
INTRODUCTION
A drug is said to be therapeutically effective when its dosage form delivers the medicament at the desired site at an appropriate quantity and rate (Brahmankar and Jaiswal, 2011). A number of classes of drugs, such as therapeutic peptides and nucleic acids, are hydrolyzed by pro-degrading enzymes. Enzymes such as trypsin, chymotrypsin, and pepsin effectively degrade teriparatide in the human intestine (Werle et al., 2006). Bioenhancers are substances that are administered to increase the therapeutic efficacy of drugs without altering the properties of drugs. Increased use of bioenhancer drugs has improved the bioavailability of a number of drugs administered intravenously. The system works by modifying enzymes, stimulating the Gamma-Glutamyl Transferase (GGT) enzyme, improving the absorption of the gastrointestinal tract, and targeting drugs (Jain and Patil, 2015; Looareesuwan and Chulay et al., 1999). Relenza is a synthetic drug prescribed by physicians to treat patients with flu. When the viral enzyme is formed by a docking protein, the virus is released into the cell. Relenza competes with the protein binding and blocks the docking of the antibody. Due to the lack of viral release from infected cells, the virus spreads less via airborne means (Amtul et al., 2002). Bioenhancers come from a variety of botanical components, mostly herbs. A wide range of medicinal plants are considered to contain abundant bioenhancers, including piperine, naringin, gallic acid, quercetin and many more (Gupta et al., 2015; Patil et al., 2015; Wadhwa et al., 2014). Naringin occurs naturally in citrus fruits, particularly in grapefruit. It is found in foods such as apples, onions, and tea. It has therapeutic properties, including anti-aging, anti-ulcer, and anti-allergic properties (Alam et al., 2014). Oral ingestion of naringin 3.3 mg/kg and 10 mg/kg prior to the 3 mg/kg dose of paclitaxel significantly increases, Area under the curve (AUC) of paclitaxel. Naringin significantly increases the bioavailability of paclitaxel at a dose of 3.3 mg/kg. Other types of drugs, such as diltiazem, verapamil, saquinavir, and cyclosporine, may also be increased (Oladimeji et al., 2018). Naringin inhibits CYP-450 enzymes, particularly CYP-3A4. CYP3A4 enzymes located in the small and large intestines and the liver metabolize a large number of drugs to a great extent (Burkina et al., 2016). The research topic intends to develop and evaluate the bilayer tablet of atorvastatin calcium and bioenhancer naringin for better absorption of atorvastatin calcium.
MATERIALS AND METHODS
Atorvastatin calcium (ATC) was received as a gift sample from Reltsen Health Care, Pondicherry. Naringin (NG) was purchased from Sigma Aldrich, crospovidone from ACS Chemicals. Polyvinylpyrrolidone (PVP) K-30, croscarmellose sodium, and microcrystalline cellulose 102 were purchased from Yarrow Chem Products. Talc, lactose monohydrate, and starch were purchased from NICE Chemicals. Magnesium stearate was purchased from LOBA Chemie and Neelilake Tartrazine Lake was received as a gift sample from Neelikon Food Dyes and Chemicals Ltd.
Methods
In vivo bioavailability study
The concept of combining an enzyme inhibitor with a statin to improve the bioavailability of the statin was studied and confirmed in an in vivo study in Sprague-Dawley rats (Wu et al., 2016). Animal testing was carried out with prior approval of the Institutional Animal Ethics Committee (Approval No: KMCRET/PhD/28/2016-17). They were divided into 11 groups of six each. All the animals were housed and fed under proper conditions. Group 1 received 8.27 mg/kg and Group 2 received 1.03 mg/kg ATC. (equivalent to 80 and 10 mg of human ATC dose). Group 3 ATC and NG at doses of 1.03 and 0.5 mg/kg, respectively. Groups 4–7 were pre-treated with 0.5 mg/kg NG, followed by 1.03 mg/kg ATC in 15, 30, 60, and 120 minutes after treatment with NG. Groups 8–11 were given 1.0, 1.5, 2.0, and 2.5 mg/kg of NG, and then all four groups were treated with 1.03 mg/kg of ATC 15 minutes later. Blood samples were collected by retro-orbital puncture (Kumar et al., 2017) at different time intervals of 0.25, 0.5, 1, 3, 6, 12, and 24 hours. The filtered plasma was separated and analyzed for drug content by High-performance liquid chromatography (HPLC) using acetonitrile C18 column: distilled water (85:15) (Sangshetti et al., 2016) as a mobile phase at a flow rate of 1.0 ml/minute (Cao et al., 2006).
