fikes J, Chesnut RW, Paulson J, Bundle DR, DeFrees S, Sette A. Linear PADRE T helper epitope and carbohydrate B cell epitope conjugates induce specific high titer IgG antibody responses. J Immunol, 2000; 164:1625–33. CrossRef
Angala SK, Belardinelli JM, Huc-Claustre E, Wheat WH, Jackson M. The cell envelope glycoconjugates of Mycobacterium tuberculosis. Crit Rev Biochem Mol Biol, 2014; 49:361–99.
Andersen P, Doherty TM. The success and failure of BCG—implications for a novel tuberculosis vaccine. Nat Rev Microbiol, 2005; 3:656–62. CrossRef
Axelsson-Robertson R, Ju JH, Kim HY, Zumla A, Maeurer M. Mycobacterium tuberculosis-specific and MHC class I-restricted CD8+ T-cells exhibit a stem cell precursor-like phenotype in patients with active pulmonary tuberculosis. Int J Infect Dis, 2015; 32:13–22. CrossRef
Bendtsen JD, Kiemer L, Fausbøll A, Brunak S. Non-classical protein secretion in bacteria. BMC Microbiol, 2005; 5; doi;10.1186/1471-2180-5-58 CrossRef
Bendtsen JD, Jensen LJ, Blom N, Von Heijne G, Brunak S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel, 2004; 17:349–56. CrossRef
Barry CE, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, Schnappinger D, Wilkinson RJ, Young D. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol, 2009; 7:845–55. CrossRef
Castan P, De Pablo A, Fernández-Romero N, Rubio JM, Cobb BD, Mingorance J, Toro C. Point-of-care system for detection of Mycobacterium tuberculosis and rifampin resistance in sputum samples. J Clin Microbiol, 2014; 52:502–7. CrossRef
Chauhan V, Rungta T, Goyal K, Singh MP. Designing a multi-epitope based vaccine to combat Kaposi Sarcoma utilizing immunoinformatics approach. Sci Rep, 2019; 9:1–15. CrossRef
Cheng J, Randall AZ, Sweredoski MJ, Baldi P. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res, 2005; 33:72–6. CrossRef
Chen F. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res, 2006; 34:D363–8. CrossRef
Cresswell FV, Te Brake L, Atherton R, Ruslami R, Dooley KE, Aarnoutse R, Van Crevel R. Intensified antibiotic treatment of tuberculosis meningitis. Expert Rev Clin Pharmacol, 2019; 12:267–88. CrossRef
Darrah PA, DiFazio RM, Maiello P, Gideon HP, Myers AJ, Rodgers MA, Hackney JA, Lindenstrom T, Evans T, Scanga CA, Prikhodko V, Andersen P, Lin PL, Laddy D, Roederer M, Seder RA, Flynn JAL. Boosting BCG with proteins or rAd5 does not enhance protection against tuberculosis in rhesus macaques. NPJ Vaccines, 2019; 4:1–3. CrossRef
Dimitrov I, Naneva L, Doytchinova I, Bangov I. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics, 2014; 30:846–51. CrossRef
Donovan J, Figaji A, Imran D, Phu NH, Rohlwink U, Thwaites GE. The neurocritical care of tuberculous meningitis. Lancet Neurol, 2019; 18:771–83. CrossRef
Dookie N, Rambaran S, Padayatchi N, Mahomed S, Naidoo K. Evolution of drug resistance in Mycobacterium tuberculosis: a review on the molecular determinants of resistance and implications for personalized care. J Antimicrob Chemother, 2018; 73:1138–51. CrossRef
Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform, 2007; 8:1–7. CrossRef
