
INTRODUCTION
Palbociclib (PB) is an orally administrated, highly selective inhibitor of cyclin-dependent kinase 4/6 (CDK). PB is authorized for the use in conjunction with letrozole as a first line endocrine-based treatment for postmenopausal women with estrogen receptor positive, human epidermal growth factor receptor 2 (HER2)-negative advanced breast cancer (Ehab and Elbaz, 2016). The drug is available in the market is formulated as hard capsules containing 75, 100, or 125 mg of PB as active ingredient under the brand name IBRANCE. As mentioned by the innovator, PB belongs to Biopharmaceutics Classification System Class II molecule (low solubility and high permeability) (Parylo et al., 2019).
Oral cancer chemotherapy with protein kinases such as CDK inhibitors involves administration of a variety of supportive medications such as analgesics, anti-helminthics, and acid-reducing agents (ARA), such as proton pump inhibitors (PPIs) (Cazzaniga et al., 2019). Supportive medicines are prescribed to alleviate the adverse effects of anticancer treatments and to address comorbidities associated with chemotherapy. PPIs are presently utilized in the top 20% of drugs worldwide to decrease gastroesophageal reflux and toxicity-related comorbidities associated with cancer therapy (Sharma et al., 2019). In clinical practice, PPIs significantly elevate stomach pH, which may impair the absorption of other therapeutic agents, particularly those that are weakly basic and show low solubility at higher pH. This type of pH-dependent drug-drug interaction (DDI) with PPIs decreases systemic drug exposure and has a significant impact on these medicines’ therapeutic outcomes (Patel et al., 2020; Zhang et al., 2014). After 1–3 hours, a single oral dose of esomeprazole or lansoprazole achieves effective serum concentration and raises the stomach pH in the majority of patients from 2.0 to above 6.0. Increased stomach pH produced by ARAs has been found to decrease the bioavailability and exposure of weakly basic drugs with pH-dependent solubility (Gay et al., 2017). Early identification of such physiochemical factors is critical for predicting the drug’s pharmacodynamic behavior (in vivo), and the establishment of in vitro in vivo correlation. In preliminary solubility analysis, 125 mg of PB was found to be completely soluble at equilibrium in 250 ml of dissolution media at pH < 4.3 with solubility surpassing 0.5 mg/ml, as observed in new drug application report submitted by innovator to U.S. Food and Drug Administration. However, the solubility fell substantially below 0.5 mg/ml when pH was raised over 4.5, which is the stomach pH commonly reached after administration of ARA (Sun et al., 2017).
Experimental and computational techniques, such as in vitro dissolution studies in various biorelevant media, solubility profiling in gastrointestinal pH, and pharmacokinetic data, have been developed to either anticipate or eliminate the danger of pH impact. In silico techniques that frequently incorporate dissolution/absorption simulation and modelling with inputs from in vitro biorelevant dissolution have been used in drug discovery and development to simulate the effects of solubility and dissolution on drug absorption (Hamed et al., 2016; Mitra et al., 2020; Varma et al., 2012; Zhu et al., 2016). The ideal experimental model would include physiologically acceptable volumes of relevant dissolving media, as well as fluid transfer and pH changes in a dynamic process. To the authors’ knowledge, there are relatively few published researches that consider the pH-effect risk across a variety of drug category, with a focus on predicting the magnitude of the risk and understanding the essential pharmacological features that drive it. The pH-modifying drug’s complex effects, fluctuating pH, and transit time make clinical pH effects challenging. Due to the constraints of clinical studies, the DDI cannot be assessed with all ARA categories. Dissolution properties of chemically diverse compounds show a wide range of clinical pH-effects. To accomplish this, in vitro micro-dissolution assay as described by Mathias et al. (2013) was used to investigate dissolution and precipitation kinetics of PB by mimicking the effects of a high dosage of PPI (Mathias et al., 2013). The purpose of this work was to determine the pH-dependent solubility and absorption of PB in biorelevant simulated media using a micro-dissolution technique. The use of biorelevant media with a pH shift effect to replicate the circumstances shown in the GI tract has been proposed for determining the dissolution and solubility of drugs in vivo and predicting their absorption. Built with suitable simulated media and hydrodynamics are beneficial in formulating strategies and establishing in vitro in vivo correlation, which will result in a decrease in animal experiments, bioavailability, and bioequivalence studies (Klein, 2010). The clinical pH impact was also seen to be linked to in vitro dissolution experiment’s pH effect. The ratio of human Cmax/AUC with and without ARA will be calculated from literature and experimental in vitro solubility data was compared. This ratio helps to determine the impact of pH on drug solubility and can be correlated between in vitro dissolution profile and in vivo AUCinf or Cmax data.
