
INTRODUCTION
Diacerein (Diacetylrhein, DCN) is anthraquinone derivative of 9, 10- dihydro-4, 5 bis (acetyl)-9, 10-dioxo-2- anthracene carboxylic acid (Fig. 1). It possesses anti-inflammatory, anticatabolic, and pro-anabolic properties on cartilage and synovial membranes (Pavelka et al., 2016) and potentiates the production of proteoglycan and hyaluronic acid (Solignac, 2004), which are the structural components of cartilage. Additionally, DCN and its metabolite rhein block the production of cytokine (IL-1, IL-6, and IL-16 TNFa) (Deshmukh et al., 2010).
 | Figure 1. The structure of diacerein. [Click here to view] |
DCN is very sparingly soluble in water (0.01 mg/ml). The poor solubility and wettability of DCN pose problems in pharmaceutical formulation meant for oral or parenteral use, which result in variation in bioavailability (Maski et al., 2009). The solubility of ineffectively water-soluble drugs has been improved by numerous approaches, the solid dispersion technique the most widely used one (Dalvi et al., 2015). The oral absorption of poorly water-soluble drugs was enhanced by solid dispersion technique due to the increases in the dissolution rate by using water-soluble inert carriers (Bhirud and Phalak, 2016; Delahaye et al., 1997). The possible mechanisms for solubility enhancement by solid dispersion involve particle size reduction, surface area increasing, and increasing the wettability of the poorly soluble drug with surrounding hydrophilic carrier to improve dissolution rate (Patil et al., 2010).
Fast dissolving tablets (FDTs) have picked up much consideration as a perfect elective to routine tablets and capsules. FDTs can replace conventional tablets to allow for intraoral drug absorption and bypass gastrointestinal absorption (Seager, 1998). They are uncoated (Fu et al., 2004); they disintegrate rapidly releasing the active pharmaceutical ingredient which is dispersed or dissolved within saliva and absorbed within oral cavity, pharynx, or oesophagus. Thus, improving the bioavailability of drug over conventional tablets (Luca, 2002). They disintegrate and dissolve in the oral cavity in seconds without the requirement of water (Bradoo et al., 2001; Porter, 2001). Therefore, in this study, the poor solubility of diacerein was improved by solid dispersion strategy, then the formulated solid dispersion was formulated into FDTs for improvement of dissolution, absorption of DCN, and hence its bioavailability and improving patient compliance. The FDTs were prepared by direct compression strategy, utilizing different approaches, including the disintegrant addition, the combination of superdisintegration and sublimation approach. Pre- and post-compression assessments were carried for powder blend and tablets, respectively. The formulation with the lowest disintegration and wetting times and highest amount of DCN released after 15 minutes was selected for further investigations. Kinetics and stability studies were carried on the selected formulation.
MATERIALS AND METHODS
Materials
Diacerein powder kindly supplied from Global Napi Pharmaceuticals, Egypt. Polyethylene glycols 4000, WINLAB, laboratory chemical, UK. Polyvinylpyrolidone K25, FLUKA Chemika, Switzerland. Sorbitol, SIGMA ALDRICH, Germany. Potassium dihydrogen orthophosphate anhydrous and dipotassium hydrogen orthophosphate anhydrous, Acetone and Methyl alcohol, EL-Nasr pharm. Chem. Co., Cairo, Egypt. Crospovidone (CP) was kindly provided by Sigmatec Company, Egypt. Camphor, Menthol, Magnesium stearate, and anhydrous lactose were purchased from El-Nasr pharmaceutical chemicals Co., Egypt. Aspartame was obtained from Alpha Chemika, India. All other materials and solvents were of analytical grade.
Determination of ÊŽmax of DCN
Five milligrams of DCN powder was dissolved in 50 ml acetone to give a stock solution of 100 μg/ml. Two milliliters of this solution was placed in a volumetric flask (10 ml), then complete to the mark with distilled water, phosphate buffer of pH 7.4 and 6.8. The prepared solution was scanned (Double beam spectrophotometer, UV-1601 (Shimadzu Co., Japan) within the ultraviolet region 200–800 nm to decide the wave length of the maximum absorption in the used medium.
Construction of calibration curves of DCN in different media
Five milligrams mg of DCN was dissolved in 10 ml acetone. The volume was adjusted to 50 ml (with distilled water, phosphate buffer pH 7.4 or 6.8) to give stock solution of 100 μg/ml, from this stock solution, serial dilutions were prepared with 1:4 acetone:distilled water, phosphate buffer pH 7.4 or 6.8 to obtain concentrations between 10 and 60 μg/ml The absorbance of each solution was measured at ÊŽmax specific for each medium against a blank. Each experiment was carried out in triplicate and the absorbance was plotted versus the concentration to construct the standard calibration curve.