Evans TG, Schrager L, Thole J. Status of vaccine research and development of vaccines for tuberculosis. Vaccine, 2016; 34:2911–4. CrossRef
Faksri K, Xia E, Ong RTH, Tan JH, Nonghanphithak D, Makhao N, Thamnongdee N, Thanormchat A, Phurattanakornkul A, Rattanarangsee S, Ratanajaraya C, Suriyaphol P, Prammananan T, Teo YY, Chaiprasert A. Comparative whole-genome sequence analysis of Mycobacterium tuberculosis isolated from tuberculous meningitis and pulmonary tuberculosis patients. Sci Rep, 2018; 8:1–10. CrossRef
Gagneux S, DeRiemer K, Van T, Kato-Maeda M, De Jong BC, Narayanan S, Nicol M, Niemann S, Kremeri K, Gutierrez MC, Hilty M, Hopewell PC, Small PM. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A, 2006; 103:2869–73. CrossRef
Gasteiger EAR, Hoogland C, Gattiker A, Duvaud S, Wilkins MR. Protein identification and analysis tools on the ExPASy. Humana Press, 2005. CrossRef
Ghajavand H, Kamakoli MK, Khanipour S, Dizaji SP, Masoumi M, Jamnani FR, Fateh A, Siadat SD, Vaziria F. High prevalence of bedaquiline resistance in treatment-naive tuberculosis patients and verapamil effectiveness. Antimicrob Agents Chemother, 2019; 63:e02530–18. CrossRef
Goldberg DE, Siliciano RF, Jacobs WR Jr. Outwitting evolution: fighting drug-resistant TB, malaria, and HIV. Cell, 2012; 148:1271–83. CrossRef
Grzelak EM, Choules MP, Gao W, Cai G, Wan B, Wang Y, McAlpine JB, Cheng J, Jin Y, Lee H, Suh JW, Pauli GF, Franzblau SG, Jaki BU, Cho S. Strategies in anti-Mycobacterium tuberculosis drug discovery based on phenotypic screening. J Antibiot (Tokyo), 2019; 72:719–28. CrossRef
Hatolkar SM, Misra RN, Mahato R, Jadhav S. Whole-genome sequencing and annotation of a drug-resistant extrapulmonary clinical isolate of Beijing genotype Mycobacterium tuberculosis from Pune, India. Genome Announc, 2018; 6:e00504–18. CrossRef
He Y, Xiang Z, Mobley HLT. Vaxign: the first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J Biomed Biotechnol, 2010; 2010:297505. CrossRef
Henrik N. Predicting secretory proteins with SignalP. Methods Mol Biol, 2017; 1611:73.
Honda M, Matsuo K, Kanekiyo M, Promkhatkaew D. Recombinant BCG vaccine. US7670610B2 (Google Patents). 2008. Available via https://patents.google.com/patent/US7670610B2/en (Accessed 2 January 2020).
Kavvas ES, Catoiu E, Mih N, Yurkovich JT, Seif Y, Dillon N, Heckmann D, Anand A, Yang L, Nizet V, Monk JM, Palsson BO. Machine learning and structural analysis of Mycobacterium tuberculosis pan-genome identifies genetic signatures of antibiotic resistance. Nat Commun, 2018; 9:1–9. CrossRef
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc, 2015; 10:845–58. CrossRef
Khan F, Srivastava V, Kumar A. Computational identification and characterization of potential T-Cell epitope for the utility of vaccine design against enterotoxigenic Escherichia coli. Int J Pept Res Ther, 2019; 25:289–302. CrossRef
Krogh A, Larsson B, Von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol, 2001; 305:567–80. CrossRef
Laal S, Zolla-Pazner S. Immunodominant Mycobacterium tuberculosis peptides from cell wall proteins for early diagnosis and immunization. EP2281198B1 (Google Patents). 2010. (Accessed 31 December 2019).