Since drug solubility is the most important aspect of this experiment, the developed analytical method should be sensitive enough to detect relatively minor changes in drug solubility accurately. Design and development of analytical methods are critical in order to quantify minor solubility changes in drug solution. Various analytical techniques based on different formulations and biological fluids are available in the literature for PB quantification. However, the described analytical techniques’ sensitivity, relatability, and use of a high-cost organic solvent, as well as high-end instrumental analysis utilizing mass spectrometry, among other factors, limited their applicability (Al-Shehri et al., 2020; Dange et al., 2018; Posocco et al., 2020). Furthermore, PB is a hydrophobic and nonpolar aromatic hetero polycyclic molecule, so minor changes in critical method parameters (CMPs) such as pH or mobile phase organic content have a significant influence on the analytical method. As a result, using the analytical quality by design (QbD) approach, a highly specific, accurate, efficient, and robust reverse phase high performance liquid chromatography (RP-HPLC) technique for quantification of PB was established in the present investigation. In the development of analytical methods, analytical QbD was used to ensure that the desired development approach was adequate. It aids in identifying important quality features that have a major influence on the final outcome, in addition to knowing particular variables (Fukuda et al., 2018; Peraman et al., 2015a). To the best of authors’ knowledge, this is the first study to disclose full development as well as optimization of PB by RP-HPLC method using a QbD approach, which will provide a wide range of analytical applications as well as a method operable region for overcoming out-of-specification findings.
MATERIALS AND METHODS
Chemicals and reagents
PB was kindly gifted by MSN laboratories private limited, Hyderabad. Simulated intestinal/gastric fluid powder used to make fasted state simulated intestinal fluid and fasted state simulated gastric fluid was acquired from Biorelevant.com (Croydon, Surrey, United Kingdom). Buffers and salts like potassium dihydrogen phosphate, sodium phosphate dibasic, hydrochloric acid, sodium chloride, and orthophosphoric acid were supplied by Merck. Water was purified by Milli-Q UV plus systems (Millipore Co., Bedford, MA). All remaining chemicals, if used, were of analytical grade.
Instrumentation
HPLC system
The optimized chromatographic method was implemented utilizing a Shimadzu Prominence HPLC system with a configuration of LC-20AD quaternary pumps, DGU-20A5 degasser unit, SPD-M 10A Photo Diode Array detector, SIL 20AC HT autosampler, and CTO-10AS column oven. The data analysis was performed using LC solution software.
Software
Design-Expert software (Version 12.0; Stat-Ease, Minneapolis, MN) was used for statistical analysis.
Chromatographic conditions
The chromatographic method optimization was performed with integrated analytical QbD technique. The chromatographic separation was achieved using a Unison RP C8 (150 × 4.6 mm, 5 µ) column as the stationary phase and an optimized mobile phase consisting of mixture Acetonitrile, methanol and 10 mM ammonium acetate (pH 4.5) in the ratio 50:10:40. The column temperature was maintained at 25°C. The injection volume was 20 µl, with a 10-minute run time with an optimized 0.8 ml/minute flow rate at detection wavelength of 264 nm.
Analytical design of experiment (DoE) method optimization
Defining analytical target profile (ATP) and critical analytical attributes (CAAs)
The first stage in developing a QbD-based RP-HPLC technique is to design an ATP (Karmarkar et al., 2011; Krishna et al., 2016; Peraman et al., 2015b). The analytical method’s quality, safety, and applicability will be assured by defining ATP. Different CAAs might be chosen to fulfil the ATP criteria. Out of these CAAs, the ATPs selected for this technique are (i) drug retention time (Rt) and (ii) tailing factor (Tf), both of which have a significant impact on the method’s performance.
Risk assessment for screening of factors affecting the method development
A risk assessment study was initially conducted to determine the possibility of risks or failures. The Ishikawa fish-bone model was used to create a cause–effect link between critical material attributes (CMAs) and critical process parameters. It is utilized to discover the probable parameters that might have tremendous potential to impact the detected CAAs and are classified as CMPs.
Method development and optimization using Box–Behnken design (BBD)
BBD with three variables was designed to optimize CMA of analytical method (Table 1). Based on the screening results, three independent variables were chosen for further optimization: mobile phase pH (X1), % organic within mobile phase (X2), and flow rate (X3). The design matrix provided 15 trial runs utilizing a combination of CAA. All tests were carried out at a drug concentration of 10 µg/ml of PB. Analysis of variance (ANOVA) statistical analysis and response surface analysis were used to further evaluate the model created by design using DoE software.
METHOD VALIDATION
The developed analytical method was validated as per ICH (International Conference of Harmonization) Q2 (R1) guidelines (Harron, 2013) over the parameters of specificity, linearity, limit of detection (LOD), limit of quantification (LOQ), accuracy, and precision.