Formulation development
The bilayer tablets were prepared using the Ridhi® multi-station tablet press with a round punch of 6 mm (Badawy et al., 2019). Among the two layers, one layer was prepared by wet granulation (Qu et al., 2017) and the other by direct compression (Swamy et al., 2008). The tablets were tested using the prescribed official procedure for disintegration time, hardness, and compared to the standard marketed atorvastatin calcium tablets. Experiments were conducted in different phases. Phase I involved the development of atorvastatin calcium layer, Phase II involved the development of naringin layer, and Phase III involved the combination of atorvastatin calcium and naringin into a bilayer tablet (Yohannes et al., 2017). Design of experiments method (DoE) (Bhujbal and Darkunde, 2019), were carried out using Design-Expert® version 11 (Visser et al., 2015). The significance of the test was assessed using a one-way analysis of variance (ANOVA). The administration of 0.91 mg/kg of atorvastatin and 2 mg/kg of naringin resulted in human doses of 9 mg/kg and 19 mg/kg, respectively.
Phase I: development of atorvastatin calcium layer
Phase I study focused mainly on the development of the atorvastatin calcium (Akbar et al., 2017) layer and the optimization of the various factors responsible for the timely delivery of atorvastatin calcium (Desai et al., 2016). Based on recent findings in preclinical rat models, pre-treatment with naringin was most effective in increasing the absorption of atorvastatin calcium. This concept was used in the formulation as the atorvastatin calcium layer of the bilayer tablet would disintegrate within the Gastrointestinal tract (GIT) only after 15 minutes. Thus, in order to meet this target, the dissolution time of the atorvastatin calcium layer was calibrated by 15 minutes and the formulation was designed with this in mind. Based on the prediction of this study, a two-way factorial design with two factors and two responses was used (Sanchez et al., 2018). The concentration of disintegrants and binders was a key factor in the development of the calcium layer of atorvastatin. The response chosen was the time of the tablet’s disintegration and hardness. Based on the factors and responses mentioned above, a two-level factorial design model was selected in Design-Expert® as shown in Table 1. The formula for the preparation of the tablet is provided in Table 2. The tablets were prepared in a batch size of 250, and their quality was assessed. The results were fitted to the Design-Expert; the model proved to be statistically significant and the results were tested and confirmed by ANOVA (Ainurofiq et al., 2016). Various solutions were derived on the basis of the set constraints in order to achieve the target responses. The most desirable solution (Rahimi and Mahmoudi, 2017), i.e., 3.313 mg PVP K30 as a binder without any disintegrants, was selected for the confirmatory study.
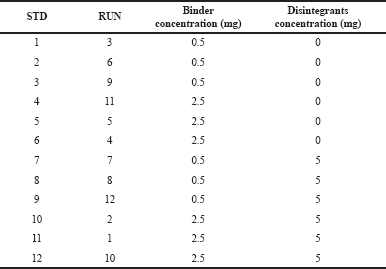 | Table 1. Two-level two-factorial design model for the development of atorvastatin calcium layer. [Click here to view] |
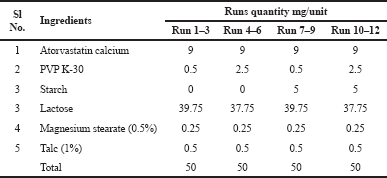 | Table 2. The unit formula for the development of atorvastatin calcium layer. [Click here to view] |
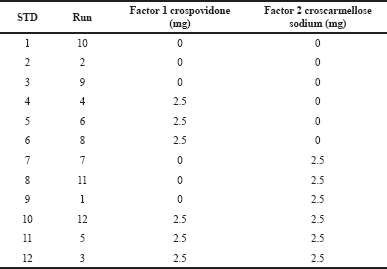 | Table 3. The two-way two-factorial design in the development of naringin layer. [Click here to view] |
Phase II: development of naringin layer for atorvastatin calcium and Naringin bi-layer tablet
The Phase II study was designed to develop naringin layers and investigate whether the desired responses could be achieved. Based on preclinical animal data, this layer was prepared as an immediate layer with a target disintegration time of 2 minutes to provide pre-treatment with naringin. Two different disintegrants (crospovidone and croscarmellose sodium) (Ibrahim and Abou El Ela, 2017) were tested at −1, +1 and measured using DoE. The two-way factorial design for the development of the naringin layer is shown in Table 3 and the unit formula in Table 4. The tablets were prepared in a batch size of 250 and tested. The results were well aligned with the Design-Expert® model and the significance of the results was statistically tested with ANOVA. By considering the set of constraints, multiple solutions have been chosen to meet the target. For the confirmatory study, a solution containing 2,987 mg of crospovidone alone was selected as a disintegrant.