Formulation of DCN solid dispersion by Co-evaporation strategy
Different hydrophilic carriers (PEG4000, PVPk25, and Sorbitol) were employed for preparing DCN solid dispersions (co-evaporates). They were prepared by co-evaporation method in three ratios of drug: polymer (1:1, 1:2, and 1:3 w/w). The calculated amount of DCN and the polymer was dissolved in a minimum amount of acetone. The solvent was allowed to evaporate at room temperature. The obtained co-evaporates was kept in a desiccator for 48 hours at room temperature. The dried mass was pulverized and sieved through 250 μm sieve and collected for further investigation (Thenmozhi and Yoo, 2017).
Preparation of DCN physical mixture
The drug and the previously specified polymers or carriers was gently triturated in the same ratio using a glass mortar to prepare a physical mixture. The obtained physical mixture was kept in desiccators for 48 hours at room temperature, pulverized, and sieved as earlier described.
Drug content
Precisely weighed amount equivalent to 20 mg of DCN from solid dispersions and their comparing physical mixture was dissolved in acetone and phosphate buffer pH 7.4 employing a sonicator. The absorbance of each solution was measured spectrophotometrically at 345 nm, and then the corresponding concentration was calculated (Deshmukh et al., 2010).
In vitro dissolution study
Dissolution studies of Solid dispersions and their PMs were carried out utilizing Dissolution test apparatus, SR II, 6 flasks (paddle type) (Hanson Research Co., USA). A precisely weighed amount of the prepared systems identical to 20 mg DCN were weighed and sprinkled over the surface of the dissolution medium of 900 ml phosphate buffer pH 7.4 maintained at 37oC ±1 and 50 rpm, samples of 5 ml were withdrawn at time interval of 5, 10, 15, 20, 30, 45, 60, 90, 120, 150, and 180 minutes and replaced promptly with the same volume of phosphate buffer pH 7.4 and analyzed spectrophotometrically at 345 nm. The cumulative amount of medicate dissolved was calculated. The test was conducted in triplicate and the average was recorded (Borgmann et al., 2008; Maski et al., 2009).
Fourier Transform Infrared (FT-IR) spectroscopy
The systems that possess higher dissolution rate were subjected to a qualitative IR analysis. Samples of 5 mg of a drug, carriers, the prepared solid dispersion and their corresponding PMs were compressed into transparent disc individually under pressure using KBr powder and scanned from 4,000 to 400 cm−1 using Shimadzu SSP-CoA IR compression machine and Infrared spectrophotometer, IR-476 (Shimadzu Co., Japan) (Chaulang et al., 2009; Gupta and Singhvi, 2015).
Differential scanning calorimetric (DSC) studies
The system that gives best dissolution rate was also subjected to differential scanning calorimetric to examine any possible interaction between DCN and carrier used. Thermograms of the samples (DCN, polymer, solid dispersions and PMs respectively) were obtained by using a DSC DSC-60, Shimadzu, Kyoto, Japan). Samples comparable to 2 mg of the medicate were stacked into aluminum dish and the tops were pleated. The thermal behavior of samples was investigated under nitrogen with a heating rate of 10oC/minute and temperature ranges of 25–400oC (Gupta and Singhvi, 2015; Xie et al., 2008)
X-ray diffraction study (XRPD)
The system that gives best dissolution rate was further evaluated with X-ray diffractometer for characterization of the crystalline phases. The XRPD of the chosen samples was gotten utilizing Philips 1710 diffractometer (Germany). The powder diffraction patterns were achieved using scan mode of 2θ ranging from 4o to 60o at a rate of 0.5o/minute (Gupta and Singhvi, 2015).
Scanning electron microscope (SEM)
DCN powder, Sorbitol, DCN-Sorbitol solid dispersion and DCN-Sorbitol physical mixture were covered with a lean gold layer by sputter coater unit (SPI, sputter, USA). At that point, SEM photos were taken by a scanning electron microscope (Joel JSM 5400LV SEM, Japan) worked at an increasing speed voltage of 15 kV.
