
INTRODUCTION
The development of new pharmaceutical agents is one of the greatest challenges in overcoming widely spread epidermics and drug resistance. Furthermore, the ineffectiveness of medications such as antimicrobial drugs cause an increase in mortality, morbidity, and public health problems. The major components of resistance to many classes of antimicrobials are efflux mechanisms (Reygaert, 2018). In these mechanisms, efflux pump inhibitors are used to stop bacteria from pumping antibiotics out of cells, and it has been reported that 5-nitro-2-phenylindole ‘…..(INF55) (1) (Fig. 1) participates in the NorA efflux pump of Gram-positive bacteria and increases the susceptibility of Staphylococcus aureus to antibiotics (Markham et al., 1999). Substituents on the phenyl ring in INF55 (1) are also important for biological activities; that is, the 2,5-dimethoxyl group on the aryl ring (2) increases the inhibitory activity of NorA (Samosorn et al., 2006). This indicated that the substituents on the phenyl ring of the 2-arylindole nucleus affect biological activity. Similarly, the substitutions of the benzene ring of the indole nucleus diverted the biological activity, which were reported in the literature (Lal and Snape, 2012; Naik et al., 2014; Williams et al., 2013). Among these, 5-methoxy-2-phenylindole (3) was reported to be a significant antibacterial activity against Gram-positive Bacillus cereus minimum inhibitory concentrations (MIC) = 3.9 μg/ml. Furthermore, 5-amidine-2-arylindole bearing a 3,5-disubstituted aryl ring (4) remarkably affects the inhibition of acid-sensing ion channels (Kuduk et al., 2009). In addition, the 2-aryl-substituted indole derivative (5) is a bacterial histidine kinase inhibitor (Deschenes et al., 1999).
With the wide-ranging biological activities, the 2-arylindole scaffold is among the privileged frameworks to study the antibacterial activity and its inhibited NorA efflux pump-resistant mechanism. Thus, the goal of this study is to investigate the 2-arylindoles with various substituents on the aryl ring with their antibacterial activities and the ability to potentiate the activity of commercially available drugs and overcome drug resistance.
Indole is synthesized using various synthesis methods, including Fischer indole synthesis (Cho et al., 2009), Bischler synthesis (Cossío et al., 2008), Madelung cyclization (Imanishi et al., 1996), and transition metal catalyzation (Heck and Terpko, 1979; Larock and Yum, 1991). However, in terms of synthesis efficiency, these methods are limited by their requirement of several steps to be completed (Dai et al., 2001; Ezquerra et al., 1996; Rudisill and Stille, 1989; Taylor et al., 1985). Furthermore, only a few methods are available to substitute the indole ring at the C2 position because the C3 position is more reactive than the C2 position for electrophilic aromatic substitution. Nevertheless, an N-protected indole can undergo organometallic substitution at the C2 position.
In this study, Fischer indole synthesis was applied as a simple and effective method to prepare 2-substituted indoles. The antibacterial activities of the synthesized compounds and their synergistic antibacterial potential with tetracyclines against the clinical isolate of methicillin-resistant S. aureus (MRSA) Sp6 were evaluated and described. Their synergistic mechanism was also investigated through molecular docking.
MATERIALS AND METHODS
General information
Melting points (m.p.) were determined using a Stuart Scientific SMP 2 m.p. apparatus and uncorrected. 1H- and 13C-NMR spectra were obtained with (D) d6-DMSO solutions at 300 MHz for 1H and 75 MHz for 13C with a Bruker AVANCE 300 spectrometer. Tetramethylsilane was used as an internal standard. Mass spectra were recorded with a Polaris Q or Hewlett Packard 5973 mass spectrometer. Reagents and solvents were supplied by Acros, Aldrich, Fluka, and TCI.
General procedure for the synthesis of 2-substituted indoles (4a–m)
Phenylhydrazine (1) (1.5 eq.) was mixed with ketone (2a–m) (1.0 eq.) in 50% acetic acid (20 ml). The mixture was added to a beaker containing polyphosphoric acid (1.0 eq.), stirred while being heated in an oil bath for 30 minutes. The reaction mixture was cooled to room temperature and added to iced water (50 ml) to yield a black precipitate. The precipitate was crystallized in ethanol to obtain 2-substituted indoles (4a–m).
2-Methylindole (4a)
The title compound was synthesized from 1 (0.50 ml, 5.1 mmol) and acetone (2a) (0.5 ml, 6.8 mmol) to afford 4a (0.44 g, 66% yield) as a pale yellow substance with m.p. of 55.3°C–56.5°C [(Nakazaki, 1976); 58°C–60°C]; 1H NMR (300 MHz, CDCl3); δ: 7.83 (br, N), 7.50 (d, J = 7.4 Hz, 1H), 7.28 (d, J = 7.4 Hz, 1H), 7.11 (td, J = 1.4, 7.1 Hz, 1H), 7.06 (td, J = 1.4, 7.1 Hz, 1H), 6.21 (s, 1H), 2.45 (s, 3H) ppm.
2-tert-Butyl indole (4b)
The title compound was synthesized from 1 (0.60 ml, 6.1 mmol) and 1,1,1-trimethylacetone (2b) (0.90 ml, 6.1 mmol) to afford 4b (0.49 g, 46% yield) as dark solids; m.p. 71.3°C–72.8°C [(Fuher et al., 2019); 75°C–76°C]; 1H NMR (300 MHz, CDCl3); δ: 7.94 (br, NH), 7.54 (d, J = 7.6 Hz, 1H), 7.32 (d, J = 7.6 Hz, 1H), 7.12 (t, J = 7.6 Hz, 1H), 7.06 (t, J = 7.6 Hz, 1H), 6.26 (s, 1H),1.40 (s, 9H) ppm.
2-Phenyl-1H-indole (4c)
The title compound was synthesized from 1 (0.60 ml, 60 mmol) and acetophenone (2c) (0.50 ml, 40 mmol) to afford 4c (6.49 g, 84% yield) as yellow crystals; m.p. 187.9°C–188.3°C [(Bremner et al., 2008); 190.5°C–191.0°C]; 1H NMR (300 MHz, DMSO-d6); δ: 6.90 (1H, s), 6.99 (1H, t, J = 7.5 Hz), 7.10 (1H, t, J = 7.5 Hz), 7.3 (1H, t, J = 7.8 Hz), 7.41 (1H, d, J = 7.5 Hz), 7.46 (2H, t, J = 7.8 Hz), 7.53 (1H, d, J = 7.8 Hz), 7.86 (2H, d, J = 7.8 Hz), 11.53 (1H, s); 13C-NMR (DMSO-d6); δ: 100.0, 110.9, 120.3, 120.7, 112.4, 125.2, 127.7, 129.9, 129.3, 132.4, 136.8, 137.9; HRES-MS m/z calcd for [M]+ C14H11N: 194.0970, found: 194.0966.