Phase III-A: the development of bilayer tablets
In Phases I and II, the amount of binders and disintegrants required for the ATC and NG layers was identified. In Phase III, bilayer tablets were prepared using the best formulation identified in Phase I and Phase II studies. 3.313 mg PVP K-30 was used as binding agent for the ATC layer and 2.987 mg crospovidone was used as disintegrating agent for the NG layer. The amount of additional ingredients for the ATC and NG layers is the same as shown in Tables 2 and 4. The bilayer tablets were prepared and evaluated using a wet granulation method with a batch size of 250 numbers.
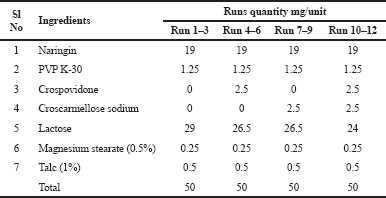 | Table 4. The unit formula for the development of naringin layer. [Click here to view] |
Phase III-B: optimizing the granular property by changing the diluent in bilayer tablets
Based on the results of Phase III-A, granules have been known to be hygroscopic. Phase III-B was conducted to reduce the hygroscopic nature of granules by converting the diluent from lactose to microcrystalline cellulose 102. The other components for the ATC and NG layers were the same as shown in Tables 2 and 4. The bilayer tablets were prepared and evaluated using a wet granulation method with a batch size of 250 numbers.
Phase III-C: optimizing the binder levels and method of tablet compression
Based on the results of Phase III-B to achieve the target disintegrating time for atorvastatin, Phase III-C increased the concentration of PVP K-30 to 5 mg/unit and the direct compression process for both layers was used. The quantity of additional ingredients was equal to the amount shown in Tables 2 and 4. The bilayer tablets were produced and tested with 250 units each.
Phase III-D: optimization of tablet compression method
Based on the Phase III-C results, the atorvastatin calcium layer was prepared using a wet granulation technique and the naringin layer was prepared using a direct compression method with a batch size of 250 tablets. The final formula can be found in Table 5. In order to differentiate the layers, Neellilake Tartrazine Lake (0.1%) was also incorporated into the naringin layer.
RESULTS AND DISCUSSION
In vivo bioavailability study
Animal studies conducted to determine the effect of naringin on the bioavailability of atorvastatin show that naringin has time- and dose-dependent actions to improve the bioavailability of atorvastatin. The study conducted to assess the effect of pre-treatment with naringin concludes that 15-minute pre-treatment with naringin increases the AUC of atorvastatin from 434.0 ± 8.77 to 624.2 ± 28.45 μg • hour/Ml. The study conducted to determine the optimal dose of naringin proved that a dose of 2 mg/kg 15 minutes before administration of atorvastatin increased the AUC of atorvastatin to 1,667.4 ± 29.68 μg • hour/Ml, which is higher than the AUC of atorvastatin alone at a dose of 8.27 mg/kg. This concluded that 0.91 mg/kg of atorvastatin and 15 minutes of 2 mg/kg of naringin produced an equivalent result of 8.27 mg/kg (= 80 mg human dose) of atorvastatin alone in the rat model.
Results of phase I study: development of atorvastatin calcium layer
Response to regular 2-level factor design studies has been conducted to optimize the two factors (binder concentration and disintegrant) for the development of the atorvastatin calcium layer. For both factors, 12 runs were carried out at two levels (−1, +1) with three replicates. Both factors were assessed on the basis of hardness and time of disintegration. The concentration of the binder and the hardness of the binder have been found to have a linear relationship. Disintegrants have also contributed to the increase in hardness but have not been proportionate. A lower concentration of binders and higher disintegrants resulted in less disintegration time (Steele et al., 2002). The model was tested for its relevance and it can be concluded that the two factors had a significant impact on reactions such as hardness (p-value = 0.0157) and disintegration time (p-value < 0.0001).