Lee SJ, Shin SJ, Lee MH, Lee MG, Kang TH, Park, Soh BY, Park JH, Shin YK, Kim HW, Yun CH, Jung ID, Park YM. A potential protein adjuvant derived from Mycobacterium tuberculosis Rv0652 enhances dendritic cells-based tumor immunotherapy. PLoS One, 2014, 9:e104351. CrossRef
Majid M, Andleeb S. Designing a multi-epitopic vaccine against the enterotoxigenic Bacteroides fragilis based on immunoinformatics approach. Sci Rep, 2019; 9:1–15. CrossRef
Magnan CN, Randall A, Baldi P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics, 2009; 25:2200–7. CrossRef
Magnan CN, Zeller M, Kayala MA, Vigil A, Randall A, Felgner PL, Baldi P. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics, 2010; 26:2936–43. CrossRef
Marti P, Truffer R, Stadler MB, Keller-Gautschi E, Crameri R, Mari A, Schmid-Grendelmeier P, Miescher SM, Stadler BM, Vogel M. Allergen motifs and the prediction of allergenicity. Immunol Lett, 2007; 109:47–55. CrossRef
McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics, 2000; 16:404–5. CrossRef
Nguipdop-Djomo P, Heldal E, Rodrigues LC, Abubakar I, Mangtani P. Duration of BCG protection against tuberculosis and change in effectiveness with time since vaccination in Norway: a retrospective population-based cohort study. Lancet Infect Dis, 2016; 16:219–26. CrossRef
Nguyen TNA, Anton-Le Berre V, Bañuls AL, Nguyen TVA. Molecular diagnosis of drug-resistant tuberculosis; a literature review. Front Microbiol, 2019; 10:794.
Nieuwenhuizen NE, Kaufmann SHE. Next-generation vaccines based on Bacille Calmette-Guérin. Front Immunol, 2018; 9:121. CrossRef
Ocampo M, Curtidor H, Vanegas M, Patarroyo MA, Patarroyo ME. Specific interaction between Mycobacterium tuberculosis lipoprotein-derived peptides and target cells inhibits Mycobacterial entry in vitro. Chem Biol Drug Des, 2014; 84:626–41. CrossRef
Pahil S, Taneja N, Ansari HR, Raghava GPS. In silico analysis to identify vaccine candidates common to multiple serotypes of Shigella and evaluation of their immunogenicity. PLoS One, 2017; 12:e0180505. CrossRef
Palucci I, Camassa S, Cascioferro A, Sali M, Anoosheh S, Zumbo A, Minerva M, Iantomasi R, De Maio F, Di Sante G, Ria F, Sanguinetti M, Palù G, Brennan MJ, Manganelli R, Delogu G. PE_PGRS33 contributes to Mycobacterium tuberculosis entry in macrophages through interaction with TLR2. PLoS One, 2016; 11:e0150800. CrossRef
Phelan JE, Coll F, Bergval I, Anthony RM, Warren R, Sampson SL, Gey van Pittius NC, Glynn JR, Crampin AC, Alves A, Bessa TB, Campino S, Dheda K, Grandjean L, Hasan R, Hasan Z, Miranda A, Moore D, Panaiotov S, Perdigao J, Portugal I, Sheen P, de Oliveira Sousa E, Streicher EM, van Helden PD, Viveiros M, Hibberd ML, Pain A, McNerney R, Clark TG. Recombination in pe/ppe genes contributes to genetic variation in Mycobacterium tuberculosis lineages. BMC Genomics, 2016; 17:1–12. CrossRef
Pierleoni A, Martelli P, Casadio R. PredGPI: a GPI-anchor predictor. BMC Bioinform, 2008; 9:1–11. CrossRef
Polsfuss S, Hofmann-Thiel S, Merker M, Krieger D, Niemann S, Rüssmann H, Schönfeld N, Hoffmann H, Kranzer K. Emergence of low-level delamanid and bedaquiline resistance during extremely drug-resistant tuberculosis treatment. Clin Infect Dis, 2019; 69:1229–31. CrossRef
Rohlwink UK, Figaji A, Wilkinson KA, Horswell S, Sesay AK, Deffur A, Enslin N, Solomons R, Van Toorn R, Eley B, Levin M. Tuberculous meningitis in children is characterized by compartmentalized immune responses and neural excitotoxicity. Nat Commun, 2019; 10:1–8. CrossRef
Sable SB, Posey JE, Scriba TJ. Tuberculosis vaccine development: progress in clinical evaluation. Clin Microbiol Rev, 2020; 33:e00100–19. CrossRef
