
INTRODUCTION
As per WHO, more than 235 million people have been detected with severe acute respiratory syndrome caused by novel coronavirus 2 (SARS-CoV-2), and nearly 4.80 million deaths as of October 3, 2021 (WHO, 2021). At first, SARS-CoV-2 was reported in China by 2019, and as of now, Alpha, Beta, Gamma, Delta, Lambda, and Mu variants of SARS-CoV-2 had been reported in different countries. The first mutant variety was identified in the UK by September 2020. The second variant, Beta, was identified in South Africa by May 2020, and the third variant, Gamma, was identified in Brazil by November 2020. The Delta variant was reported initially in India by October 2020, and the Lambda variant was identified in Peru by December 2020. Recently, the Mu variant was detected in Colombia by January 2021 [https://www.who.int/en/activities/tracking-SARS-CoV-2-variants (Accessed on 06 October 2021)].
Many factors influence the precision of the diagnosis, including the increased rate of spreading, the high number of asymptomatic individuals, and genetic variants; hence, it is decisive to diagnose an individual for SARS-CoV-2 infection more accurately (Bokam et al., 2021). Initially, nasopharyngeal/sputum samples are the most preferred sampling method for diagnosis. To avoid bewildering negative results, WHO has streamlined the diagnostic method using Reverse Transcription-Polymerase Chain Reaction (RT-PCR) for more accuracy. As a result of whole-genome sequencing of SARS-CoV-2, exclusive primers that target spike genes (S), RdRp gene, E gene, and N gene had been developed. In recent times, RT-PCR followed by radiological imaging using computerized tomography is the complementary diagnostic approach for SARS-CoV-2 infection. The ground-glass opacities in specific lung segments are considered as a characteristic patterned feature of SARS-CoV-2 infection. In addition, serological screening of patients with elevated interleukins, IL-6, IL-2, IL-7, IL-10, and C-reactive protein is also considered an important biochemical signal for SARS-CoV-2 (Sharma et al., 2021).
Next to diagnosis, finding out a suitable antiviral drug for the treatment is an essential step. Finding an efficient antiviral agent in this hectic pandemic situation is the foremost objective in modern drug discovery and development. Until discovering the novel antiviral drug, screening the existing approved drugs against SARS-CoV-2 is an alternate option for emergency management. The repurpose screening has the advantage of skipping the essential steps of usual drug development processes like understanding the properties like pharmacodynamics, pharmacokinetics, absorption, distribution, metabolism, excretion, and toxicity since the selected drugs are already characterized and approved. Many chemical representatives have been explored as antiviral drugs that may directly impact the life of viruses (Das et al., 2021).
The development of a vaccine against SARS-CoV-2 is also in a rapid phase. So far, 218 candidate vaccines have been developed and out of which 26 candidate vaccines are under phase 1–3 trials. Different molecular platforms are investigating a more efficient vaccine based on DNA/RNA, peptide, virus-like particle, inactivated virus, and viral vector formulations (Sarma et al., 2021).
Protease inhibitors are the most predominant division of antivirals next to the inhibitors of polymerase. Protease inhibitors target a crucial maturation step in polyprotein processing that applies to most viruses with the help of viral proteases (Chaudhuri et al., 2018). In the available protein targets of SARS-CoV-2, the main protease (Mpro) has gained massive attention since it has a direct role in processing polyproteins complexes of SARS-CoV-2, ppla, and pplb, translated from viral RNA (Boopathi et al., 2021). Only in the dimer form, Mpro is biologically active, and the dimer structure is not explored thoroughly. The main protease is the key enzyme that cleaves the polyprotein obtained from viral mRNA at 11 different sites and produces proteins essentially required for the replication machinery (Zhang et al., 2020). Hence, discovering a drug as an inhibitor for Mpro will be the most significant barrier for viral replication. Structurally, Mpro is made up of 306 amino acids arranged in three different domains. Most of the amino acids in domains I and II are arranged in beta-barrel form, while the third domain primarily comprises alpha helices (Khan et al., 2021b).
With the available antiviral drugs, most countries have initiated trial-based research observations to know the efficacy of the existing approved drugs. In a few cases, positive results have been obtained in patients of SARS-CoV-2, treated with a mixture of lopinavir and ritonavir, potential anti-HIV drugs. Oseltamivir and hydroxychloroquine were also tried in the US, India, and so on. The familiar anti-HIV drugs, lopinavir, ritonavir, and ribavirin, were used in combined form in South Korea, which successfully revokes the viral clearance (Chen et al., 2020a). In recent times, many in silico research has reported that Food and Drug Administration (FDA)-approved drugs lopinavir (Pant et al., 2021), setrobuvir (Elfiky, 2021), saquinavir (Al-Khafaji et al., 2021), remdesivir, saquinavir, darunavir (Khan et al., 2021a), and disulfiram (Lobo-Galo et al., 2021), which have strong bonding with SARS-CoV-2. Nevertheless, the need for the potential drug candidate is sky high due to the acceleration in the spreading rate of SARS-CoV-2 universally. In this context, more FDA-approved drugs should be screened with Mpro of SARS-CoV-2.
In this study, 42 FDA-approved nonprotein antiviral drugs were selected to screen against Mpro of SARS-CoV-2 for the possibility of repurposing. Out of 42, 19 drugs are in use for the treatment of HIV-1, 4 drugs are in use for herpes simplex virus, 3 drugs are in use against hepatitis B virus, 10 drugs are in use against hepatitis C virus (HCV), 4 drugs are used against influenza, and 2 drugs for cytomegalovirus infection. Most of these drugs mainly act as inhibitors to the viral protease, polymerase, and integrase enzymes involved in the virus multiplication processes (Chaudhuri et al., 2018).