 | Table 5. The final optimised unit formula for the development of Atorvastatin calcium Naringin bilayer tablet. [Click here to view] |
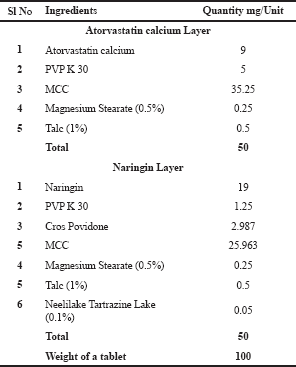
 | Figure 1. Contour plot explaining the effect of binder concentration and disintegrants on hardness of the atorvastatin calcium layer. [Click here to view] |
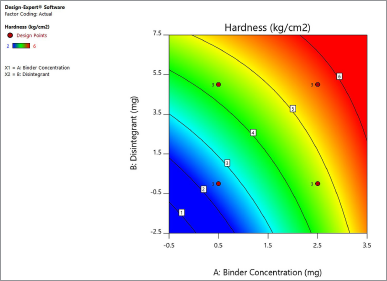
Based on the contour plot of the hardness shown in Figure 1, 3 mg of PVP K-30 is required for a 50 mg tablet to achieve a hardness of more than 5 kg/cm2. Binder concentrations ranging from 1 to 2.5 mg yield a hardness of 3–5 kg/cm2. Disintegrants have also contributed slightly to hardness, but have minimal effects and are less powerful. The contour plot for the effect of disintegrants and concentration of binders on disintegrating time is shown in Figure 2. As a result, a minimum of 2 mg disintegrants with a lower concentration of binders may result in a disintegration period of less than 5 minutes. It increases accordingly, however, with an increase in the concentration of binders. A minimum of 2.5 mg PVP K-30 without any disintegrants is required for a higher disintegration time of more than 15 minutes for a 50 mg tablet.
 | Figure 2. Contour plot explaining the effect of binder concentration and disintegrants on disintegration time of the atorvastatin calcium layer. [Click here to view] |
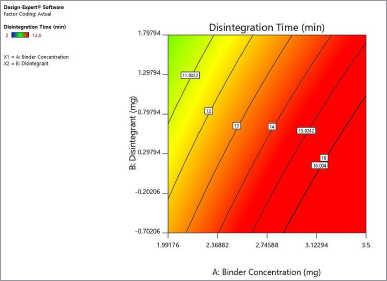
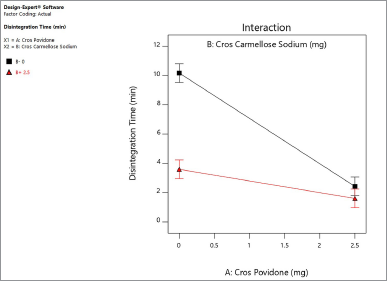 | Figure 3. Interactive effect of crospovidone and croscarmellose sodium on disintegration time of the naringin layer. [Click here to view] |
Based on the above observation of hardness and disintegration time, the formula was optimized under certain constraints to achieve the target of 15 minutes of disintegration time. Limits set for yield results are binding concentrations ranging from 2.5 to 3.5 mg, avoiding tablet disintegrants. Based on the set constraints, a 3.313 mg binder is required to achieve a 15-minute disintegration time result. The 3.313 mg binder tablets produced a disintegration time of 16 minutes and a hardness of 5.56 kg/cm2.
Results for phase II studies: development of naringin layer
Based on the raw data on the disintegration test, crospovidone’s contribution is much higher than croscarmellose sodium in decreasing disintegration time. Crospovidone alone reduced the disintegration time from 6.87 to 2.011 minutes at −1, +1, while croscarmellose sodium decreased the disintegration time from 6.2 to 2.5 minutes at −1, +1. Figure 3 shows the interaction between two disintegrants at the disintegrating time of the naringin layer. Both disintegrants reduced the disintegration time in proportion. Crospovidone produced the lowest disintegration time at a higher level of 2.5 mg. Crospovidone reduced the disintegration time from 10.16 to 2.42 minutes at its −1, +1 level in the absence of croscarmellose sodium, whereas at higher levels of croscarmellose sodium, crospovidone decreased the disintegration time from 3.59 to 1.6 minutes at −1, +1. Based on these data, the effect of crospovidone is found to be more significant than the effect of croscarmellose sodium (Tomar et al., 2018) since the time of disintegration of croscarmellose sodium alone decreased from 10.1 to 3.5 at −1, +1 levels.