Micro-dissolution pH shift studies of PB
The in vitro micro-dissolution test is divided into two stages—gastric and intestinal test. Considering clinical dose of PB as 125 mg, an equivalent dose of PB was added to 14 ml of pH 1.2 FaSSGF buffer taken in a glass beaker, and samples were withdrawn for 20 minutes at various time intervals. To mimic the pH change from stomach to small intestine, 21 ml of pH 6.5 fasted state simulated intestinal fluid (FaSSIF) buffer was added to the same beaker, and sampling was performed for another 180 minutes. A similar arrangement was created in another beaker with drug being added to 14 ml of pH 6.5 FaSSIF buffer, and sampled for 20 minutes at various time intervals before adding 21 ml of pH 6.8 FaSSIF buffer to the same assembly and sampled at intervals till 180 minutes. To maintain sink conditions, the withdrawn samples were replaced with equal quantity of fresh buffer. The two setups were kept stirring continuously at 350 rpm throughout the experiment with the aid of magnetic stirrers. Sampling was performed at time intervals of 0, 2, 5, 10, 15, 20, 21, 25, 30, 60, 90, 120, 150, and 180 minutes. The withdrawn samples were centrifuged at 10,000 rpm for 15 minutes to separate the precipitated drug and the supernatant was analyzed using a novel and optimized RP-HPLC technique supplemented by analytical QbD approach.
RESULTS AND DISCUSSION
Analytical DoE method optimization
To design an analytical technique that is both effective and sensitive, experiments with various combinations of mobile phase solvents, buffers, and stationary phases were undertaken. The buffer system is evaluated at several pH values ranging from 3 to 6 combining phosphate and acetate buffers. These combinations indicated that acetonitrile with 10% methanol as a mobile phase modifier and 10 mM ammonium acetate as the aqueous phase suited the mobile phase the best. It provided enhanced selectivity with peak symmetry and a low tailing factor, as well as increased sensitivity and theoretical plate count.
Risk assessment
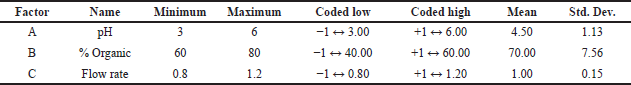
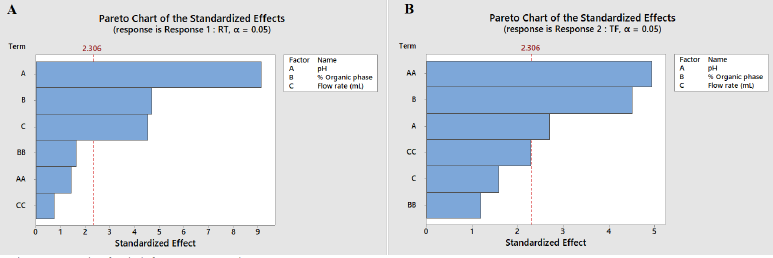
Using a three level three factorial BBD, the impact of three independent method factors, mobile phase pH (X1), % organic within the mobile phase (X2), and flow rate (X3), on two responses variables drug Rt (R1) and Tf (R2) of PB was assessed. The results were assessed by statistical analysis using Design Expert 12.0 software, and the proposed chromatographic experimental conditions by DoE were carried out using HPLC. The Ishikawa fish-bone diagram was used to do initial risk assessments for CMA of techniques like column chemistry, temperature, and HPLC system. The Ishikawa fish-bone diagram summaries the possible risk variables that have a significant impact on the ATP of methods. The model’s riskiest CMA was identified using a Pareto chart. Figure 1A shows that all three independent factors have a greater impact on PB retention, with factor X1: mobile phase pH and X2: % organic being the most relevant factors for PB tailing factor (Fig. 1B). The Tf has been impacted by the pH of the aqueous mobile phase, since acidic pH has no effect on the ionization of the silanol groups of the C18 stationary phase and increases the retention of basic drugs.