The main objective of this study was to screen the selected 42 FDA-approved drugs against the main protease of SARS-CoV-2 in silico. The active site amino acids of the main protease involved in bonding are to be revealed. Molecular dynamics simulation (MDS) analysis using GROMACS has also been performed to understand the stability of the Mpro-drug complex, and the results are analyzed thoroughly. The best drug that makes strong interactions with the main protease of SARS-CoV-2 would be revealed.
MATERIALS AND METHODS
Retrieval and preparation of protein receptors
The three-dimensional structure of SARS-CoV-2 main protease [Protein Data Bank (PDB) ID: 7BUY], complexed with carmofur, with a resolution of 1.60 Å obtained by XRD, is retrieved from PDB (Fig. 1). The water molecules and ligand (inhibitor) were removed from the structure, and the essential polar hydrogen was added to the structure using Discovery Studio Visualizer (version 20.1.0.19295) developed by Dassault Systemes BIOVIA Corporation. The prepared protein structure was further finalized using an online server of the Centre for Molecular and Biomolecular Informatics, Radboud University, Netherland https://swift.cmbi.umcn.nl/servers/html/index.html (Accessed on 24 March 2021), and then saved for docking in the form of PDB format.
Retrieval of ligands
The selected 42 US-FDA-approved antiviral drugs (Table 1) such as abacavir, acyclovir, adefovir, amprenavir, boceprevir, cidofovir, darunavir, delavirdine, didanosine, docosanol, efavirenz, elvitegravir, emtricitabine, entecavir, famciclovir, glecaprevir, lamivudine, ledipasvir, letermovir, lopinavir, nevirapine, ombitasvir, oseltamivir, paritaprevir, penciclovir, peramivir, ribavirin, rilpivirine, rimantadine, ritonavir, saquinavir, sofosbuvir, stavudine, telaprevir, telbivudine, tenofovir, tipranavir, velpatasvir, voxilaprevir, zalcitabine, zanamivir, and zidovudine were downloaded from PubChem in 2D sdf format and converted into 3D mol2 using PyMOL. These drugs were chosen based on their proven antiviral effects (Chaudhuri et al., 2018).
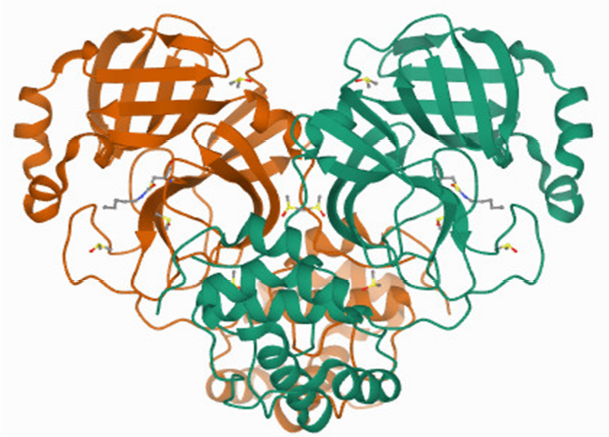 | Figure 1. 3D structure of main protease of SARS-CoV-2 (PDB ID: 7BUY). [Click here to view] |
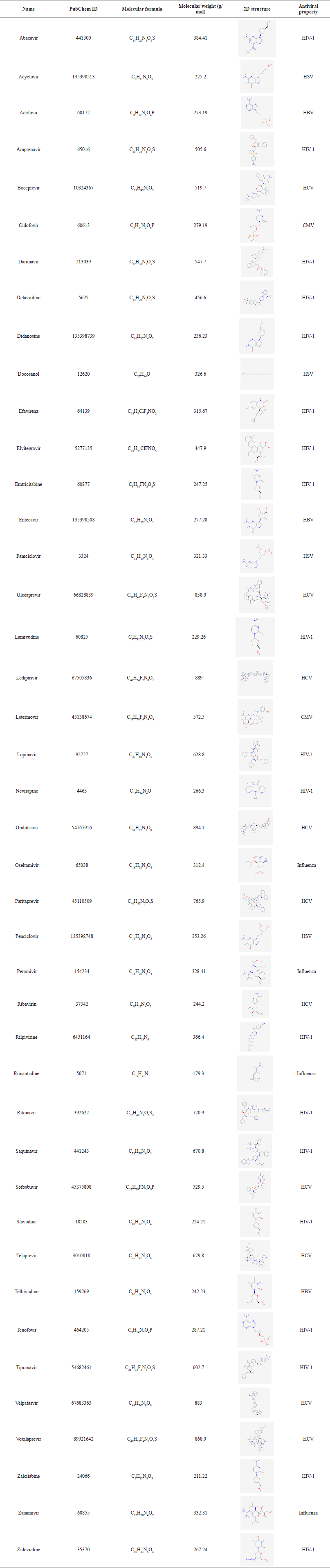 | Table 1. Details of selected US-FDA-approved antiviral drugs. [Click here to view] |
Analysis of molecular docking
Molecular docking of protein with selected ligands was performed using the PyRx-Vina (version 0.8). The clumped final output file was obtained in pdbqt format. All the eight individual poses for each ligand are separated by Vina split, and the best pose was selected based on binding affinity (Kroemer, 2007). The 3D dock view and 2D pose views were prepared using Discovery Studio Visualizer 2020. The details of amino acids involved in hydrogen bonding and other interactions were analyzed and tabulated.