Figure 4 shows the contour plot. The disintegration time of the naringin layer showed a minimum requirement of 2 mg for crospovidone and 2.3 mg for croscarmellose sodium to produce a disintegration time of less than 4 minutes. The combination of 2.5 mg crospovidone and 3 mg croscarmellose sodium may produce a disintegration time of less than 2 minutes. Figure 5 shows that the 3D surface area, croscarmellose sodium (−1), and crospovidone (+1) produced the least disintegrating time of 2.7 minutes compared to croscarmellose sodium (+1) and crospovidone (−1) which produced a disintegration time of 3.37 minutes (Figure 6). A combination of croscarmellose sodium and crospovidone at +1, +1 level is shown in Figure 7. The disintegration time is 1.3 minutes. The combination of these two superdisintegrants can result in a time of targeted disintegration. However, the total consumption of superdisintegrants is 5 mg, which is above the acceptable limit. Thus, the combination of these two superdisintegrants at the highest concentration cannot proceed. The target can be achieved by crospovidone 3 mg alone. In the case of croscarmellose sodium alone, the target minimum of 4 mg is required. As a result, the target disintegration time with minimum disintegrants was optimized for crospovidone. The amount of crospovidone required to produce the desired effect is determined under certain constraints by the Design-Expert and is given as 2,987 mg, which will produce a disintegration time of 0,913 minutes, with a desirability of 1. This was tested in the confirmatory study and the results show that the batch had a disintegration time of 0.873 minutes, which was at a 95% low and high predictive interval of 0.163 and 2.60 minutes, respectively.
Results of phase III-A: development of bilayer tablets
In Phase III, bilayer tablets were prepared using the best formulation identified in Phases I and II. The values for the calcium layer of atorvastatin have been confirmed using different statistical parameters at α = 0.05 at a two-sided interval. Obtained disintegration means 16.53 ± 1.03 minutes in the 95% predicted interval at two levels and closer to the predicted mean 16.79 ± 1.01 minutes. The mean data for hardness was determined to be 6.46 ± 1 kg/cm2. This mean was higher than the predicted value of 5.5 ± 0.90 kg/cm2. The hardness increased more than the results of the previous experiment. This could be due to the multilayer of the tablet compared with a single layer in previous experiments (Yaakub et al., 2018).
In the case of the naringin layer, the predicted disintegration time was 1.22 ± 0.67 with a 95% predictive range of 0.16 and 2.60 at −1, +1. The mean disintegration time obtained was 1.783 ± 0.30 minutes, which was within the predictive interval, whereas it was higher than the predictive mean value obtained for the naringin layer when compressed as a 50 mg single-layer tablet for the optimization process (Phase II). In this study, both the naringin and atorvastatin calcium layers were compressed together as a single 100 mg tablet. As a result, the one-third effective surface area of the bilayer tablet for the naringin layer was reduced. The decrease in surface area increased the disintegration time of the naringin layer to 1.783 ± 0.30 minutes. The granules prepared were hygroscopic in nature, and compression was difficult.
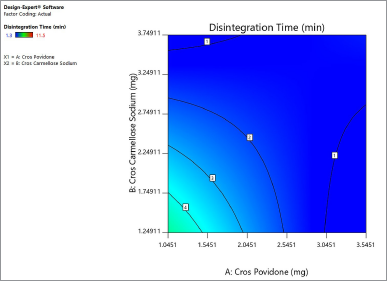 | Figure 4. Contour plot explaining the effect of superdisintegrants crospovidone and croscarmellose sodium on disintegration time of naringin layer. [Click here to view] |
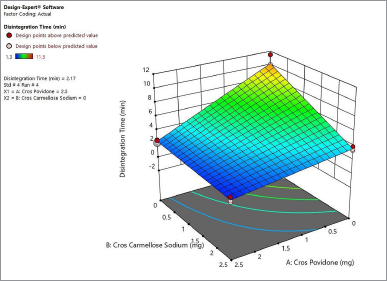 | Figure 5. 3 D surface area produces the least disintegration time of 2.7 minutes for naringin layer. [Click here to view] |
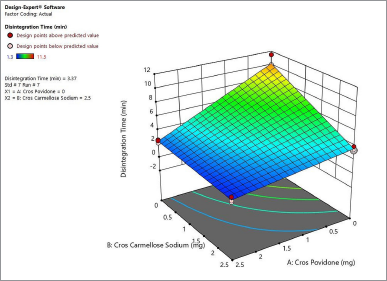 | Figure 6. 3-D surface area produces a disintegration time of 3.37 minutes for naringin layer. [Click here to view] |
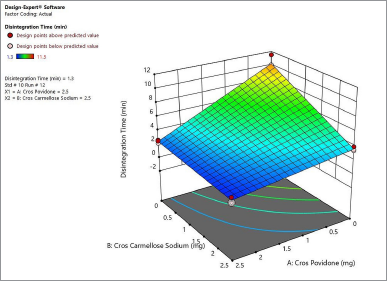 | Figure 7. 3-D surface area produces a disintegration time of 1.3 minutes for naringin layer. [Click here to view] |
Results of phase III-B: optimizing the granular property
Another set of experiments was conducted to improve the granulation properties by altering the diluents of lactose to microcrystalline cellulose 102 without changing any other parameters. Granules prepared with lactose have more hygroscopic properties under certain climatic conditions and are affected by the tablet process. That is why lactose was replaced by microcrystalline cellulose 102. This has a higher adsorbing capacity. The tablet was prepared according to the optimized process and formula. As per the point forecast, the predicted mean was 16.79 ±1.01 with 95% confidence interval values of 14.81 and 18.77 at−1, +1 and % tolerance intervals of 10.71 and 22.87 at −1, +1 at 0.05 and tolerances of 0.99. In response data, the hardness increased to 11.5 kg/cm2 and the disintegration time of the atorvastatin calcium layer decreased to 10.5 minutes. Confirmatory values at two-sided intervals were obtained using α = 0.05. The mean disintegration obtained was less than 95% confidence interval, predictive interval, and tolerance interval. The results are not within the limits. In the case of the naringin layer, the predicted mean is 1.22 minutes with a 95% CI of 0.16 and 2.6 at−1,+1 levels and 95% tolerance intervals of −2.6 and 5.6 at −1, +1 in the response data of the naringin layer; the disintegration time increased to 3.36 minutes. Even though it is within the acceptable time limit, it is beyond the target time point.