 | Table 1. Independent variables and their levels. [Click here to view] |
Statistical analysis
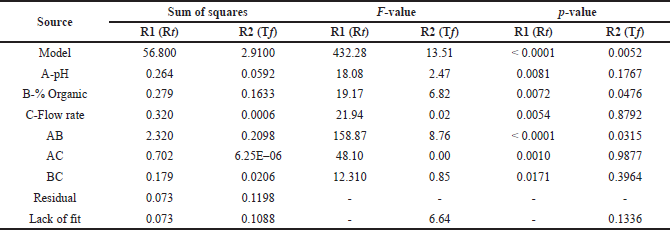
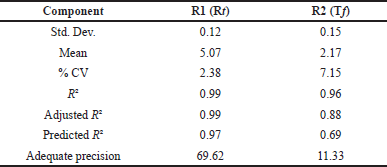
The DoE design was validated using ANOVA, which revealed that the model was statistically significant with a p-value of < 0.001. The responses R1 and R2 have model F-values of 432.28 and 13.51, respectively, indicating that the model is significant. Design Expert software recommended a quadratic model with process order for major effects and a factorial design for both responses as the best fitting model. Individual model variables such as mobile phase pH and % organic content had p-values > 0.005, indicating that these factors had a substantial influence on response R1. Similarly, the p-value for each individual model for response R2 is more than 0.005 for all independent factors, showing that these CMA have a substantial influence on method CQA (Table 2). Table 3 shows that the predicted R-squared for all responses is reasonably close to the adjusted R-squared values, with a difference of less than 0.2 in each case. The ratio of 69.62 to 11.33 indicates that the signal is acceptable (ratio > 4.0). The model’s desirability was estimated to be 1.
Perturbation plot analysis and 2D response surface plot analysis
Perturbation plots and contour plots were used to examine the visual impact of independent factors and their interactions on the response variables. Due to the existence of quadratic components in the regression model, the pH of the mobile phase has a considerable impact on tailing factor, and the output response seen in the contour plot is a curvature response surface. However, the pH of the mobile phase has less effect on retention time; therefore the model is expanded to include both quadratic and linear factors. The % of organic moiety in the mobile phase has a significant impact on the tailing factor but only a little impact on the drug’s retention time. The tailing factor was unaffected by the mobile phase flow rate (Fig. 2). According to perturbation plots with smallest variation from reference point, the independent factor pH has a negative correlation, but % organic phase and flow rate have a positive correlation towards tailing factor and PB retention time (Fig. 3).
 | Figure 1. Pareto plot of analysis for responses R1 and R2. [Click here to view] |
 | Table 2. ANOVA results for responses R1: Retention time and R2: Tailing factor. [Click here to view] |
 | Table 3. Model Fit summary for the responses R1: Retention time and R2: Tailing factor. [Click here to view] |
Optimized chromatographic condition
After examining these interactions using contour plot analysis, a method operable design region (MODR) was established. Statistical analysis verification was used to determine the optimal experimental settings and model’s desirability was found within the acceptance limit. The desirability function of Derringer was utilized to optimize two parameters to distinct goals. Desirability function, which generates D values between 0 and 1. In Figure 4, the response surface plot corresponding to the d-value with optimized area is indicated in yellow. Desirability coordinates denoted optimal circumstances. Working inside the MODR observed region will yield satisfactory results for our targeted criteria. In Table 4, the anticipated and actual results for responses R1 and R2 are summarized. Figure 5 represents an optimized chromatogram of PB by RP-HPLC method.
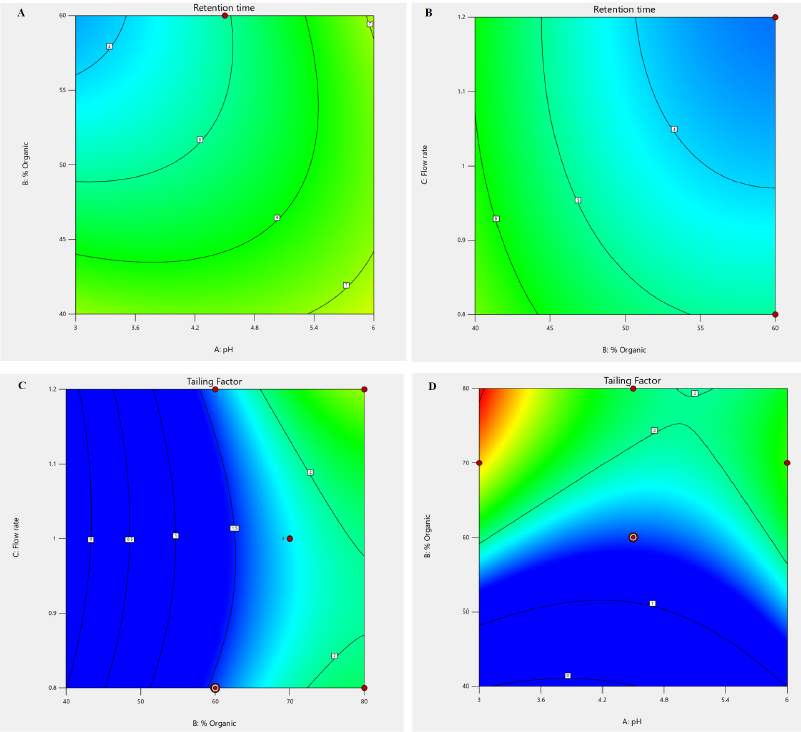 | Figure 2. Contour plot of interaction effect of factor X1 and X2 on responses. [Click here to view] |
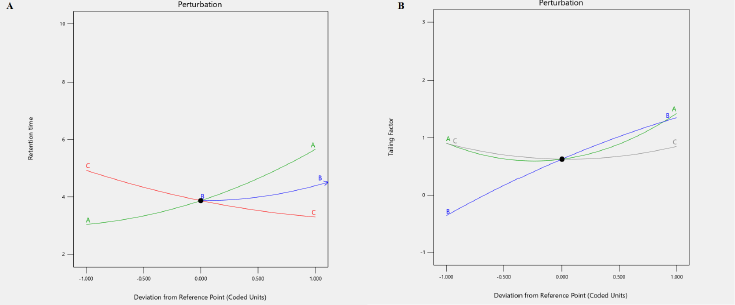 | Figure 3. Perturbation plot of analysis for responses R1 and R2. [Click here to view] |