MDS using GROMACS
MDS was carried out with the main protease (7buy) and drug candidate complex in the solvent system using GROMACS 5.1.2 and GROMOS96 54a7 force field (Schmid et al., 2011). The PDBQT files of the main protease and drug molecules of docked complexes were saved as PDB files using Discovery Studio (BIOVIA, 2020), and the PDB files were then refined by using Swiss PDB Viewer (Guex and Peitsch, 1997). The topology file for the main protease was created using GROMACS, while the topology file for drug candidates was generated using the PRODRG2 (Schuttelkopf and van Aalten, 2004). Protein–ligand complex and topology files were built. Dodecahedron box was created with 1 nm in X, Y, and Z dimensions, the simulation box was filled with water as a solvent, and 4 Na ions were added to maintain the electrostatic charge balance of the system. Energy minimization was done at 1,000 steps using the steepest descent algorithm. Then position restraints were applied to the drug candidates and main protease, but the solvent could diffuse freely. The constant number of particles, volume and temperature (NVT) and number of particles, pressure and temperature (NPT) equilibration were achieved with the following conditions. The output analog coordinates were set to 500 steps. The Particle Mesh Ewald was used as the calculation method (Schmid et al., 2011), and the cut-off value of electrostatic action was set to 1.2 nm. After NVT and NPT simulations, by keeping the temperature at 300K and the pressure at 1 bar, production MD runs were then performed with the step size of 2 fs. A 20 ns simulation was performed.
Energy calculations based on the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) method
MM-PBSA method was used to calculate the binding free energy by g_mmpbsa and Adaptive Poisson-Boltzmann Solver (APBS) packages (Baker et al., 2001; Eisenhaber et al., 1995; Kumari et al., 2014; Pronk et al., 2013) over a 20 ns time stamp in steps of 50 ps for the MD simulation.
RESULTS AND DISCUSSIONS
Docking analysis with main protease
The three-dimensional structure of the main protease was docked with the 42 ligands (US-FDA-approved drugs) individually. Each drug has effectively interacted with the main protease with varied binding affinity. The binding affinity of all the drugs was compared (Fig. 2). The amino acids of the main protease involved in bonding with ligands were analyzed (Table 2).