2-(4-Methylphenyl)-1H-indole (4d)
The title compound was synthesized from 1 (6.0 ml,60 mmol) and 4-methylacetonephenone (2d) (6.56 g, 40 mmol) to afford 4d (6.88 g, 83% yield) as yellow crystals; m.p. 218.4°C–219.7°C; 1H NMR (300 MHz, DMSO-d6); δ: 2.34 (3H, s), 6.83 (1H, s), 6.98 (1H, t, J = 7.8 Hz), 7.08 (1H, t, J = 7.8 Hz), 7.28 (2H, d, J = 8.1 Hz), 7.39 (1H, d, J = 7.8 Hz), 7.56 (1H, d, J = 7.8 Hz), 7.75 (2H, d, J = 8.1 Hz), 11.47 (1H, s);); 13C-NMR (DMSO-d6); δ: 20.9, 98.2, 111.3, 119.5, 120.0, 121.5, 125.0, 128.8, 129.5, 129.6, 136.9, 137.1, 137.9 [(Shi et al., 2008); 1H NMR (300 MHz, acetone-d6); δ: 2.29 (3H, s), 6.80 (1H, s), 7.00–6.95 (1H, m), 7.08–7.03 (2H, m), 7.23 (1H, d, J = 7.8), 7.38–7.34 (1H, m), 7.52 (1H, d, J = 9.0 Hz), 7.71 (2H, t, J = 8.4 Hz), 10.56 (1H, s). 13C NMR (75 MHz, acetone-d6); δ: (21.0, 99.2, 111.8, 120.2, 120.8, 122.3, 125.7, 130.2, 130.8, 137.1, 138.1)]; HRES-MS m/z calcd for [M]+ C15H14N: 208.1126, found: 208.1131.
2-(3-Methoxyphenyl)-1H-indole (4e)
The title compound was synthesized from 1 (6.0 ml, 60 mmol) and 3-methoxyacetophenone (2e) (6.56 g, 40 mmol) to afford 4e (7.41 g, 83% yield) as dark yellow crystals; m.p. 128.9°C–130.0°C; 1H NMR (300 MHz, DMSO-d6); δ: 3.84 (3H, s), 6.88 (1H, d, J = 8.1 Hz), 6.92 (1H, s), 7.00 (1H, t, J = 7.8 Hz), 7.10 (1H, t, J = 8.1 Hz), 7.36 (1H, t, J = 8.1 Hz), 7.40 (1H, d, J = 8.1 Hz), 7.44 (1H, s), 7.45 (1H, d, J = 7.8 Hz), 7.53 (1H, d, J = 7.8 Hz), 11.53 (1H, s); 13C-NMR (DMSO-d6); δ: 55.6, 99.3, 110.5, 111.6, 113.4, 117.8, 119.8, 120.4, 122.0, 128.5, 130.4, 133.8, 137.3, 137.7, 160.0 [(Shi et al., 2008); 1H NMR (300 MHz, CDCl3); δ: 3.85 (3H, s), 6.74 (1H, s), 6.84 (1H, d, J = 8.4 Hz), 7.08 (1H, s), 7.43–7.24 (4H, m), 7.61 (2H, d, J = 7.2 Hz), 8.23 (1H, s). 13C NMR (75 MHz, CDCl3); δ: 55.8, 99.8, 102.2, 111.6, 112.6, 125.0, 127.5, 128.9, 129.7, 138.6, 152.0, 154.4]; HRES-MS m/z calcd for [M]+ C15H14NO: 224.1075, found: 224.1060.
2-(4-Methoxyphenyl)-1H-indole (4f)
The title compound was synthesized from 1 (6.0 ml, 60 mmol) and 4-methoxyacetophenone (2f) (6.56 g, 40 mmol) to afford 4f (7.14 g, 80% yield) as dark yellow crystals; m.p. 221.0°C–222.5°C; 1H NMR (300 MHz, DMSO-d6); δ: 3.83 (3H, s),6.75 (1H, s), 6.97 (1H, t, J = 6.9 Hz), 7.03 (2H, d, J = 6.9 Hz), 7.06 (1H, t, J = 9.0 Hz), 7.37 (1H, d, J = 6.9 Hz), 7.49 (1H, d, J = 6.9 Hz), 7.79 (2H, d, J = 9.0 Hz), 11.42 (1H, s);); 13C-NMR (DMSO-d6); δ: 55.4, 97.5, 111.2, 114.5, 119.4, 119.8, 121.2, 125.0, 126.5,1 29.0, 137.1, 138.0, 159.0 [(Shi et al., 2008); 1H NMR (300 MHz, acetone-d6); δ: 3.86 (3H, s), 6.76 (1H, s), 7.01–6.97 (4H, m),7.38 (1H, d, J = 9.0), 7.53 (1H, d, J = 7.5 Hz), 7.85–7.77 (2H, m),10.55 (1H, s). 13C NMR (75 MHz, acetone-d6); δ: 60.4, 103.5, 116.6, 120.0, 125.1, 125.5, 126.9, 131.1, 132.1, 135.2, 143.1, 143.8, 165.1]; HRES-MS m/z calcd for [M]+ C15H14NO: 224.1075, found: 224.1088.
2-(2-Hydroxyphenyl)-1H-indole (4g)
The title compound was synthesized from 1 (6.0 ml, 60 mmol) and 2-hydroxyacetophenone (2g) (4.80 ml, 40 mmol) to afford 4g (5.01 g, 60% yield) as dark green crystals; m.p. 221.0°C–223.0°C; 1H NMR (300 MHz, DMSO-d6); δ: 6.82–6.91 (3H, m), 7.04 (1H, d, J = 8.7 Hz), 7.06 (1H, d, J = 8.7 Hz), 7.21 (1H, t, J = 8.0 Hz), 7.28 (1H, t, J = 8.7 Hz), 7.29 (1H, t, J = 8.7 Hz), 7.54 (1H, d, J = 8.0 Hz), 9.43 (1H, s), 12.80 (1H, s); 13C-NMR (DMSO-d6); δ: 112.6, 116.7, 117.7, 119.0, 119.4, 120.4, 120.6, 127.5, 129.5, 131.6, 136.5, 145.0, 147.9, 157.4 [(Snape et al., 2013); 1H NMR (300 MHz; CDCl3); δ: 6.0–5.0 (1H, br s), 6.87 (1H, m, Ar), 6.91 (1H, dd, J = 0.9 and 8.1 Hz, Ar), 7.04 (1H, td, J = 1.1 and 7.6 Hz, Ar), 7.26–7.11 (3H, m, Ar), 7.42 (1H, d, J = 8.1 Hz, Ar), 7.66 (1H, d, J = 7.8 Hz, Ar), 7.70 (1H, dd, J = 1.6 and 7.8 Hz, Ar), 9.22 (1H, br s); 13C-NMR (75 MHz; CDCl3); δ: 100.2, 116.6, 119.1, 120.1, 120.4, 121.5, 122.2, 128.4, 128.9, 134.8, 136.4, 152.0]; HRES-MS m/z calcd for [M]+ C14H12NO: 210.0919, found: 210.0908.
2-(3-Hydroxyphenyl)-1H-indole (4h)
The title compound was synthesized from 1 (6.0 ml, 60 mmol) and 3-hydroxyacetophenone (2h) (5.44 g, 40 mmol) to afford 4h (5.02 g, 60% yield) as violet crystals; m.p. 220.0°C–222.0°C; 1H NMR (300 MHz, DMSO-d6); δ: 6.84 (1H, s), 6.97 (1H, t, J = 7.8 Hz), 7.07 (1H, t, J = 7.8 Hz), 7.13 (1H, d, J = 7.8 Hz), 7.35 (1H, t, J = 7.8 Hz), 7.38 (1H, t, J = 7.8 Hz), 7.51 (1H, d, J = 7.8 Hz), 7.55 (1H, d, J = 7.8 Hz), 7.67 (1H, s), 11.67 (1H, s); 13C-NMR (DMSO-d6); δ: 99.0, 111.6, 120.1, 114.7, 116.8, 119.6, 120.2, 121.7, 128.7, 129.8, 133.5, 137.3, 137.6, 157.9 [(Kim et al., 2016); 1H NMR (400 MHz, DMSO-d6); δ: 6.73 (1H, d, J = 7.6 Hz), 6.79 (1H, s), 7.38 (1H, dd, J = 8.0, 2.4 Hz), 6.98 (1H, td, J = 7.6, 2.6 Hz), 7.08 (1H, td, J = 7.5 Hz, 2.3 Hz), 7.31–7.20 (3H, m), 7.51 (1H, dd, J = 7.5, 2.0 Hz), 9.55 (1H, s), 11.44 (1H, s); 13C NMR (100 MHz, DMSO-d6); δ: 98.5, 111.3, 111.9, 114.5, 116.0, 119.3, 120.0, 121.4, 128.6, 129.9, 133.5, 137.0, 127.8, 157.8]; HRES-MS m/z calcd for [M]+ C14H12NO: 210.0919, found: 210.0934.