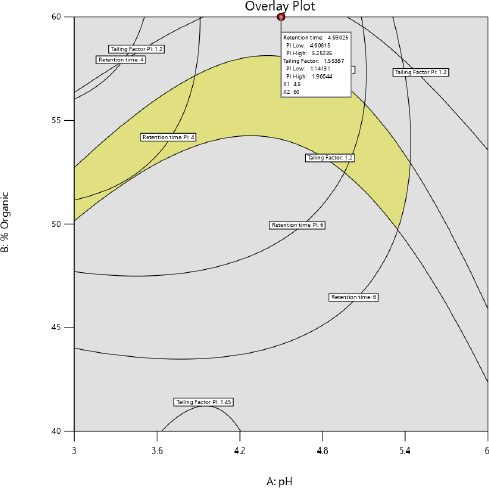 | Figure 4. Design space and operable region for the independent factors. [Click here to view] |
METHOD VALIDATION
System suitability was performed to assess the reproducibility of the chromatographic method. relative standard deviation (%RSD) for peak area of six repeated injections of PB was found to be 0.59%. The developed method was found to be highly specific as there was no interfering peaks around the retention time of PB, as shown in Figure 5. The peak purity was found to be greater than the peak purity threshold, indicating that PB in both standards as well as micro dissolution processed samples was pure, and there was no interference of impurities, biorelevant dissolution components or degradants. The linearity for PB was established, and the concentration versus peak area calibration curve was plotted as linear regression analysis. The method was found to be linear for PB over the concentration range of 0.1–150 µg/ml. The correlation coefficient (R2) was found to be 0.999. The LOD and LOQ for PB were found to be 0.9 and 2.71 µg/ml, respectively. The mean percent recovery for PB was found to be within the limit of 98%–105% and the recovery data is shown in Table 5. The precision of the given method was determined by intermediate precision and repeatability. The results were reported in terms of %RSD as shown in Table 6. The method was found to be robust while deliberately modifying mobile phase, wavelength, column temperature, flow rate. %RSD was found to be less than 2%.
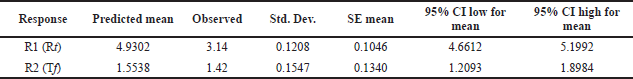 | Table 4. Predicted and actual point prediction at two-sided 95% confidence interval. [Click here to view] |
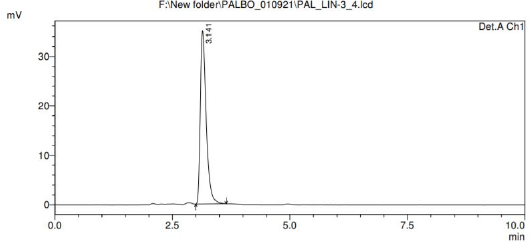 | Figure 5. Representative chromatogram of PB. [Click here to view] |
Micro-dissolution pH shift studies of PB
The effect of pH change on PB solubility and dissolution profile in biorelevant medium is shown in Figure 6 (FaSSGF and FaSSIF). FaSSGF was used to dissolve the initial drug concentration, where solubility of PB reached 0.5 mg/ml. As the pH of the experimental setup was increased from 1.2 to 6.5, the solubility of the drug declined drastically from 0.7 to 0.07 mg/ml. The concentration of PB in the micro-dissolution assembly steadied at 0.068 mg/ml after 180 minutes. The solubility of PB in the second set up, FaSSIF pH 6.5 to 6.8 was observed to be 0.04 and 0.02 mg/ml, respectively, while equilibrating at 0.022 mg/ml at the end of 180 minutes. The substantial drop in drug solubility after the pH shift from FaSSGF buffer highlights the influence of pH on drug solubility and validates the drug’s pH sensitivity. With initial poor solubility in FaSSIF, the solubility dips even further as the pH rises, indicating a change in solubility as the pH increases. Because the change from stomach to intestinal pH is rapid in this test, intestinal supersaturation occurs quickly upon transfer from FaSSGF pH 1.2 to FaSSIF pH 6.8. Supersaturation causes immediate precipitation and a reduction in its inherent solubility, which may be accurately determined by this in vitro technique at the maximum dosage. The entire research is done utilizing fasting simulation fluid to connect the dosage regime with PPIs. FaSSGF is a dissolving medium that properly mimics the contents of the stomach in a fasting state and contains physiologically relevant amounts of pepsin, bile salts, and lecithin to create a surface tension comparable to that observed in vivo. As PB has no dietary impact on medication absorption (Fotaki and Vertzoni, 2010), FaSSIF 6.5 has been effectively used to predict exact pH shift effects on PB absorption towards basic pH. The solubility of PB in several buffer pH ranges has been provided by the inventor, but