The results indicate that, out of 42 ligands, ledipasvir displayed the maximum binding affinity of −10.4 kcal/mol, followed by paritaprevir with −9.1 kcal/mol and velpatasvir with −8.8 kcal/mol. The least binding affinity was observed in darunavir with −2.6 kcal/mol. Another drug, voxilaprevir, has also shown a binding affinity similar to that of velpatasvir. However, due to the similarity in the amino acids involved in interactions, velpatasvir was considered the top third drug for further analysis. All the 42 drug-Mpro complexes were analyzed individually using Discovery Studio Visualizer 2020, and the amino acids involved in those interactions are shown in Table 2. The number of bonds, the nature of the bond, and the respective amino acid involved are mentioned in Table 2. The three-dimensional dock view of the drug-Mpro complex and their two-dimensional pose view were made using the Discovery Studio Visualizer.
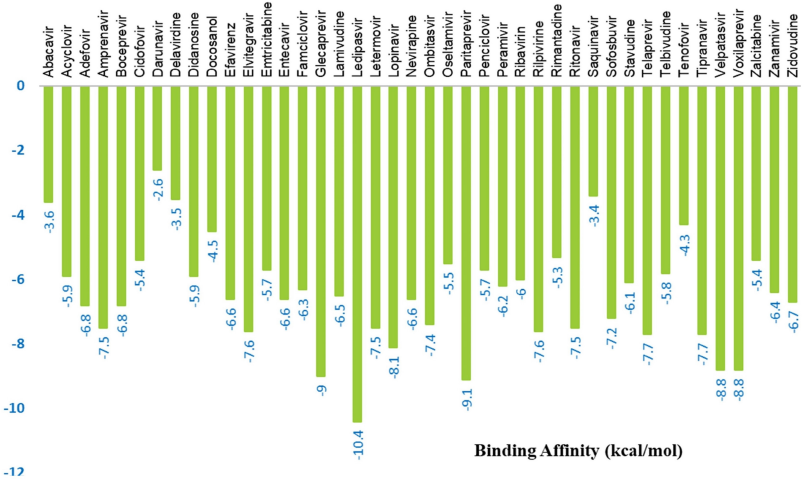 | Figure 2. The binding affinity of selected drugs with main protease. [Click here to view] |
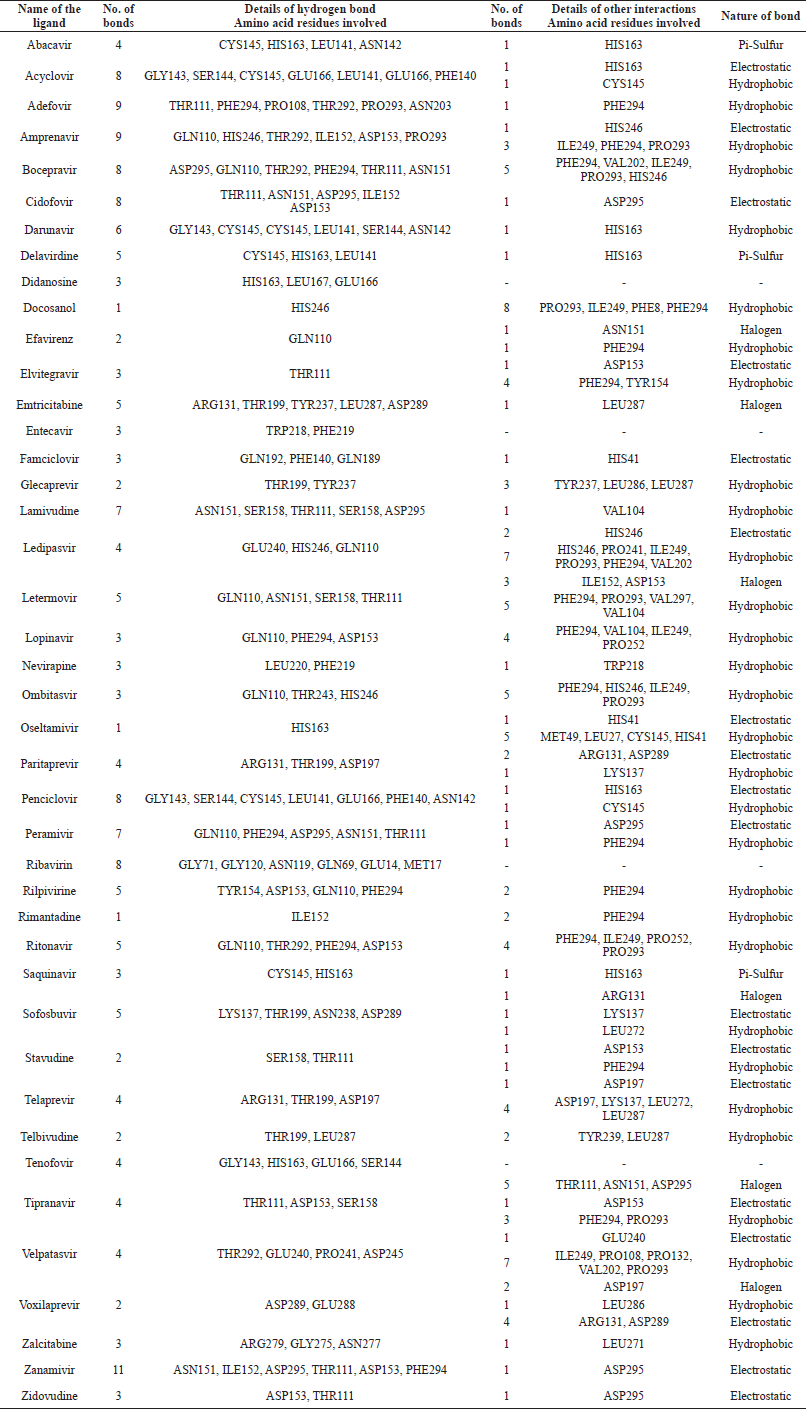 | Table 2. Amino acids of main protease involved in bonding with ligands. [Click here to view] |
The further analysis revealed that ledipasvir (Fig. 3) had made four hydrogen bonds with Mpro at GLU240, HIS246, and GLN110, two electrostatic bonds with HIS246, and seven hydrophobic interactions at HIS246, PRO241, ILE249, PRO293, PHE294, and VAL202. In all the 42 US-FDA-approved drugs screened, ledipasvir has shown a huge binding affinity. According to a study (Cheng et al., 2016), ledipasvir has been reported as a potent inhibitor of NS3 protease and NS5B polymerase along with sofosbuvir in HCV infection. It has been evident that ledipasvir showed the picomolar antiviral effect on genotype 1a and 1b replicons of HCV with the EC50 value of 0.031 and 0.004 nM, respectively. A recent study reported another anti-HCV drug, sofosbuvir, as a choice of drug repurposed to treat SARS-CoV-2 (Sayad et al., 2020). So, it is essential to screen the competitor, anti-HCV, ledipasvir against the protein targets of SARS-CoV-2. Chen et al. (2020b) have positively stated that ledipasvir has shown a huge possibility of repurposing against SARS-CoV-2 by inhibiting the 3CL protease of SARS-CoV-2. Another repurposing study Joshi et al. (2020) also reported that ledipasvir has inhibitory potential against the same enzyme. The results of in silico analysis of our results are on par with these study and hence ledipasvir can be a mighty drug choice for repurposing. Further, it should also be noted that ledipasvir showed inhibition against the main protease enzyme. Still, there is not much information available about the interaction details of the ledipasvir with the major protein targets of the SARS-CoV-2. Hence, our results will be a keystone among researchers, and further dynamic simulation analysis confirmed the stability of the ledipasvir-Mpro complex without any major conformational changes. The results of the simulation analysis of ledipasvir justify it as a drug with maximum binding affinity.