2-(4-Hydroxyphenyl)-1H-indole (4i)
The title compound was synthesized from 1 (6.0 ml, 60 mmol) and 4-hydroxyacetophenone (2i) (5.44 g, 40 mmol) to afford 4i (5.10 g, 61% yield) as yellow crystals; m.p. 240.0°C–241.5°C; 1H NMR (300 MHz, DMSO-d6); δ: 6.68. (1H, s), 6.86 (1H, d, J = 8.7 Hz), 6.96 (1H, t, J = 7.2 Hz), 7.04 (1H, t, J = 7.2 Hz), 7.21 (1H, s),7.37 (1H, d, J = 7.2 Hz), 7.47 (1H, d, J = 7.2 Hz), 7.68 (1H, d, J = 8.7 Hz), 11.31 (1H, s) [(Tsuchimoto et al., 2005); 1H NMR (300 MHz, acetone-d6); δ: d 6.71 (1H, dd, J = 2.1, 0.9 Hz,), 6.91 (2H, dt, J = 9.0, 2.4 Hz), 6.97 (1H, ddd, J = 7.8, 6.8, 1.2 Hz), 6.97 (1H ddd, J = 8.0, 7.0, 1.2 Hz), 7.33–7.38 (1H, m), 7.48–7.53 (1H, m),7.69 (2H, dt, J = 8.7, 2.5 Hz), 8.52 (1H, s), 10.47 (1H, bs)]; 13C-NMR (DMSO-d6); δ: 97.0, 114.2, 115.3, 119.7, 121.0, 123.6, 126.6, 126.7, 128.8, 136.0, 137.0, 157.2; HRES-MS m/z calcd for [M]+ C14H12NO: 210.0919, found: 210.0903.
2-(3-Nitrophenyl)-1H-indole (4j)
The title compound was synthesized from 1 (6.0 ml, 60 mmol) and 3-nitroacetophenone (2j) (6.56 g, 40 mmol) to afford 4j (7.05 g, 74% yield) as dark yellow crystals; m.p. 169.2°C–171.3°C; 1H NMR (300 MHz, DMSO-d6); δ: 7.06 (1H, t,J = 7.8 Hz), 7.13 (1H, s), 7.16 (1H, t, J = 7.8 Hz), 7.40 (1H, d, J = 8.1 Hz), 7.57 (1H, d, J = 7.8 Hz), 7.73 (1H, d, J = 7.8 Hz), 8.13 (1H, d, J = 8.1 Hz), 8.83 (1H, d, J = 8.1 Hz), 8.70 (1H, s), 11.42 (1H, s); 13C-NMR (DMSO-d6); δ: 87.1, 111.7, 117.1, 118.5, 120.8, 123.3, 125.1, 130.2, 132.8, 138.7, 141.7, 146.5, 148.5, 161.0 [(Shi et al., 2008); 1H NMR (300 MHz, acetone-d6); δ: 6.65 (1H, s), 7.38–7.16 (4H, m), 7.66–7.58 (2H, m), 7.81 (1H, d, J = 9.0), 8.21 (1H, d, J = 9.0), 8.35 (1H, s), 10.53 (1H, s); 13C-NMR (75 MHz, acetone-d6); δ: 103.2, 109.8, 120.3, 120.8, 122.6, 123.6, 127.6, 129.5, 134.4, 134.9, 138.7, 148.3]; HRES-MS m/z calcd for [M]+ C14H11N2O2: 239.0821, found: 239.0843.
2-(4-Nitrophenyl)-1H-indole (4k)
The title compound was synthesized from 1 (6.0 ml, 60 mmol) and 4-nitroacetophenone (2k) (6.56 g, 40 mmol) to afford 4k (7.60 g, 80% yield) as dark yellow crystals; m.p. 173.2°C–175.3°C [(Westwell et al., 2013); 168°C–172°C]; 1H NMR (300 MHz, DMSO-d6); δ: 6.87 (1H, s), 6.99 (1H, t, J = 7.5 Hz), 7.09 (1H, t, J = 7.5 Hz), 7.31 (2H, d, J = 9.0 Hz), 7.38 (1H, d, J = 7.5 Hz), 7.53 (1H, d, J = 7.5 Hz), 7.90 (2H, d, J = 9.0 Hz), 11.42 (1H, s); 13C-NMR (DMSO-d6); δ: 102.5, 111.7, 119.9, 120.8, 123.1, 124.36, 125.5, 128.4, 135.2, 137.9, 138.6, 145.8; HRES-MS m/z calcd for [M]+ C14H11N2O2: 239.0821, found: 239.0803.
2-(4-Fluorophenyl)-1H-indole (4l)
The title compound was synthesized from 1 (6.0 ml, 60 mmol) and 4-fluoroacetophenone (2l) (0.50 ml, 40 mmol) to afford 4l (6.80 g, 80% yield) as dark green crystals; m.p. 172.9°C–173.5°C; 1H NMR (300 MHz, DMSO-d6); δ: 6.87 (1H, s),6.99 (1H, t, J = 7.5 Hz), 7.09 (1H, t, J = 7.5 Hz), 7.31 (2H, dd, J = 9.0, 9.0 Hz), 7.38 (1H, d, J = 7.5 Hz), 7.53 (1H, d, J = 7.5 Hz), 7.90 (2H, dd, J = 9.0, 5.4 Hz), 11.42 (1H, s); 13C-NMR (DMSO-d6); δ: 99.2, 111.8, 116.2, 116.5,120.0, 120.6, 122.2, 127.4, 127.5, 129.1, 137.2, 137.69 [(Shi et al., 2008); 1H NMR (300 MHz, acetone-d6); δ: 6.84 (1H, s), 7.24–7.01 (3H, m), 7.42 (1H, d, J = 10.2 Hz), 7.56 (1H, d, J = 8.1 Hz), 7.89–7.84 (1H, m), 10.58 (1H, s); 13C NMR (75 MHz, acetone-d6); δ: 99.6, 111.7, 116.1, 116.3, 120.2, 122.4, 127.5, 127.6, 129.9, 138.1, 164.3]; HRES-MS m/z calcd for [M]+ C14H11FN: 212.0876, found: 212.0853.