because PB is a weakly soluble drug, it would not have been feasible to predict in vivo performance of PB in an increased stomach pH without biorelevant dissolving media. The ratio of the dissolution AUC profiles (AUC of FaSSIF pH 6.5 to FaSSIF pH 6.8/ AUC of FaSSGF pH 1.2 to FaSSIF pH 6.8) was used to calculate the risk of in vitro pH impact. This ratio aids in determining the effect of pH on drug solubility and may be linked between in vitro dissolution profiles and in vivo data. The estimated clinical pH effect ratio of PB in a human clinical study with and without ARA (AUCtreated / AUCuntreated) is reported as 0.205 and 0.367 for Cmax and AUCinf, respectively. The micro-dissolution experiment yields a value of 0.532 for the in vitro pH-effect risk that illustrates a parallel link between in vitro dissolution and clinical pH impact. The pH impact, in vitro and the clinical Cmax pH effect are linearly related. In practice, the smaller the ratio, the more significant is the pH impact. A ratio of 1 indicates that there is no clinical impact of pH. Thus, PB exhibits a significant pH-dependent absorption and DDI in both in vitro and clinical Cmax pH-effects. Thus, the pH shift effect causes alterations in solubility which in turn has considerable influence on PB absorption, resulting in clinically significant interaction and poor bioavailability.
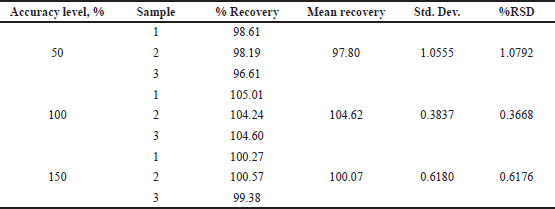 | Table 5. Accuracy and % recovery of PB. [Click here to view] |
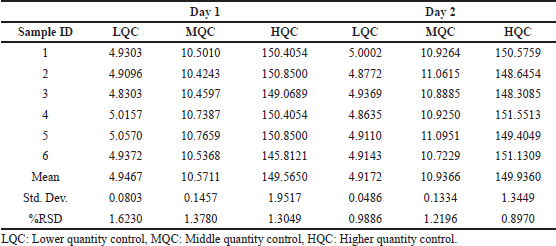 | Table 6. Interday and intraday precision of PB at quality control levels. [Click here to view] |
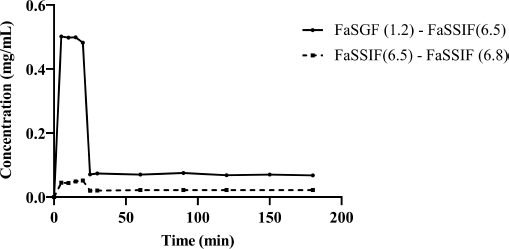 | Figure 6. Kinetics of pH dependant dissolution from in vitro micro-dissolution test for PB [Click here to view] |
CONCLUSION
The impact of pH on the solubility of the drug is investigated in this experiment, as solubility is a crucial factor in the early stage of pharmacokinetic behavior, i.e., absorption. Despite the fact that computational approaches are used to predict potential interactions, this in vitro procedure is simple, accurate, and relevant to real-world circumstances, as well as produces accurate results. When the drug’s solubility value across the physiological pH range (1.2–6.8) is less than dose/250 ml, FDA requires an in vivo clinical investigation (Patel et al., 2020; Zhang et al., 2014). Early understanding of pH-effect risk assessment is critical to influencing clinical trial design to include a pH-modifier treatment arm and obtaining a clinical report on the pH impact of high-risk medications. PPIs generally have a longer duration of acid secretion suppression effect than H2 blockers and local acting antacids, and interfere with the intestinal absorption of weakly basic drugs to a greater extent. Use of PPIs may be considered a worst-case scenario in the in vivo evaluation of the clinical pH effect on novel investigational drugs. The present study comprehensively characterizes pH-dependent DDIs using an in vitro micro-dissolution test, which is a new, efficient, practical, and active pharmaceutical ingredient sparing technique for predicting dissolution and precipitation kinetics. This study highlights the critical need of conducting early research and interaction profiling prior to initiating clinical trials for new molecular entities and medications that are weakly basic in nature and exhibit pH-dependent solubility. While it was applied retrospectively to this compound after the effect was observed in a clinical setting, the same experimental sequences and analytical reasoning could be used to assess the risk of a pH-effect prior to clinical evaluation, thereby providing direction for physical form development and formulation strategies early in clinical development.