The results of this experiment paved the way for the following observation, the mean disintegration time of the atorvastatin calcium layer decreased and the naringin layer increased, while the hardness also increased to 11.5 kg/cm2. Based on these observations, we concluded that the use of microcrystalline cellulose 102 for wet granulation may increase the hardness of the tablet above the limit (Tomar et al., 2018), making it the most suitable for direct compression (Osamura et al., 2018). This may be the reason for the increase in disintegration time for the naringin layer. The disintegration time for atorvastatin calcium decreased. This may have happened due to the following reasons. The concentration of the binder is not sufficient to granulate with microcrystalline cellulose 102 (Pishnamazi et al., 2019). Microcrystalline cellulose 102 also has some disintegration properties (Osamura et al., 2018). The effective surface area of the atorvastatin calcium layer has increased following the disintegration of the naringin layer. Thus, to modify the results, the layer of atorvastatin calcium requires more binder quantity (Markl and Zeitler, 2017).
Results of phase III-C: optimizing the binder levels and method of tablet compression
In order to improve the disintegration properties of the tablet in such a way as to achieve a disintegration time of 15 minutes for the atorvastatin calcium layer and less than 2 minutes for naringin layer, this experiment was carried out by increasing the concentration of atorvastatin calcium to 5 mg and the preparation process as a direct compression method (Moghal et al., 2016). The mean disintegration time observed for the atorvastatin calcium layer was 10.5 ± 4.3 minutes, which is less than the 95% predicted interval. The standard deviation is also observed to be high (n = 3). The mean tablet hardness is 10.3 ± 0.85 kg/cm2 (n = 3). Even after the addition of an extra binder, the disintegration time did not increase, but the standard deviation increased. The lower disintegration time is assumed to have occurred due to the penetration of superdisintegrants present in the naringin layer into the atorvastatin calcium layer during compression.
In the case of the naringin layer, the mean disintegrating time was 1.833 ± 0.35 minutes, i.e., within 95% of the two-sided predicted interval with α = 0.05 and the mean hardness was also observed as 10.3 ± 0.854 kg/cm2. Based on the current experimental results, the disintegration time of the atorvastatin calcium layer has decreased and the naringin layer has reached the limit with good hardness. As a result, the formula and process for the preparation of the Naringin layer is finalized as 2.98 mg crospovidone for the 50 mg naringin layer to make 100 mg bilayer tablet by direct compression method.
Results of phase III-D: optimization of tablet compression method
In order to improve the disintegration time of the atorvastatin calcium layer, a further experiment was carried out by altering the process of preparation of the atorvastatin calcium layer as wet granulation and the naringin layer as previously optimized. Confirmation data show the tablet hardness as 9.33 ± 1.02 kg/cm2 and the disintegration time as 16.27 ± 1.0 minutes for the atorvastatin calcium layer and 1.5 ± 0.305 minutes for the naringin layer. Both responses are within the 95% confidence interval.
CONCLUSION
Atorvastatin calcium/naringin was prepared as a bilayer tablet to enhance the bioavailability of atorvastatin calcium. The experiments were designed using the Design-Expert® as per the DoE model. The effects of various factors (disintegrants and binder conc.) on atorvastatin calcium and the naringin layers have been optimized on the basis of two-level factorial designs. Based on the experimental confirmatory test, atorvastatin calcium/naringin bilayer tablets were prepared under the following conditions: the target disintegration time of 15 minutes for atorvastatin calcium layer (50 mg) was achieved by a wet granulation method using 5 mg PVP K-30 and microcrystalline cellulose pH 102 as binders and diluents. The formula for the atorvastatin calcium layer was optimized without disintegrants. The target disintegration time of 2 minutes for the naringin layer (50 mg) was achieved by direct compression of the tablet using 2.987 mg crospovidone, 1.25 mg PVP K-30, and microcrystalline cellulose 102 as disintegrants, binders, and diluents. The formula has been optimized and validated.