2-(4-Aminophenyl)-1H-indole (4m)
The title compound was synthesized from 1 (6.0 ml, 60 mmol) and 4-aminoacetophenone (2m) (5.40 g, 40 mmol) to afford 4m (6.99 g, 84% yield) as dark green crystals; m.p. 261.4°C–262.8°C [(Westwell et al., 2013); 168°C–172°C]; 1H NMR (300 MHz, DMSO-d6); δ: 6.71 (1H, s), 6.67–7.00 (2H, br), 6.96 (1H, t, J = 7.8 Hz), 6.97 (2H, d, J = 7.8 Hz), 7.04 (1H, t, J = 7.8 Hz), 7.36 (1H, d, J = 7.8 Hz), 7.46 (1H, d, J = 7.8 Hz), 7.77 (2H, d, J = 7.8 Hz), 11.39 (1H, s) [(Westwell et al., 2013); 1H NMR (500 MHz, DMSO-d6); δ: 5.27 (2H, s), 6.57 (1H, s), 6.64 (2H, d, J = 8.4 Hz), 6.93 (1H, d, J = 8.5 Hz), 7.00 (1H, m), 7.32 (1H, d, J = 8.2 Hz), 7.43 (1H, d, J = 8.8 Hz), 7.52 (2H, d, J = 8.4 Hz), 11.16 (1H, s)]; 13C-NMR (DMSO-d6); δ: 95.9, 111.1, 114.6, 119.3, 119.5, 120.6, 120.7, 126.3, 129.3, 136.9, 139.3, 148.0; HRES-MS m/z calcd for [M]+ C14H13N2: 209.1079, found: 209.1090
General procedure for the amidation of 4m with acid bromide derivatives (6a–f)
Acid bromide (5) (1.7 eq.) was added to a solution of 2-(4-aminophenyl)-1H-indole (4m) (1.0 eq.) and Et3N (0.8 eq.) in dry THF (2.5 ml) at 0°C. The mixture was stirred for 2 hours and water (25 ml) was added. The mixture was extracted with CH2Cl2 (3 × 20 ml). The organic phase was washed with sat. NaHCO3 and dried with anh. Na2SO4. Then, the solvent was evaporated under vacuum and purified via column chromatography (silica gel, 1:2 hexane:EtOAc) to give the amide derives (6a–f).
2-(4-Acetamidophenyl)-1H-indole (6a)
The title compound was synthesized from 4m (75 mg, 0.36 mmol), Et3N (0.05 ml, 0.36 mmol), and acetyl bromide (0.05 ml, 0.6 mmol) to obtain 2-(4-acetamidophenyl)-1H-indole (6a) (47.8 mg, 53% yield) as a brown solid; m.p. 286.5°C–288.2°C [(Ackermann et al., 2009); 283.9°C–286.8°C]; 1H NMR (300 MHz, DMSO-d6); δ: 11.42 (br, NH), 10.04 (br, NH), 7.75 (d, J = 8.7 Hz, 2H), 7.65 (d, J = 8.7 Hz, 2H), 7.49 (d, J = 7.7 Hz, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.07 (t, J = 8.0 Hz, 1H), 6.97 (t, J = 7.9 Hz, 1H), 6.79 (s, 1H), 2.07 (s, 3H) ppm; 13C-NMR (DMSO-d6); δ: 21.4 (CH3), 96.5, 111.9, 114.5 (×2), 118.6, 119.3, 120.7, 121.7, 127.7 (×2), 128.9, 136.0, 139.5, 147.8, 172.5 [(Ackermann et al., 2009); 1H-NMR (300 MHz, DMSO-d6); δ: 2.10 (3H, s), 6.80 (1H, m), 7.02–6.96 (1H, m), 7.11–7.06 (1H, m), 7.42–7.40 (1H, m), 7.52 (1H, d, J = 7.7 Hz), 7.71 (2H, d, J = 8.6 Hz), 7.81 (2H, d, J = 8.6 Hz), 10.04 (1H, s), 11.42 (1H, s). 13C-NMR (126 MHz, DMSO-d6); δ: 24.0, 97.8, 111.1, 119.2, 119.2, 119.7, 121.2, 125.4, 127.0, 128.8, 137.0, 137.7, 138.6, 168.3]; HRES-MS m/z calcd for [M]+ C16H14N2O: 250.1106, found 250.1079.
2-(4-Propioamidophenyl)-1H-indole (6b)
The title compound was synthesized from 4m (64 mg, 0.31 mmol), Et3N (0.05 ml, 0.4 mmol), and propanoyl chloride (0.05 ml, 0.6 mmol) to obtain 2-(4-propioamidophenyl)-1H-indole (6b) (50.0 mg, 62% yield) as a pale orange solid; m.p. 256.1°C–257.2°C; 1H NMR (300 MHz, DMSO-d6); δ: 1.11 (3H, t, J = 7.5 Hz), 2.35 (2H, q, J = 7.5 Hz), 6.81 (1H, s), 6.98 (1H, t, J = 7.4 Hz), 7.07 (1H, t, J = 8.0 Hz), 7.38 (1H, d, J = 8.0 Hz), 7.50 (1H, d, J = 8.0 Hz), 7.68 (2H, d, J = 8.4 Hz), 7.79 (2H, d, J = 8.4 Hz), 9.97 (1H, br, NH), 11.45 (1H, br, NH); 13C-NMR (DMSO-d6); δ: 9.5, 26.6, 96.8, 111.1, 114.8 (×2), 119.4, 119.8, 120.7, 121.5, 127.6 (×2), 129.4, 135.6, 138.5, 148.2, 173.2; HRES-MS m/z calcd for [M]+ C17H16N2O: 264.1263, found 264.1222.
2-(4-Octanamidophenyl)-1H-indole (6c)
The title compound was synthesized from 4m (70 mg, 0.34 mmol), Et3N (0.05 ml, 0.36 mmol), and octanoyl chloride (0.10 ml, 0.59 mmol) to obtain 2-(4-octanamidophenyl)-1H-indole (6c) (50 mg, 44% yield) as a white solid; m.p. 239.2°C–240.2°C; 1H NMR (300 MHz, DMSO-d6); δ: 0.88 (3H, t, J = 6.9 Hz), 1.35–1.20 (8H, m), 1.70–1.55 (2H, m), 2.33 (2H, t, J = 7.4 Hz, 2H), 6.80 (1H, s), 6.98 (1H, t, J = 7.0 Hz), 7.07 (1H, t, J = 7.0 Hz), 7.38 (1H, d, J = 8.0 Hz), 7.50 (1H, d, J = 7.8 Hz), 7.68 (2H, d, J = 8.7 Hz), 7.78 (2H, d, J = 8.7 Hz), 9.97 (1H, br, NH), 11.43 (1H, br, NH); 13C-NMR (DMSO-d6); δ: 14.1, 25.5, 29.2, 29.5 (×2), 31.7, 33.7, 40.5, 111.1, 114.8 (×2), 119.4, 119.8, 120.7, 121.5, 127.6 (×2), 129.4, 135.6, 138.5, 148.2, 173.2; HRES-MS m/z calcd for [M]+ C21H24N2O: 320.1889, found 320.1875.
2-(4-Benzamidophenyl)-1H-indole (6d)
The title compound was synthesized from 4m (92.3 mg, 0.44 mmol), Et3N (0.1 ml, 0.72 mmol), and benzoyl chloride (0.10 ml, 0.86 mmol) to obtain 2-(4-benzamidophenyl)-1H-indole (6d) (0.12 g, 90% yield) as a brown solid; m.p. 281.7°C–282.8°C; 1H NMR (300 MHz, DMSO-d6); δ: 6.01 (1H, s), 6.14 (1H, t, J = 7.5 Hz), 6.24 (1H, t, J = 7.3 Hz), 6.55 (1H, d, J = 9.0 Hz), 6.81–6.64 (4H, m), 7.07–6.98 (4H, m), 7.14 (2H, d, J = 7.2 Hz), 9.51 (1H, br, NH), 10.63 (1H, br, NH); 13C-NMR (DMSO-d6); δ: 96.8, 111.1, 114.7 (×2), 119.6, 119.8, 120.7, 121.7, 126.9 (×2), 128.6 (×2), 130.0, 132.1, 134.2, 136.0, 136.3, 137.6, 137.9, 148.5, 171.6; HRES-MS m/z calcd for [M]+ C21H16N2O: 312.1263, found 312.1249.