The implement of QbD paradigms has been entering the industrial, academic, and government sectors at an alarming rate with the objective to gain full process and product knowledge. As a consequence, for sample analysis of PB randomized order integrated QbD trials for estimate were carried out to decrease the influence of uncontrolled factors that could bias the outcome. Implementation of QbD technique has led to more robust method which can generate consistent, dependable, and quality data throughout the process and also saves time & expense.
ACKNOWLEDGMENT
Prajakta Harish Patil would like to express his gratitude for the fellowship and financial support provided by the Indian Council of Medical Research, Government of India. Mrunal Desai wishes to express her gratitude to Manipal College of Pharmaceutical Sciences and Manipal Academy of Higher Education for awarding her the TMA Pai fellowship (IMF) and providing the research facilities.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
DATA AVAILABILITY
All data generated and analyzed are included within this research article.
FUNDING
This research has been supported by the ICMR India (Ad-hoc research Grant no.2020-4462).
ETHICAL APPROVAL
The study does not have involvement of experiments on humans or animals.
REFERENCES
Al-Shehri M, Hefnawy M, Abuelizz H, Alzamil A. Evaluation of the pharmacokinetics of the simultaneous quantification of letrozole and palbociclib in rat plasma by a developed and validated HPLC-PDA. Acta Chromatogr, 2020; 32:170–8; doi:10.1556/1326.2019.00635 CrossRef
Cazzaniga ME, Danesi R, Girmenia C, Invernizzi P, Elvevi A, Uguccioni M, NetworkER+. Management of toxicities associated with targeted therapies for HR-positive metastatic breast cancer: a multidisciplinary approach is the key to success. Breast Cancer Res Treat, 2019; 176:483–94; doi:10.1007/s10549-019-05261-5 CrossRef
Dange Y, Bhinge S, Salunkhe V. Optimization and validation of RP-HPLC method for simultaneous estimation of palbociclib and letrozole. Toxicol Mech Methods, 2018; 28:187–94; doi:10.1080/15376516.2017.1388458 CrossRef
Ehab M, Elbaz M. Profile of palbociclib in the treatment of metastatic breast cancer. Breast Cancer Targets Ther, 2016; 8:83–91; doi:10.2147/BCTT.S83146 CrossRef
Fotaki N, Vertzoni M. Biorelevant dissolution methods and their applications in in vitro in vivo correlations for oral formulations. Open Drug Deliv J, 2010; 4:2–13; doi:10.2174/1874126601004020002 CrossRef
Fukuda IM, Pinto CFF, Moreira CDS, Saviano AM, Lourenço FR. Design of experiments (DoE) applied to pharmaceutical and analytical quality by design (QbD). Brazilian J Pharm Sci, 2018; 54:1–16; doi:10.1590/s2175-97902018000001006. CrossRef
Gay C, Toulet D, Le Corre P. Pharmacokinetic drug-drug interactions of tyrosine kinase inhibitors: a focus on cytochrome P450, transporters, and acid suppression therapy. Hematol Oncol, 2017; 35:259–80; doi:10.1002/hon.2335 CrossRef
Hamed R, Awadallah A, Sunoqrot S, Tarawneh O, Nazzal S, AlBaraghthi T, Al Sayyad J, Abbas A. pH-dependent solubility and dissolution behavior of carvedilol—case example of a weakly basic BCS class II drug. AAPS PharmSciTech, 2016; 17:418–26; doi:10.1208/s12249-015-0365-2 CrossRef
Harron DWG. Technical requirements for registration of pharmaceuticals for human use: the ICH process. Textb Pharm Med, 2013; 1994:447–60; doi:10.1002/9781118532331.ch23 CrossRef
Karmarkar S, Garber R, Genchanok Y, George S, Yang X, Hammond R. Quality by design (QbD) based development of a stability indicating HPLC method for drug and impurities. J Chromatogr Sci, 2011; 49:439–46. CrossRef
Klein S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J, 2010; 12:397–406; doi:10.1208/s12248-010-9203-3 CrossRef