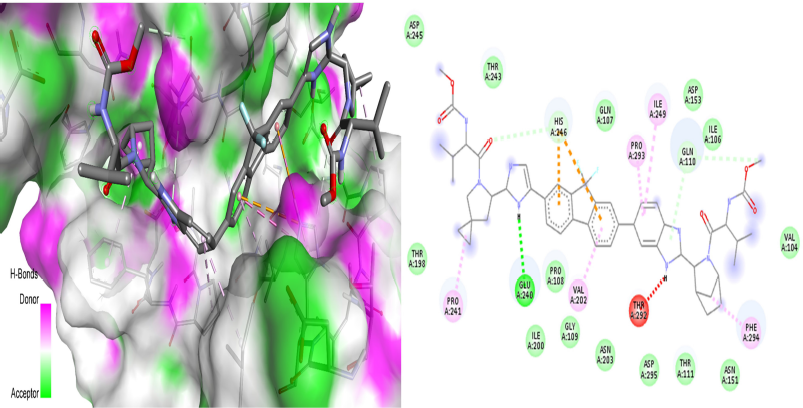 | Figure 3. 3D dock view and 2D pose view of Mpro-ledipasvir complex. [Click here to view] |
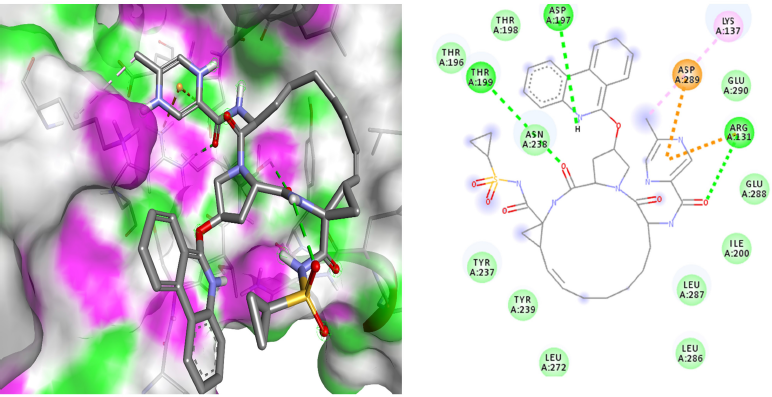 | Figure 4. 3D dock view and 2D pose view of Mpro-paritaprevir complex. [Click here to view] |
The drug with the second-highest affinity score, paritaprevir (Fig. 4), has formed four hydrogen bonds at ARG131, THR199, and ASP197, two electrostatic interactions at ARG131 and ASP289, and one hydrophobic bond at LYS137. A lot of research and screening are going for the repurposing of this drug. According to a recent study (Shaha et al., 2020), methisazone and paritaprevir have been noted to be promising inhibitory drugs against SARS-CoV-2. Another study (Guan et al., 2020) has confirmed the inhibitory potential of the paritaprevir against 3CL protease of SARS-CoV-2, and this result was on par with the previous study (Rameez et al., 2020). We have obtained similar results in virtual screening, and the dynamic simulation analysis further showed the stability information of the drug-protein complex. Like ledipasvir, there is not much research available about the anti-SARS-CoV-2 potential of paritaprevir, and hence our results will be in the limelight in this context repurposing.
Next to ledipasvir and paritaprevir, velpatasvir showed the maximum affinity in the virtual screening. Velpatasvir (Fig. 5) has made four hydrogen bonds at THR292, GLU240, PRO241, and ASP245, seven hydrophobic bonds at ILE249, PRO108 PRO132, VAL202, and PRO293, and one electrostatic interaction at GLU240. According to a recent report (Sayad et al., 2020), it has been revealed that velpatasvir, along with sofosbuvir, can be tried in clinical trials due to its huge binding affinity with the major protein targets in SARS-CoV-2. Recently, a study (Jockusch et al., 2020) has reported that, along with sofosbuvir, velpatasvir could inhibit the lifecycle of SARS-CoV-2 by inhibiting the RNA-dependent DNA polymerase. Two more in silico investigations (Chen et al., 2020b; Elfiky, 2020) have also reported the inhibitory potential of velpatasvir.
It is also evident that both ledipasvir and velpatasvir have made successful interactions with Mpro, specifically at GLU240, PRO241, ILE249, PRO293, and VAL202. These five amino acids might present in the binding pocket of Mpro where ledipasvir and velpatasvir have successfully interacted.
Molecular dynamics simulation
The docked complex of the main protease with drugs was subjected to MDS analysis for 20 ns to study the steadiness of the protein-drug complex. The root means square deviation plot for the main protease complex with three antiviral drugs, namely, ledipasvir (red), paritaprevir (orange), and velpatasvir (blue), and the free form of main protease (green) was depicted in Figure 6.
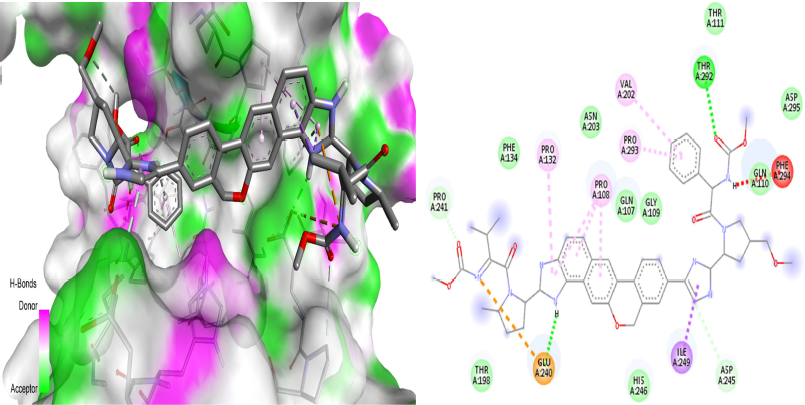 | Figure 5. 3D dock view and 2D pose view of Mpro-velpatasvir complex. [Click here to view] |
The main protease with ledipasvir (red) showed unchanging between 0.13 nm and 0.33 nm. The main protease with paritaprevir (orange) showed steady RMSD between 0.11 nm and 0.29 nm.
The free main protease (green) showed constant RMSD between 0.12 nm and 0.36 nm. The main protease with velpatasvir (blue) showed stable RMSD between 0.13 nm and 0.29 nm. The antiviral compound ledipasvir (red) showed deviation from the free main protease form (green) from 5 ns to 10 ns range, and it may be due to the binding interaction of the ledipasvir to the main protease. The main protease with paritaprevir (orange) showed fluctuations from the free main protease form (green) from 3 ns to 20 ns range, and it may be due to the binding interaction of the paritaprevir to the main protease. The antiviral compound velpatasvir (blue) showed deviation from the free main protease form (green) from 5 ns to 20 ns range, and it may be due to the binding interaction of the velpatasvir to the main protease. The result proved that paritaprevir and velpatasvir induce more conformational change in the main protease than ledipasvir.
The root mean square fluctuation (RMSF) plot (Fig. 7) also confirms these results by local changes in docked complexes’ residues. The main protease with ledipasvir (red) does not affect the structure of the main protease during simulation, whereas the main protease with paritaprevir (orange) and velpatasvir (blue) affects the structure during the simulation.