2-(2-Nitrobenzamidophenyl)-1H-indole (6e)
The title compound was synthesized from 4m (75.0 mg, 0.36 mmol), Et3N (0.05 ml, 0.36 mmol, 0.8 eq.), and 4-nitrobenzyl chloride (0.11 g, 0.63 mmol) to obtain 2-(2-nitrobenzamidophenyl)-1H-indole (6e) (0.10 g, 88% yield) as a yellow solid; 1H NMR (300 MHz, DMSO-d6); δ: 6.91 (1H, s), 7.03 (1H, t, J = 7.5 Hz), 7.12 (1H, t, J = 7.7 Hz), 7.43 (1H, d, J = 9.0 Hz), 7.56 (1H, d, J = 9.0 Hz), 7.92 (4H, s), 8.26 (2H, d, J = 9.0 Hz), 8.43 (2H, d, J = 9.0 Hz), 10.74 (1H, br, NH), 11.55 (1H, br, NH); 13C-NMR (DMSO-d6); δ: 107.9, 111.1, 113.2, 113.8, 120.2, 120.5, 121.0, 122.2, 126.1, 129.0, 136.92, 136.97, 147.2, 152.0, 171.9; HRES-MS m/z calcd for [M]+ C21H15N3O3: 357.1113, found 357.1100.
2-(4-Nitrobenzamidophenyl)-1H-indole (6f)
The title compound was synthesized from 4m (0.11 g, 0.52 mmol), Et3N (0.05 ml, 0.36 mmol), and 4-nitrobenzyl chloride (0.05 g, 1 mmol) to obtain 2-(4-nitrobenzamidophenyl)-1H-indole (6f) (88.0 mg, 47% yield) as a yellow solid; m.p. 263.4°C–264.4°C; 1H NMR (300 MHz, DMSO-d6); δ: 6.86 (1H, s), 7.00 (1H, t, J = 7.4 Hz), 7.10 (1H, t, J = 7.5 Hz), 7.40 (1H, d, J = 8.1 Hz), 7.53 (1H, d, J = 7.8 Hz), 7.84–7.71 (4H, m, 4H), 7.95–7.84 (3H, m), 8.18 (1H, d, J = 7.5 Hz), 10.79 (1H, br, NH), 11.48 (1H, br, NH); 13C-NMR (DMSO-d6); δ: 107.8, 110.9, 113.1, 113.8, 120.0, 120.4, 121.0, 122.1, 126.1, 128.9, 136.8, 136.9, 147.1, 152.0, 171.8; HRES-MS m/z calcd for [M]+ C21H15N3O3: 357.1113, found 357.1105.
Determination of antibacterial activity
The MICs of 4a–4 m and 6a–6f were tested against S. aureus ATCC 25932, B. cereus ATCC 7064, Bacillus subtilis ATCC 6633, Escherichia coli ATCC 10536, and Salmonella typhi ATCC 19430 and determined with National Committee for Clinical Laboratory Standards (NCCLS) microbroth dilution methods (National Committee for Clinical Laboratory Standards, 2000). Kanamycin and chloramphenicol were used as references for antibacterial activity. The test samples were dissolved in DMSO with a range of 512–0.98 μg/ml in nutrient broth supplemented with 10% glucose and 0.05% phenol red (NBGP). Then, 100 μl of each concentration was added to each well (96-well microplate) containing 95 μl of NBGP and 5 μl of inoculum (standardized at 1.5 × 106 cfu/ml by adjusting the optical density to 0.1 at 600 nm. The final concentration of DMSO in the well was less than 1% preliminary analyses with 1% (v/v) DMSO/NBGP affected neither the growth of the test organisms nor the change in color because of this growth). The negative control well contained 195 μl of NBGP and 5 μl of the standard inoculum. The plates were covered with a sterile plate sealer, agitated using a plate shaker to mix the content of the wells, and incubated at 37°C for 24 hours. The assay was repeated twice, and microbial growth was determined by observing the change in the color of the content of the wells (i.e., red = without growth, yellow with growth). The lowest concentration showing no color change was considered the MIC.
Synergistic antibacterial assay
MRSA Sp6 was isolated from pus samples of patients in Nakhon Pathom Hospital (Taechowisan et al., 2018). The drug resistance of this strain was detected via a cefoxitin paper method of the American Association of Clinical and Laboratory Standards Institute (CLSI, 2014). Its drug resistance and efflux pump genes were also detected through PCR amplification. The primers of the species-specific S. aureus (femA) gene, the penicillin-binding protein 2 (mecA) gene, and the active efflux pump of the plasmid-located tetracycline (tetK) gene were utilized for PCR assay as previously described (Taechowisan et al., 2018).
The MICs of methicillin and tetracycline were determined using a twofold dilution method in accordance with the guidelines of the National Committee for Clinical Laboratory Standards (1997). In this procedure, 100 ml of MRSA Sp6 (105 cfu/ml) was mixed with different concentrations of methicillin and tetracycline (16, 32, 64, and 128 mg/ml) in a 96-well plate and incubated at 37°C for 24 hours. A control group was prepared by culturing MRSA Sp6 with 0 mg/ml methicillin and tetracycline. Various concentrations (16, 32, 64, and 128 mg/ml) were applied to 100 ml of MRSA Sp6 (105 cfu/ml) cells alone or in combination with either methicillin or tetracycline (16, 32, 64, and 128 mg/ml) in 96-well plates to elucidate the effect of methicillin and tetracycline activities in the presence of 2-arylindole derivatives. The plates were incubated at 37°C for 24 hours. A blank control was also prepared by culturing MRSA Sp6 cells in media only without methicillin and tetracycline or 2-arylindole derivatives.
Molecular modeling
The homology model of NorA from S. aureus was constructed using a Swiss-Model server and the protein EmrD efflux pump from E. coli (PDB ID: 2GFP) because the crystal structure of NorA from S. aureus remained unavailable (Zárate et al.,2019). Molecular docking was performed with iGEMDOCK v2.1 (Hsu et al., 2011) to explore the protein-substrate interactions and binding position of 4g–k and 4 m in the active site of NorA from S. aureus. Ciprofloxacin, which is known as a good inhibitor of NorA efflux pumps, was also docked into the NorA efflux pump, and its binding energy was compared with those of our compounds.
Prediction of ADMET by computational analysis
The computational prediction of the compounds was performed through the online software SwissADME (http://swissadme.ch) in order to evaluate the pharmacokinetics of the synthesized molecules. The interaction of the synthesized molecules with CYP was used to predict the toxicity properties. Passive human gastrointestinal absorption (HIA) and blood–brain barrier (BBB) were used to predict the pharmacokinetic behaviors of these molecules. Caco-2-permeability was considered as drug-likeness property.
RESULTS AND DISCUSSION
Chemistry
2-Arylindoles were prepared in a manner similar to previously reported Fischer indole synthesis (Hughes, 1993) (Scheme 1). After the reaction parameters were screened, the reaction of 1 (1.5 eq.) and methyl ketone derivatives (2; 1.0 eq.) in 50% acetic acid (20 ml) produced moderate to high yields of indole derivatives (4) via hydrazone derivatives (3). The reaction time and temperature of each product under the optimized conditions are shown in Table 1. The heating duration of most of the aromatic derivatives of 2 at high temperatures should be prolonged. Unidentified products except nitrobenzene substitution (entries 10–11) were obtained at high temperatures (Scheme 2). The structures of all known compounds were confirmed by m.p. and spectroscopic analysis.