Krishna MV, Dash RN, Reddy BJ, Venugopal P, Sandeep P, Madhavi G. Quality by design ( QbD ) approach to develop HPLC method for eberconazole nitrate : application to hydrolytic , thermal, oxidative and photolytic degradation kinetics. J Saudi Chem Soc, 2016; 20:S313–22; doi:10.1016/j.jscs.2012.12.001 CrossRef
Mathias NR, Xu Y, Patel D, Grass M, Caldwell B, Jager C, Mullin J, Hansen L, Crison J, Saari A, Gesenberg C, Morrison J, Vig B, Raghavan K. Assessing the risk of pH-dependent absorption for new molecular entities: a novel in vitro dissolution test, physicochemical analysis, and risk assessment strategy. Mol Pharm, 2013; 10:4063–73; doi:10.1021/mp400426f CrossRef
Mitra A, Parrott N, Miller N, Lloyd R, Tistaert C, Heimbach T, Ji Y, Kesisoglou F. Prediction of pH-dependent drug-drug interactions for basic drugs using physiologically based biopharmaceutics modeling: industry case studies. J Pharm Sci, 2020; 109:1380–94; doi:10.1016/j.xphs.2019.11.017 CrossRef
Parylo S, Vennepureddy A, Dhar V, Patibandla P, Sokoloff A. Role of cyclin-dependent kinase 4/6 inhibitors in the current and future eras of cancer treatment. J Oncol Pharm Pract, 2019; 25:110–29; doi:10.1177/1078155218770904 CrossRef
Patel D, Bertz R, Ren S, Boulton DW, Någård M. A systematic review of gastric acid-reducing agent-mediated drug–drug interactions with orally administered medications. Clin Pharmacokinet, 2020; 59:447–62; doi:10.1007/s40262-019-00844-3 CrossRef
Peraman R, Bhadraya K, Reddy YP. Analytical quality by design: a tool for regulatory flexibility and robust analytics. Int J Anal Chem, 2015a; 2015:868727. CrossRef
Peraman R, Bhadraya K, Reddy YP, Reddy CS, Lokesh T. Analytical quality by design approach in rp-hplc method development for the assay of etofenamate in dosage forms. Indian J Pharm Sci, 2015b; 77:751–7. CrossRef
Posocco B, Buzzo M, Poetto AS, Orleni M, Gagno S, Zanchetta M, Lacuzzi V, Guardascione M, Puglisi F, Basile D, Pelizzari G, Marangon E, Toffoli G. Simultaneous quantification of palbociclib, ribociclib and letrozole in human plasma by a new LC-MS/MS method for clinical application. PLoS One, 2020; 15:1–17; doi:10.1371/journal.pone.0228822 CrossRef
Sharma M, Holmes HM, Mehta HB, Chen H, Aparasu RR, Shih YCT, Giordano SH, Johnson ML. The concomitant use of tyrosine kinase inhibitors and proton pump inhibitors: prevalence, predictors, and impact on survival and discontinuation of therapy in older adults with cancer. Cancer, 2019; 125:1155–62; doi:10.1002/cncr.31917 CrossRef
Sun W, Klamerus KJ, Yuhas LM, Pawlak S, Plotka A, O’Gorman M, Kirkovsky L, Kosa M, Wang D. Impact of acid-reducing agents on the pharmacokinetics of palbociclib, a weak base with ph-dependent solubility, with different food intake conditions. Clin Pharmacol Drug Dev, 2017; 6:614–26; doi:10.1002/cpdd.356 CrossRef
Varma MV, Gardner I, Steyn SJ, Nkansah P, Rotter CJ, Whitney-Pickett C, Zhang H, Di L, Cram M, Fenner KS, El-Kattan AF. PH-dependent solubility and permeability criteria for provisional biopharmaceutics classification (BCS and BDDCS) in early drug discovery. Mol Pharm, 2012; 9:1199–212; doi:10.1021/mp2004912 CrossRef
Zhang L, Wu F, Lee SC, Zhao H, Zhang L. pH-dependent drug-drug interactions for weak base drugs: Potential implications for new drug development. Clin Pharmacol Ther, 2014; 96:266–77; doi:10.1038/clpt.2014.87 CrossRef
Zhu AZX, Ho MCD, Gemski CK, Chuang BC, Liao M, Xia CQ. Utilizing in vitro dissolution-permeation chamber for the quantitative prediction of ph-dependent drug-drug interactions with acid-reducing agents: a comparison with physiologically based pharmacokinetic modeling. AAPS J, 2016; 18:1512–23; doi:10.1208/s12248-016-9972-4 CrossRef