The radius of gyration (Rg) of the simulated systems was represented in Figure 8. It was clear that all the simulated systems have achieved results on par with the Rg values of a recent study (Khan et al., 2021b). The main protease with ledipasvir (red) showed similar Rg values that of free protease, whereas paritaprevir (orange) and velpatasvir (blue) showed deviated values from free protease. Some recent studies (Khan et al., 2021b) have reported the Rg values of antiviral compounds like remdesivir, saquinavir, and darunavir against protease of SARS-CoV-2 that are comparable to our results.
The binding strength between protein and ligand might be assessed with the help of intermolecular hydrogen bonds. The ledipasvir (red) and velpatasvir (blue) showed hydrogen bonds from 5 ns to 20 ns range of 2–4 bonds, whereas paritaprevir (orange) showed the lesser number of hydrogen bonds, mainly in the early stages of the simulation. The appearance of more hydrogen bonds from 5 to 20 ns by ledipasvir (red) and velpatasvir (blue) suggested the conformational change due to the interaction of ligands to the main protease in their binding site. At the outset, all three protein-ligand complexes showed (Fig. 9) that they are stable during simulations, while ledipasvir (red) and velpatasvir (blue) were found to be more stable than paritaprevir (orange).
Binding energy
According to earlier reports (Khan et al., 2021b; Sharma et al., 2021), 400 snapshots were used to calculate binding free energies through the g_mmpbsa tool. The binding energy values between the main protease and drugs through the MM-PBSA method are listed in Table 3. The ledipasvir (red) and velpatasvir reported higher binding energy values of −195.370 ± 1.119 and −180.778 ± 0.868, respectively, whereas paritaprevir showed less −75.679 ± 0.922 when compared to the other two drugs. The main contributors of binding energy for ledipasvir (red) and velpatasvir were Van der Waals energy, electrostatic energy, and solvent accessible surface area (SASA) energy. The deviation of binding energy between ledipasvir (red) and velpatasvir is attained through the variation of polar solvation energy.
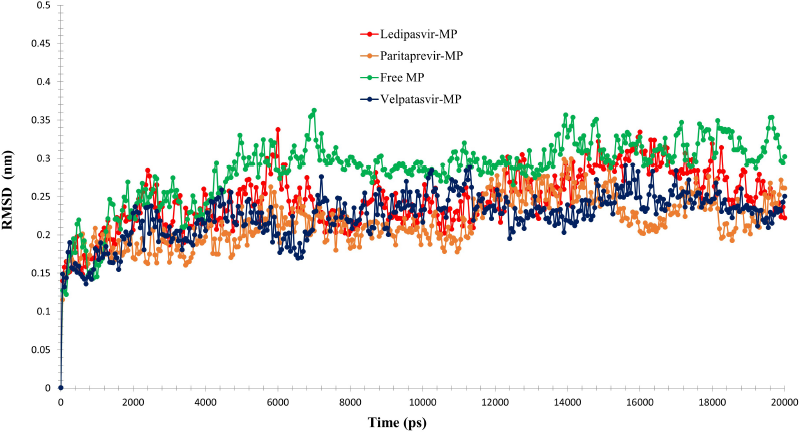 | Figure 6. RMSD analyses of MDS results of the free main protease and the main protease-drug complex. [Click here to view] |
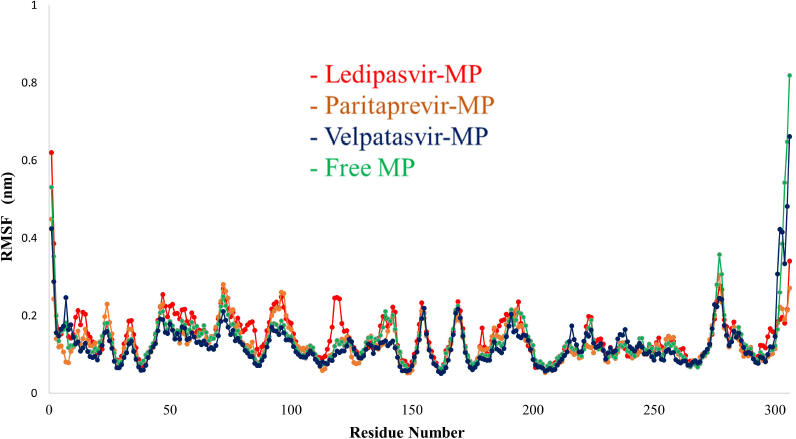 | Figure 7. RMSF analyses of MDS results of the free main protease and the main protease-drug complex. [Click here to view] |
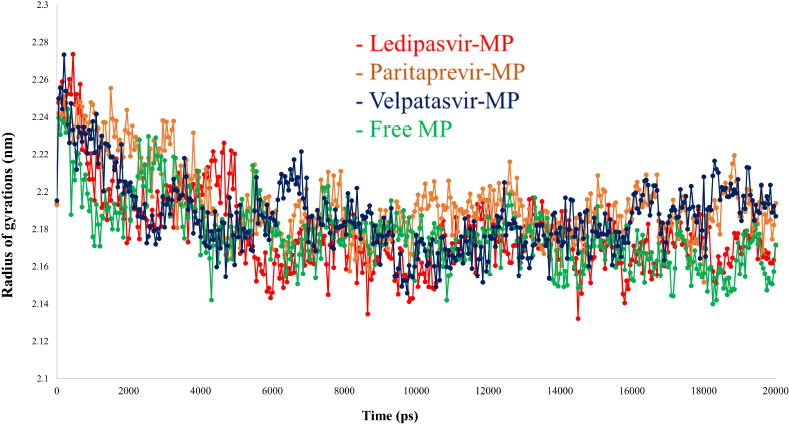 | Figure 8. RG analyses of MDS results of the free main protease and the main protease-drug complex. [Click here to view] |
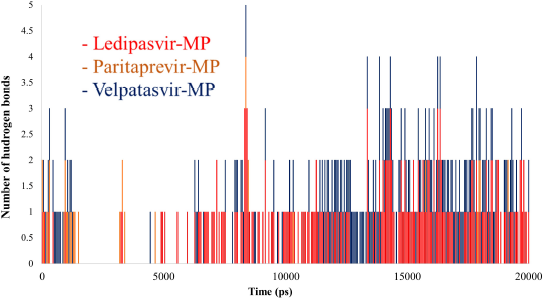 | Figure 9. Hydrogen bond analyses of MDS results of the main protease-drug complex. [Click here to view] |
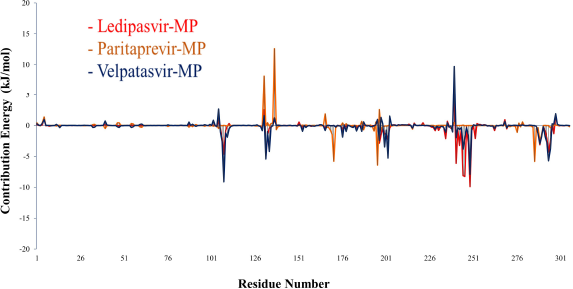 | Figure 10. MM-PBSA plot of main protease and drugs. [Click here to view] |
The protein flexibility in MDS can be used to study binding interaction on the main protease and drug complex (Kwofie et al., 2019). MM-PBSA decomposition of the binding energy was calculated, and the results were depicted in Figure 10 and Table 3.