Sajjad R, Ahmad S, Azam SS. In silico screening of antigenic B-cell derived T-cell epitopes and designing of a multi-epitope peptide vaccine for Acinetobacter nosocomialis. J Mol Graph Model, 2020; 94:107477. CrossRef
Satyam R, Bhardwaj T, Jha NK, Jha SK, Nand P. Toward a chimeric vaccine against multiple isolates of Mycobacteroides—an integrative approach. Life Sci, 2020; 250:117541. CrossRef
Singh R, Dwivedi SP, Gaharwar US, Meena R, Rajamani P, Prasad T. Recent updates on drug resistance in Mycobacterium tuberculosis. J Appl Microbiol, 2019; 128:1547–67. CrossRef
Solanki V, Tiwari M, Tiwari V. Prioritization of potential vaccine targets using comparative proteomics and designing of the chimeric multi-epitope vaccine against Pseudomonas aeruginosa. Sci Rep, 2019; 9:1–19. CrossRef
Soria J, Metcalf T, Mori N, Newby RE, Montano SM, Huaroto L, Ticona E, Zunt JR. Mortality in hospitalized patients with tuberculous meningitis. BMC Infect Dis, 2019; 19:9. CrossRef
Stamm CE, Pasko BL, Chaisavaneeyakorn S, Franco LH, Nair VR, Weigele BA, Alto NM, Shiloh MU. Screening Mycobacterium tuberculosis secreted proteins identifies Mpt64 as a eukaryotic membrane-binding bacterial effector. mSphere, 2019; 4(3):e00354–19. CrossRef
Thuong NTT, Hawn TR, Thwaites GE, Chau TTH, Lan NTN, Quy HT, Hieu NT, Aderem A, Hien TT, Farrar JJ, Dunstan SJ. A polymorphism in human TLR2 is associated with increased susceptibility to tuberculous meningitis. Genes Immun, 2007; 8:422–8. CrossRef
Thomsen M, Lundegaard C, Buus S, Lund O, Nielsen M. MHCcluster, a method for functional clustering of MHC molecules. Immunogenetics, 2013; 65:655–65. CrossRef
Thwaites GE, van Toorn R, Schoeman J. Tuberculous meningitis: more questions, still too few answers. Lancet Neurol, 2013; 12:999–1010. CrossRef
Veziris N, Bernard C, Guglielmetti L, Le Du D, Marigot-Outtandy D, Jaspard M, Caumes E, Lerat I, Rioux C, Yazdanpanah Y, Tiotiu A, Lemaitre N, Brossier F, Jarlier V, Robert J, Sougakoff W, Aubry A. Rapid emergence of Mycobacterium tuberculosis bedaquiline resistance: lessons to avoid repeating past errors. Eur Respir J, 2017; 49:1601719. CrossRef
Young C, Walzl G, Du Plessis N. Therapeutic host-directed strategies to improve outcome in tuberculosis. Mucosal Immunol, 2019; 13:190–204. CrossRef
WHO. BCG vaccines 1 report on BCG vaccine use for protection against mycobacterial infections including tuberculosis, leprosy, and other nontuberculous mycobacteria (NTM) infections. SAGE Working Group on BCG Vaccines, WHO Secretariat, 2017.
WHO. Tuberculosis. 2020a. Available via https://www.who.int/news-room/fact-sheets/detail/tuberculosis (Accessed 2 January 2020).
WHO. Country profiles for 30 high tb burden countries 20 high TB burden countries based on absolute number of incident cases 10 high TB burden countries based on severity of disease burden (incidence per capita). 2020b. Available via www.who.int/tb/data (Accessed 2 January 2020).
Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Cenk Sahinalp S, Ester M, Foster LJ, Brinkman FSL. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics, 2010; 26:1608–15. CrossRef
Yu CS, Chen YC, Lu CH, Hwang JK. Prediction of protein subcellular localization. Proteins Struct Funct Genet, 2006; 64:643–51. CrossRef
Zeng LB, Wang D, Hu NY, Zhu Q, Chen K, Dong K, Zhang Y, Yao YF, Guo XK, Chang YF, Zhu YZ. A novel pan-genome reverse vaccinology approach employing a negative-selection strategy for screening surface-exposed antigens against leptospirosis. Front Microbiol, 2017; 8:396. CrossRef