ACKNOWLEDGMENTS
We wholeheartedly acknowledge the support rendered by the management and staff of Nirmala College of Pharmacy, Muvattupuzha, Kerala, for the successful completion of the work.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
FUNDING
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
PUBLISHER’S NOTE
This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
REFERENCES
Ainurofiq A, Choiri S, Azhari MA, Siagian CR, Suryadi BB, Prihapsara F, Rohmani S. Improvement of meloxicam solubility using a β-cyclodextrin complex prepared via the kneading method and incorporated into an orally disintegrating tablet. Adv Pharm Bull, 2016; 6(3):399. CrossRef
Akbar S, Bhatta R, Rahman MA, Hossain M, Bhuiyan MS, Uddin MG. Development and characterization of atorvastatin calcium sustained release tablet using carbomer-974 and hypromellose-15000 cps. J Sci Res, 2017; 9(2):247–54. CrossRef
Alam MA, Subhan N, Rahman MM, Uddin SJ, Reza HM, Sarker SD. Effect of citrus flavonoids, Naringin and Naringenin, on metabolic syndrome and their mechanisms of action. Am Soc Nutr, 2014; 5(4):404–13. CrossRef
Amtul Z, Rahman AU, Siddiqui RA, Choudhary MI. Chemistry and mechanism of urease inhibition. Curr Med Chem, 2002; 9:1323–48. CrossRef
Badawy SIF, Narang AS, LaMarche KR, Subramanian GA, Varia SA. Mechanistic basis for the effects of process parameters on quality attributes in high shear wet granulation. Handb Pharm Wet Granulation, 2019; 1:89–118. CrossRef
Bhujbal SS, Darkunde SL. Analytical method development and optimization of sofosbuvir drug–a qbd approach. Int J Pharm Sci Res, 2019; 10(1):108–16.
Brahmankar DM, Jaiswal SB. 2011 Textbook of biopharmaceutics and pharmacokinetics. 3rd edition, New Delhi, India. Vallabh Prakashan 1-4.
Burkina V, Zlabek V, Halsne R, Ropstad E, Zamaratskaia G. In vitro effects of the citrus flavonoids diosmin, naringenin and naringin on the hepatic drug-metabolizing CYP3A enzyme in human, pig, mouse and fish. Biochem Pharmacol, 2016; 110:109–16. CrossRef
Cao X, Gibbs ST, Fang L, Miller HA, Landowski CP, Shin HC, Lennernas H, Zhong Y, Amidon GL, Yu LX, Sun D. Why is it challenging to predict intestinal drug absorption and oral bioavailability in human using rat model. Pharm Res, 2006; 23(8):1675–86. CrossRef
Desai PM, Liew CV, Heng PW. Review of disintegrants and the disintegration phenomena. J Pharm Sci, 2016; 105(9):2545–55. CrossRef
Gupta P, Authimoolam SP, Hilt JZ, Dziubla TD. Quercetin conjugated poly (β-amino esters) nanogels for the treatment of cellular oxidative stress. Acta Biomater, 2015; 27:194–204. CrossRef
Ibrahim MA, Abou El Ela AESF. Optimized furosemide taste masked orally disintegrating tablets. Saudi Pharm J, 2017; 25(7):1055–62. CrossRef
Jain G, Patil UK. Strategies for enhancement of bioavailability of medicinal agents with natural products. Int J Pharm Sci Res, 2015; 6(12):5315–24.