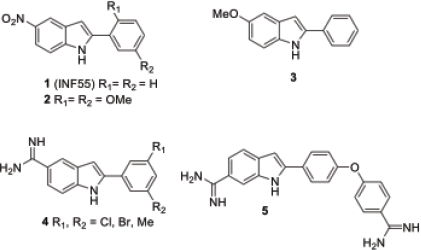 | Figure 1. Bioactive 2-arylindole. [Click here to view] |
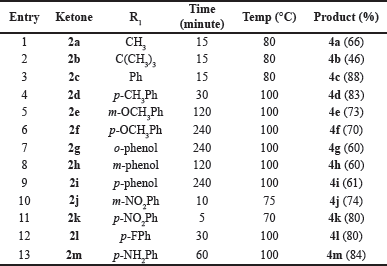 | Table 1. Synthesis of 2-substituted phenyl-1H indole derivatives. [Click here to view] |
The substitutions of amino groups on the aromatic ring were investigated to study the structure-activity relationship (Scheme 2). Amidation using acid halide (5) produced moderated yields of amides (6a–f). Then, the antibacterial activities and synergistic effects of the synthesized 2-arylindole derivatives were further evaluated.
Antibacterial activity
All the synthesized compounds 4a–m and 6a–f were tested against S. aureus ATCC 25932, B. cereus ATCC 7064,B. subtilis ATCC 6633, E. coli ATCC 10536, and S. typhi ATCC 19430 by NCCLS microbroth dilution methods (National Committee for Clinical Laboratory Standards, 1997). Kanamycin and chloramphenicol were used as positive controls, and a solvent was used as a negative control. The results are shown in Table 2 as MIC (μg/ml) unit. Among the compounds tested, 4m was the most active with an MIC of 15.6 μg/ml against B. subtilis and S. typhi. Furthermore, 4c, 4d, and 4f selectively inhibitedB. subtilis and had MICs of 31, 125, and 62 μg/ml, respectively. In addition, 4j showed antibacterial activities against S. typhi at 15.6 μg/ml. Thus, the phenyl ring, substituents, and their positions on the phenyl ring are essential for potency. Compounds 4e and 4h with phenyl-bearing electron-donating groups on the meta-position have weak or no activity, while compound 4k bearing electron-withdrawing group on the meta-position has selective activity against S. typhi. On the other hand, the phenyl-bearing hydrophilic substituents at para-position seem to be important for the activity. The protected amino derivatives (6a–f) had weak or no antibacterial activity against all the tested bacteria. This may assume that the amide linkage of 4m affected the hydrophilicity and might obstruct the interaction of these molecules and the bacterial active site.
Synergistic effect
The synthesized compounds were assayed to examine their antibacterial activities against MRSA Sp6. The results revealed that none of them exhibited an inhibitory activity at 128 mg/ml. By comparison, the MIC of tetracycline to MRSA Sp6 was 256 mg/ml. Nevertheless, the MIC of tetracycline decreased notably when the 2-arylindole derivatives were added. This result demonstrated that the sensitivity of MRSA Sp6 to tetracycline improved. Each experiment was repeated three times (Table 3). 4g, 4j, and 4k elicited significant synergistic effects with tetracycline against MRSA Sp6. Thus, the MIC of tetracycline could be reduced from 256 to 32 mg/ml when it was combined with 4g, 4j, and 4k at a concentration of 16 mg/ml.
 | Scheme 1. Synthesis of 2-substituted phenyl-1H indole derivatives. [Click here to view] |
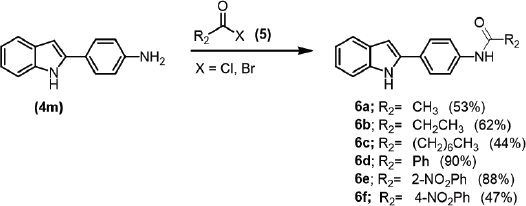 | Scheme 2. Preparation of N-substituted aminobenzene derivatives (6a–f). [Click here to view] |
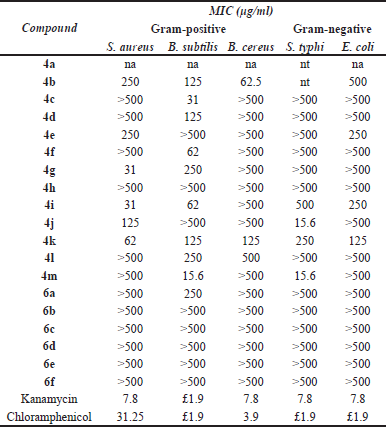 | Table 2. In vitro antimicrobial activities of compounds against the tested bacteria. [Click here to view] |
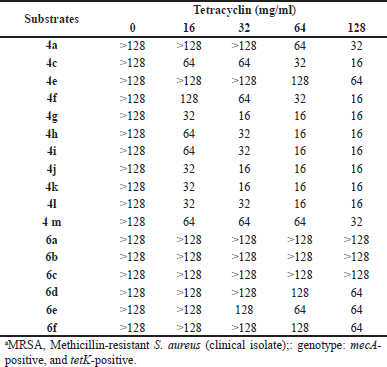 | Table 3. Synergistic effect of 2-arylindoles with tetracyclin against MRSAa. [Click here to view] |
Molecular docking
Molecular docking was conducted to explore the protein-substrate interactions and binding positions of the active synergistic compounds, namely, 4g–k and 4m, in the active site of NorA from S. aureus. These compounds were docked into the active site of the S. aureus NorA model prepared with the EmrD efflux pump as a template. Their molecular docking results were compared with those of the NorA substrate ciprofloxacin. The findings revealed that 4g–k and 4 m bound to the cavity of the NorA efflux pump in a position similar to that of ciprofloxacin (Fig. 2). The binding energies of 4g–k and 4m were −92.00 to −85.07 kcal/mol (Table 4), which were slightly higher than that of ciprofloxacin (−99.64 kcal/mol). Moreover, in the active site of the NorA efflux pump, 4g, 4h, and 4k interacted with key amino acid residues, namely, Ile240, Ala243, and Ala289, respectively, while 4i, 4j, and 4m interacted with Val44, Phe47, and Ile240, respectively (Fig. 3). Therefore, the aromatic moieties of 4g–k and 4m played important roles in the binding of these compounds to the hydrophobic core of the NorA efflux pump.