The ledipasvir (red) showed a maximum negative energy −9.8719 in the position of 249 (ILE), whereas velpatasvir (blue) contribution of showed a negative energy maximum of −9.1183 at 108 (PRO) residue. The maximum positive energy contributed by paritaprevir (orange) is 12.5964 at position 137 (LYS), whereas velpatasvir (blue) showed the next maximum value of 9.7103 at 240 (GLU) position. The ledipasvir (red) contributes mostly in the region of 240–250 range with maximum values like −6.1187 (241 PRO); −8.1143 (245 ASP); −8.2631 (246 HIS); and −9.8719 (249 ILE). Thus, it proves the binding site of the drug ledipasvir (red) to the main enzyme protease. Most of the contribution occurs at the terminal domain residues in the overall assessment because of the drug interactions.
Overall, these top three drugs, ledipasvir, paritaprevir, and velpatasvir, have shown significant stability when combined with the structure of the Mpro of SARS-CoV-2. Additionally, it was again confirmed by the binding energy calculation by the MM-PBSA method, which reveals that ledipasvir and velpatasvir showed extraordinary binding energy scores, whereas paritaprevir has a slight decline in it. Hence, when considering the huge binding affinity, stability in a complex form with Mpro, enormous binding energy, considerable contribution energy, Rg, RMSD, and RMSF values, ledipasvir is the top ligand in all aspects, followed by paritaprevir (except binding energy) and velpatasvir. Further studies on mechanisms, in vivo and in vitro screenings, and research trials may give a clear picture of the repurposing possibility.
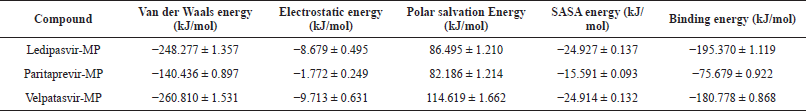 | Table 3. Binding energy between main protease and drugs through MM-PBSA method. [Click here to view] |
CONCLUSION
In this study, molecular docking analysis demonstrates that ledipasvir has more binding affinity to the Mpro of SARS-CoV-2 when compared to other drugs. Paritaprevir and velpatasvir occupy successive positions. MDS analysis exposed that all the three drug-protein complexes exhibited steadiness at 20 ns. MM-PBSA revealed the robust binding among ledipasvir and main protease with an average binding energy of −195.370 ± 1.119 kJ/mol. Simulation studies of the drugs (ledipasvir, paritaprevir, and velpatasvir) with and without a main bound protease disclosed the protein’s conformation changes. The amino acids involved in the interaction with ledipasvir were also projected, which include 241PRO, 245ASP, 246HIS, and 249ILE. Further studies and trials are warranted to understand the ability of these drugs against SARS-CoV-2 via in vivo and in vitro approaches.
ACKNOWLEDGMENTS
The authors are thankful to Dr. K. R. Venkatesan, Principal, Sri Sankara Arts and Science College (Autonomous), for his continuous encouragement and support to complete this work.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
FUNDING
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All data generated and analyzed are included within this research article.
REFERENCES
Al-Khafaji K, Al-Duhaidahawi D, Taskin Tok T. Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. J Biomol Struct Dyn, 2021; 39:3387–95. CrossRef
Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA, 2001; 98(18):10037–41. CrossRef
BIOVIA DSV. Discovery studio visualizer. San Diego, Dassault Systemes, Release, 2020, v20.1.0.19295.