Kumar M, Dandapat S, Sinha MP, Kumar A, Raipat B. Different blood collection methods from rats: a review. Balneol Res J, 2017; 8(2):46–50. CrossRef
Looareesuwan S, Wilairatana P, Glanarongran R, Indravijit KA, Supeeranontha L, Chinnapha S, Scott TR, Chulay JD. Atovaquone and proguanil hydrochloride followed by primaquine for treatment of Plasmodium vivax malaria in Thailand. Trans R Soc Trop Med Hyg, 1999; 9(3):637–40. CrossRef
Markl D, Zeitler JA. A review of disintegration mechanisms and measurement techniques. Pharm Res, 2017; 34(5):890–917. CrossRef
Moghal MM, Mazumder SC, Lira D, Rouf AS. Fabrication and in vitro evaluation of allopurinol fast dissolving tablets using croscarmellose sodium, sodium starch glycolate and crospovidone as superdisintegrants. Dhaka Univ J Pharm Sci, 2016; 15(1):73–81. CrossRef
Oladimeji FA, Adegbola AJ, Onyeji CO. Appraisal of bioenhancers in improving oral bioavailability: applications to herbal medicinal products. J Pharm Res Int, 2018; 24(4):1–23. CrossRef
Osamura T, Takeuchi Y, Onodera R, Kitamura M, Takahashi Y, Tahara K, Takeuchi H. Formulation design of granules prepared by wet granulation method using a multi-functional single-punch tablet press to avoid tableting failures. Asian J Pharm Sci, 2018; 13(2):113–9. CrossRef
Patil VS, Dziubla TD, Kalika DS. Static and dynamic properties of biodegradable poly (antioxidant β-amino ester) networks based on incorporation of curcumin multiacrylate. Polymer, 2015; 75:88–96. CrossRef
Pishnamazi M, Casilagan S, Clancy C, Shirazian S, Iqbal J, Egan D, Edlin C, Croker DM, Walker GM, Collins MN. Microcrystalline cellulose, lactose and lignin blends: process mapping of dry granulation via roll compaction. Powder Technol, 2019; 341:38–50. CrossRef
Qu L, Stewart PJ, Hapgood KP, Lakio S, Morton DAV, Zhou Q. Single-step coprocessing of cohesive powder via mechanical dry coating for direct tablet compression. J Pharm Sci, 2017; 106(1):159–67. CrossRef
Rahimi M, Mahmoudi J. Studies on optimization of efficient parameters for removal of lead from aqueous solutions by natural zeolite as a low-cost adsorbent using response surface methodology. Adv Environ Technol, 2017; 2:99–108.
Sanchez SM, Sánchez PJ, Wan H. Work smarter, not harder: a tutorial on designing and conducting simulation experiments. Winter Simul Conf, 2018; 237–51. CrossRef
Sangshetti JN, Aqeel M, Zaheer Z, Ahmed RZ, Dehghana MHG, Gonjari I. Development and validation of RP-HPLC method for determination of Atorvastatin calcium and Nicotinic acid in combined tablet dosage form. J Saudi Chem Soc, 2016; 20:S328–33. CrossRef
Steele DF, Edge S, Staniforth JN, Chen A, Woodcock PM. Powder compaction properties of sodium starch glycolate disintegrants. Drug development and industrial pharmacy. 2002;28(8):989-99. CrossRef
Swamy P, Shahidulla S, Shirsand S, Hiremath S, Ali M. Orodispersible tablets of carbamazepine prepared by direct compression method using 3 2 full factorial design. Dhaka Univ J Pharm Sci, 2008; 7(1):1–5. CrossRef
Tomar M, Sinha AR, Singh AK. Typical physical attributes of microcrystalline cellulose dried by spray, spin flash and bulk drier and their resultant effects on tablet properties. Int J Pharm Sci Res, 2018; 9(4):1545–54.
Visser JC, Dohmen WM, Hinrichs WL, Breitkreutz J, Frijlink HW, Woerdenbag HJ. Quality by design approach for optimizing the formulation and physical properties of extemporaneously prepared orodispersible films. Int J Pharm, 2015; 485(1–2):70–6. CrossRef
Wadhwa S, Singhal S, Rawat S. Bioavailability enhancement by piperine: a review. Asian J Biomed Pharm Sci, 2014; 4(36):1–8.
Werle M, Samhaber A, Bernkop-Schnürch A. Degradation of teriparatide by gastro-intestinal proteolytic enzymes. J Drug Target, 2006; 14(3):109–15. CrossRef
Wu C, Chen HC, Luo YS, Chiang SY, Wu KY. Pharmacokinetics and bioavailability of oral single-dose maleic acid in biofluids of Sprague-Dawley rats. Drug Metab Pharmacokinet, 2016; 31(6):451–7. CrossRef
Yaakub NA, Anuar MS, Tahir SM. Compaction behaviour and mechanical strength of lactose–sodium starch glycolate and lactose-croscarmellose sodium binary tablets. IOP Conf Ser Mater Sci Eng 2018; 342:012026. CrossRef
Yohannes B, Gonzalez M, Abebe A, Sprockel O, Nikfar F, Kiang S, Cuitiñoa AM. Discrete particle modelling and micromechanical characterization of bilayer tablet compaction. Int J Pharm, 2017; 529(1–2):597–607. CrossRef