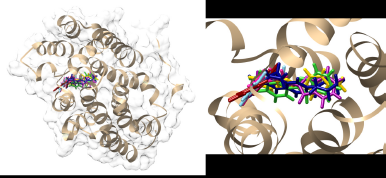 | Figure 2. Comparison of the binding positions of 4g (pink), 4h (light blue), 4i (purple), 4j (green), 4k (red), 4m (yellow), and ciprofloxacin (deep blue) in the cavity of the S. aureus NorA model prepared using the EmrD efflux pump (PDB ID: 2GFP) as a template. [Click here to view] |
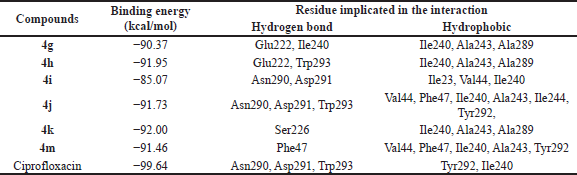 | Table 4. Binding energy (kcal/mol) and residues involved in the interaction of 4g–k, 4m, and ciprofloxacin to the NorA efflux pump model. [Click here to view] |
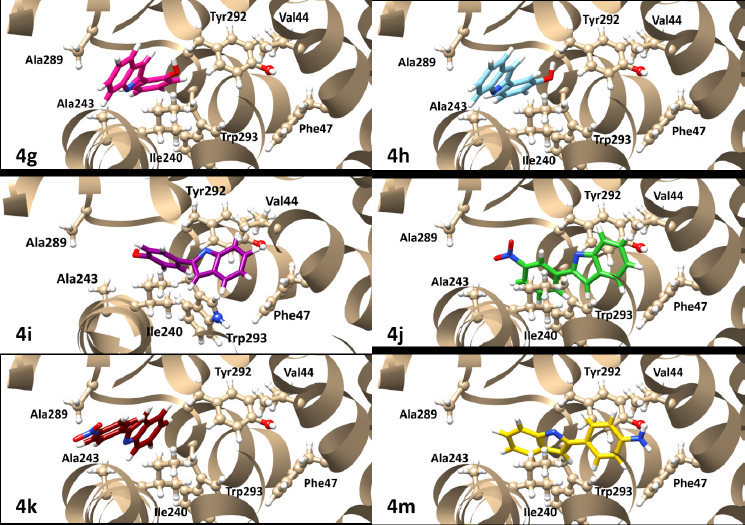 | Figure 3. Binding positions of 4g–k and 4m in the cavity of the NorA efflux pump model. [Click here to view] |
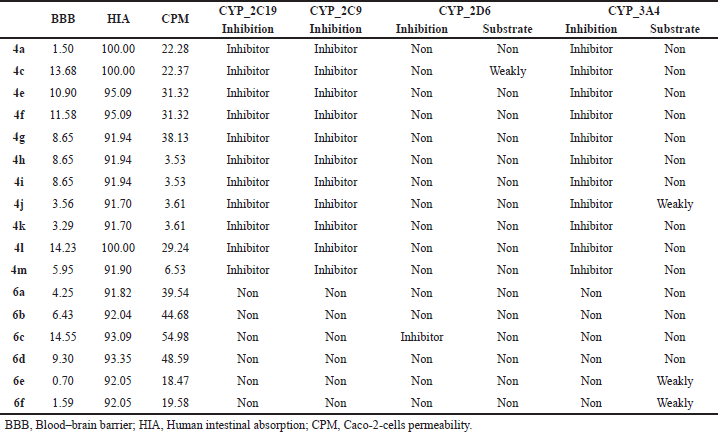 | Table 5. ADMET properties predicted for compounds 4a–m and 6a–f. [Click here to view] |
Prediction of ADMET by computational analysis
The ADMET properties of the synthesized compounds were presented in Table 5. This result suggested that most of the compounds were through to cross BBB permeation. For in vitro Caco-2 cell permeability, most of the compounds except 4h–k and 4m have high permeability and are easy to absorb. All of the compounds showed % HIA in the range of 91.70%–100.00% which were considered to be very well absorbed in the gastrointestinal tract. The toxicity predictions were considered of the cytochrome P450s (CYP), an important enzyme system for drug metabolism that influences clearance rates, toxicity, and interactions with coadministered drugs. All compounds were predicted to be not substrates and not inhibitors for four major CYP isoforms, except for compounds 4a–m, which were predicted to be CYP2C19, CYP2C9, and CYP3A4 inhibitors. This suggested that these compounds may be metabolized in the liver which may have hepatotoxicity.
CONCLUSION
2-Arylindole derivatives were synthesized, and their antibacterial activity and synergistic effects on multidrug-resistant S. aureus were evaluated. The results showed that 4j combined with tetracycline showed a potent antibacterial activity against S. typhi and exhibited a synergistic effect against MRSA Sp6 strain. Furthermore, 4 m displayed the most potent antibacterial activity against B. subtilis, whereas 4j was the most potent against S. typhi. All the synthesized compounds improved the sensitivity of MRSA Sp6 to tetracycline. Molecular docking studies on 4g, 4h, and 4k revealed that interaction with key amino acids occurred at the active site of the NorA efflux pump. However, the toxicity study by ADMET was predicted as hepatotoxicity. Therefore, the 2-arylindole derivatives could be considered as an interesting scaffold and further development of the 2-arylindole derivative with no toxicity might be the leading compounds that could potentiate the activity of commercially available drugs against multidrug-resistant bacteria.
ACKNOWLEDGEMENTS
We gratefully acknowledge the Department of Chemistry and the Department of Microbiology, Faculty of Science, Silpakorn University, for the financial support and antibacterial and synergistic effect assay. We also thank the Chulabhorn Research Institute for the measurements of the HR-ESI mass spectroscopy investigation.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit the paper to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors requirements/guidelines.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
FUNDING
There is no funding to report.
ETHICAL APPROVAL
This study does not involve experiments on animals or human subjects.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Ackermann L, Barfuesser S, Potukuchia H K. Copper-catalyzed N-arylation/hydroamin(d)ation domino synthesis of indoles and its application to the preparation of a Chek1/KDR kinase inhibitor pharmacophore. Adv Synth Catal, 2009; 351:1064–72; http://doi.org/10.1002/adsc.200900004
Bremner JB, Ambrus JI, Kelso MJ, Ball AR, Casadei G, Lewis K. Structure–activity relationships of 2-aryl-1H-indole inhibitors of the NorA efflux pump in Staphylococcus aureus. Bioorg Med Chem Lett, 2008; 18:4294–7; http://doi.org/10.1016/j.bmcl.2008.06.093
Brvar M, Perdih A, Renko M, Anderluh G, Turk D, Solmajer T. Structure-based discovery of substituted 4,5’-bithiazoles as novel DNA gyrase inhibitors. J Med Chem, 2012; 55:6413–26; http://doi.org/10.1021/jm300395d
Cho C, Park I, Suh S, Lim B. Aryl hydrazide beyond as surrogate of aryl hydrazine in the Fischer indolization: the synthesis of N-Cbz-indoles, N-Cbz-carbazoles, and N,N’-Bis-Cbz-pyrrolo[2,3-f]indoles. Org Lett, 2009; 11:5454–6; http://doi.org/10.1021/ol902250x
CLSI. Performance standards for antimicrobial susceptibility testing: 24th informational supplement, CLSI document M100-S24. Clinical and Laboratory Standards Institute, Wayne, PA, 2014.
Cossío FP, Vara Y, Aldaba E, Arrieta A, Pizarro JL, Arriortua MI. Regiochemistry of the microwave-assisted reaction between aromatic amines and α-bromoketones to yield substituted 1H-indoles. Org Biomol Chem, 2008; 6:1763–72; http://doi.org/10.1039/B719641E
Dai WM, Guo DS, Sun LP. Chemistry of aminophenols. Part 1: Remarkable additive effect on Sonogashira cross-coupling of 2-carboxamidoaryl triflates and application to novel synthesis of indoles. Tetrahedron Lett, 2001; 42:5275–8; http://doi.org/10.1016/S0040-4039(01)00965-0
Deschenes RJ, Lin H, Ault AD, Fassler JS. Antifungal properties and target evaluation of three putative bacterial histidine kinase inhibitors. Antimicrob Agents Chemother, 1999; 43(7):1700–3. http://doi.org/10.1128/AAC.43.7.1700.