Bokam YK, Guntupalli C, Gudhanti SN, Kul U, Alavala RR, Alla NR, Manne R. Importance of pharmacists as a front line warrior in improving medication compliance in COVID-19 patients. Indian J Pharm Sci, 2021; 83(2):398–401. CrossRef
Boopathi S, Poma AB, Kolandaivel P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J Biomol Struct Dyn, 2021; 39(9):3409–18. CrossRef
Chaudhuri S, Symons JA, Deval J. Innovation and trends in the development and approval of antiviral medicines: 1987–2017 and beyond. Antiviral Research, 2018; 155:76–88. CrossRef
Chen YW, Yiu CP, Wong KY. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL pro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research, 2020b; 9(129):1–16. CrossRef
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y, Yu T. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet, 2020a; 395:507–13. CrossRef
Cheng G, Tian Y, Doehle B, Peng B, Corsa A, Lee YJ, Gong R, Yu M, Han B, Xu S, Dvory-Sobol H. In vitro antiviral activity and resistance profile characterization of the hepatitis C virus NS5A inhibitor ledipasvir. Antimicrob Agents Chemother, 2016; 60(3):1847–53. CrossRef
Das S, Sarmah S, Lyndem S, Singha Roy A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J Biomol Struct Dyn, 2021; 39(9):3347–57. CrossRef
Elfiky AA. Ribavirin, remdesivir, sofosbuvir, galidesivir, and tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): a molecular docking study. Life Sci, 2020; 253:117592. CrossRef
Elfiky AA. Natural products may interfere with SARSCoV-2 attachment to the host cell. J Biomol Struct Dyn, 2021; 39:3194–203. CrossRef
Eisenhaber F, Lijnzaad P, Argos P, Sander C, Scharf M. The double cube lattice method: efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J Comp Chem, 1995; 16(3):273–84. CrossRef
Guan W, Lan W, Zhang J, Zhao S, Ou J, Wu X, Yan Y, Wu J, Zhang Q. COVID-19: antiviral agents, antibody development and traditional chinese medicine. Virol Sin, 2020; 35:685–98. CrossRef
Guex N, Peitsch, MC. SWISS-MODEL and the Swiss-Pdb viewer: an environment for comparative protein modeling. Electrophoresis, 1997; 18(15):2714–23. CrossRef
Jockusch S, Tao C, Li X, Chien M, Kumar S, Morozova I, Kalachikov S, Russo JJ, Ju J. Sofosbuvir terminated RNA is more resistant to SARS-CoV-2 proofreader than RNA terminated by Remdesivir. Sci Rep, 2020; 10:16577. CrossRef
Joshi S, Joshi M, Degani MS. Tackling SARS-CoV-2: proposed targets and repurposed drugs. Future Med Chem, 2020; 12(17):1579–601. CrossRef
Khan RJ, Jha RK, Amera GM, Jain M, Singh E, Pathak A, Singh RP, Muthukumaran J, Singh AK. Targeting SARSCoV-2: a systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 20-O-ribose methyl transferase. J Biomol Struct Dyn, 2021a; 39:2679–92. CrossRef
Khan SA, Zia K, Ashraf S, Uddin R, Ul-Haq Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J Biomol Struct Dyn, 2021b; 39(7):2607–16. CrossRef
Kroemer RT. Structure-based drug design: docking and scoring. Curr Protein Peptide Sci, 2007; 8(4):312. CrossRef
Kumari R, Kumar R, Lynn A. G-mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model, 2014; 54(7):1951–62. CrossRef
Kwofie SK, Dankwa B, Enninful KS, Adobor C, Broni E, Ntiamoah A, Wilson MD. Molecular docking and dynamics simulation studies predict Munc18b as a target of mycolactone: a plausible mechanism for granule exocytosis impairment in buruli ulcer pathogenesis. Toxins, 2019; 11(3):181. CrossRef
Lobo-Galo N, Terrazas-López M, Martínez-Martínez A, Díaz-Sánchez ÁG. FDA-approved thiol-reacting drugs that potentially bind into theSARS-CoV-2 main protease, essential for viral replication. J Biomol Struct Dyn, 2021; 39:341927. CrossRef
Pant S, Singh M, Ravichandiran V, Murty US, Srivastava HK. Peptidelike and small-molecule inhibitors against Covid-19. J Biomol Struct Dyn, 2021; 39(8):2904–13. CrossRef
Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, Van Der Spoel D, Hess B. GROMACS 4.5: a high-throughput and highly parallel open-source molecular simulation toolkit. Bioinformatics, 2013; 29(7):845–54. CrossRef
Rameez Jabeer K, Rajat KJ, Gizachew Muluneh A, Monika J, Ekampreet S, Amita P, Rashmi Prabha S, Jayaraman M, Amit Kumar S. Targeting novel coronavirus 2019: a systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 20-o-ribose methyl transferase. ChemRxiv, 2020; 1:1; doi:10.26434/chemrxiv.11888730.v1
Sayad B, Sobhani M, Khodarahmi R. Sofosbuvir as repurposed antiviral drug against COVID-19: why were we convinced to evaluate the drug in a registered/approved clinical trial? Arch Med Res, 2020; 51(6):577–81. CrossRef
Schmid N, Eichenberger AP, Choutko A, Riniker S, Winger M, Mark AE, van Gunsteren WF. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur Biophys J, 2011; 40:843. CrossRef
Schüttelkopf AW, Van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr, 2004; 60:1355–63. CrossRef
Sharma A, Ahmad Farouk I, Lal SK. COVID-19: a review on the novel coronavirus disease evolution, transmission, detection, control and prevention. Viruses, 2021a; 13:202; doi:10.3390/v13020202 CrossRef
Sarma P, Shekhar N, Prajapat M, Avti P, Kaur H, Kumar S, Singh S, Kumar H, Prakash A, Dhibar DP, Medhi B. In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). J Biomol Struct Dyn, 2021b; 39(8):2724–32. CrossRef
Shaha B, Modi P, Sagar SR. In silico studies on therapeutic agents for COVID-19: drug repurposing approach. Life Sci, 2020; 252:117652. CrossRef
WHO. Coronavirus disease (COVID-19) pandemic. 2021. Available via https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (Accessed on 22 May 2021).
Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science, 2020; 368(6489):409–12. CrossRef