Ezquerra J, Pedregal C, Lamas C, Barluenga J, Pérez M, García-Martín MA, González JM. Efficient reagents for the synthesis of 5-, 7-, and 5,7-substituted indoles starting from aromatic amines: scope and limitations. J Org Chem, 1996; 61:5804–12; http://doi.org/10.1021/jo952119+
Fuher W, Gschwend HW. Ortho functionalization of aromatic amines: ortho lithiation of N-pivaloylanilines. J Org Chem, 1979; 44(7):1133–6; http://doi.org/10.1021/jo01321a023
Heck RF, Terpko MO. Rearrangement in the palladium-catalyzed cyclization of alpha.-substituted N-acryloyl-o-bromoanilines. J Chem Soc, 1979; 101(18):5281–3; http://doi.org/10.1021/ja00512a028
Hibino S, Chosi T. Simple indole alkaloids and those with a nonrearranged monoterpenoid unit. Nat Prod Rep, 2002; 19:148–80; http://doi.org/10.1039/B007740M
Hong BC, Jiang Y, Chang Y, Lee S. Synthesis and cytotoxicity studies of cyclohepta[β]indoles, benzo[6,7]cyclohepta[1,2-β]indoles, indeno[1,2-β]indoles, and benzo[α]carbazoles. J Chin Chem Soc, 2006; 53:647–62; http://doi.org/10.1002/jccs.200600086
Hughes DL. Progress in the Fischer indole reaction. N J Chem, 1993; 6:607–32; http://doi.org/10.1080/00304949309356257
Imanishi T, Miyashita K, Kondoh K, Tsuchiya K, Miyabe H. Novel indole-ring formation by thermolysis of 2-(N-acylamino)-benzylphosphonium salts. Effective synthesis of 2-trifluoromethylindoles. J Chem Soc Perkin Trans, 1996; 1:1261–8; http://doi.org/10.1039/P19960001261
Kim BM, Kwon J, Chung J, Byun S. Efficient synthesis of indole derivatives via tandem cyclization catalyzed by magnetically recoverable palladium/magnetite (Pd-Fe3O4) nanocrystals. Asian J Org Chem, 2016; 5:470––6; http://doi.org/10.1002/ajoc.201500536
Kuduk SD, Chang RK, Wai JM, Di Marco CN, Cofre V, DiPardo RM, Cook SP, Cato MJ, Jovanovska A, Urban MO, Leitl M, Spencer RH, Kane SA, Hartman GD, Bilodeau MT. Amidine derived inhibitors of acid-sensing ion channel-3 (ASIC3). Bioorg Med Chem Lett, 2009; 19(15):4059–63; http://doi.org/10.1016/j.bmcl.2009.03.029
Lal S, Snape TJ. 2-arylindoles: a privileged molecular scaffold with potent, broad-ranging pharmacological activity. Curr Med Chem, 2012; 19:4828–37; http://doi.org/10.2174/092986712803341449
Larock RC, Yum EK Synthesis of indoles via palladium-catalyzed heteroannulation of internal alkynes. J Am Chem Soc, 1991; 113(17):6689–90; http://doi.org/10.1021/ja00017a059
Markham PN, Westhaus E, Klyachko K, Johnson EM, Neyfakh AA. Multiple novel inhibitors of the nora multidrug transporter of Staphylococcus aureus. Antimicrob Agents Chemother, 1999; 43(10):2404–8; http://doi.org/10.1128/aac.43.10.2404
Naik M, Ghorpade S, Jena LK, Gorai G, Narayan A, Guptha S, Sharma S, Dinesh N, Kaur P, Nandishaiah R, Bhat J, Balakrishnan G, Humnabadkar V, Ramachandran V, Naviri L K, Khadtare P, Panda M, Iyer PS, Chatterji M. 2-phenylindole and arylsulphonamide: novel scaffolds bactericidal against Mycobacterium tuberculosis. ACS Med Chem Lett, 2014; 5(9):1005–9; http://doi.org/10.1021/ml5001933
Nakazaki M, Yamamoto K. Direct synthesis of indole by the fischer indole synthesis. J Org Chem, 1976; 41(10):1877; http://doi.org/10.1021/jo00872a045
National Committee for Clinical Laboratory Standards. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard. NCCLS Document M7-A4, Villanova, PA, 1997.
Reygaert WC. An overview of the antimicrobial resistance mechanisms of bacteria. Microbiology, 2018; 4(3):482–501; http://doi.org/10.3934/microbiol.2018.3.482
Rudisill DE, Stille JK. Palladium-catalyzed synthesis of 2-substituted indoles. J Org Chem, 1989; 54:5856–66; http://doi.org/10.1021/jo00286a014
Samosorn S, Bremner JB, Ball A, Lewis K. Synthesis of functionalised 2-aryl-5-nitro-1H-indoles and their activity as bacterial NorA efflux pump inhibitors. Bioorg Med Chem, 2006; 14 (3):857–65; http://doi.org/10.1016/j.bmc.2005.09.019
Shi J, Yang SD, Sun CL, Fang Z, Li BJ, Li YZ. Palladium-catalyzed arylation of (Het)arenes with arylboronic acids. Synfacts, 2008; 5:0514; http://doi.org/10.1055/s-2008-1072518
Snape T, Prabhu S, Akbar Z, Harris F, Karakoula K, Lea R, Rowther F, Warr T. Preliminary biological evaluation and mechanism of action studies of selected 2-arylindoles against glioblastoma. Bioorg Med Chem, 2013; 21:1918–24; http://doi.org/org/10.1016/j.bmc.2013.01.032
Taechowisan T, Mungchukeatsakul N, Phutdhawong WS. Antimicrobial resistance pattern of Staphylococcus aureus strains isolates from clinical and hospital environment specimens and their correleation with PCR-based approaches. Res J Microbiol, 2018; 13:100–18; http://doi.org/10.3923/jm.2018.100.118
Tsuchimoto T, Matsubayashi H, Kaneko M, Shirakawa E, Kawakami Y. Easy Access to aryl-and heteroarylannulated[a]carbazoles by the indium-catalyzed reaction of 2-arylindoles with propargyl ethers. Angew Chem Int Ed, 2005; 44:1336–40; http://doi.org/10.1002/anie.200462280
Taylor EC, Katz AH, Salgado-Zamora H, McKillop AM. Thallium in organic synthesis. 68. A convenient synthesis of 2-phenylindoles from anilides. Tetrahedron Lett, 1985; 26:5963–6; http://doi.org/10.1016/S0040-4039(00)98272-8
Westwell AD, Suzen S, Coban T, Karaaslan C, Kadri H. Synthesis and antioxidant properties of substituted 2-phenyl-1H-indoles. Bioorg Med Chem Lett, 2013; 23:2671–4; http://doi.org/org/10.1016/j.bmcl.2013.02.090
Williams JD, Nguyen ST, Gu S, Ding X, Butler MM, Tashjian TF, Opperman TJ, Panchal RG, Bavari S, Peet NP, Moir DT, Terry L Bowlin TL. Potent and broad-spectrum antibacterial activity of indole-based bisamidine antibiotics: synthesis and SAR of novel analogs of MBX 1066 and MBX 1090. Bioorg Med Chem, 2013; 21(24):7790–806; http://doi.org/10.1016/j.bmc.2013.10.014
Zárate SG, Morales P, ?widerek K, Bolanos-Garcia VM, Bastida A. A molecular modeling approach to identify novel inhibitors of the major facilitator superfamily of efflux pump transporters. Antibiotics, 2019; 8(1):25; http://doi.org/10.3390/antibiotics8010025