Assessment of pre-compression parameters of the powder blends
The angle of repose was determined using fixed funnel method to determine the frictional force between the drug particles (El-Shenawy et al., 2017). Loose bulk density (LBD) and tapped bulk density (LBD) (Elkhodairy et al., 2014) were also calculated to determine the powder flowability and percent compressibility by determining Hausner's ratio (Elkhodairy et al., 2014) and carr's index (Aulton, 2002), respectively, using the following equations:
Development of DCN FDTs
The FDTs were prepared by direct compression method utilizing two different approaches, including the superdisintegrant addition method, the combination of superdisintegrant and sublimation approach; FDTs were prepared each enclosing amount of DCN: Sorbitol (1:3) SDs equivalent to 50 mg of DCN, since it showed the best dissolution rate among all SDs formulations (99.7% after 45 minutes). CP and camphor were used as superdisintegrant and subliming agent, respectively. Lactose was used as a diluent in all formulations as presented in formulations from F1 to F7 in Table 1. The drug and excipients were weighed accurately and passed through 100# screen prior to mixing. The weighed amount of medicate and lactose were blended first together then the remaining excipients were added to them to load the drug into the surface of water-soluble carrier. All ingredients were transferred to mortar in geometrical order and co-grounded for 15 minutes. The obtained mixture was compressed into tablet utilizing single punch tablet machine (Royal artist, India) (Nayak and Gopalkumar, 2004). The compression force was adjusted to provide tablet hardness within the pharmacopeia range of FDTs (2–4 kg/cm2). For FDTs containing camphor as a subliming agent, the tablets were dried at 60oC in oven till constant weight was obtained (Gohel et al., 2004). Seven formulations were designed and F1 was considered as the control formula as this formula not contains any superdisintegrant (Table 1).
 | Table 1. The composition of DCN-FDTs. [Click here to view] |
Assessment of post-compression parameters
Weight and drug content uniformity
The weight variation and drug content uniformity tests for the prepared FDTs tablets were conducted according to El Maghraby et al. (2014).
In vitro disintegration time
The disintegration time of the prepared FDTs tablets was determined employing a modified disintegration method. One tablet was placed at the center of a petri dish containing 10 ml phosphate buffer (pH7.4) at 37 ± 0.5oC. The time in seconds taken by the tablet to disintegrate completely into fine particles was recorded (El-Shenawy et al., 2017)
Wetting time
Ten milliliter phosphate buffers (pH 7.4) were placed in a Petri dish possessing an internal diameter of 10 cm. A piece of tissue paper was folded two times and moistened by placing in the dish. One milliliter of eosin dye was loaded on the upper surface of the tissue paper. One tablet was located on the center of the tissue paper and the time in seconds required for the dye color to appear on the upper surface of the tablet was determined (Bhardwaj et al., 2010).
In vitro release study
In vitro dissolution of FDTs was carried out utilizing Dissolution test apparatus, SR II, 6 flasks (paddle type) (Hanson Research Co., USA). One tablet was placed in the dissolution medium of 900 ml phosphate buffer pH 7.4 maintained at 37oC ± 1 and 50 rpm. Five-milliliter aliquots were withdrawn at specific time intervals (2, 4, 6, 8, 10, 15, 20, 25, 30, 35, 40, 45, 50, and 60 minutes) and measured at 345 nm (Damodar et al., 2014). The cumulative amount of drug dissolved was calculated. The test was conducted in triplicate and the average was recorded (Borgmann et al., 2008; Maski et al., 2009). The cumulative % release of DCN were plotted versus time in minutes as shown in Figure 9.
Percentage friability, hardness, and thickness
Roche Friabilator was used to estimate the friability of DCN FDTs following the IP 1996 specifications (Elkhodairy et al., 2014). The hardness of 10 randomly selected tablets (from each formulation) was measured in kg/cm2 using Monsanto tablet hardness tester (Phama test, Germany) (Sahitya et al., 2014). Furthermore, digimatic micrometer caliber (Mitutoyo Corporation, Japan) was employed to measure the thickness in mm of the prepared tablets.
Stability study of DCN-FDTs
Stability study was conducted for Formulation (F7). The chosen formulation was stored at 30oC/75% RH and 40oC/75% RH in firmly closed bottles, wrapped inside aluminum foil. After 12 weeks, the stored tablets were examined for their drug content, weight variation, percentage friability, hardness, wetting time, in vitro disintegration time, and amount of DCN released at 15 minutes. The results were compared to those obtained from the freshly prepared FDTs (Sahitya et al., 2014).
Kinetic study
The kinetic parameters for the in vitro dissolution results were determined by using curve-fitting method to different kinetic models of zero, first, and second orders, as well as controlled diffusion model (Auda et al., 2016; Higuchi, 1963; Varshosaz et al., 2008). The model that produced the highest correlation was chosen to describe the release rate.
Zero-order release Mt = Mo + Kot
First-order Log C = Log Co K1t / 2.303
Second-order 1/C = 1/Co + K2t
Higuchi-Model Mt = Kt0.5
In which Mo and Mt are the amount of DCN released initially and at time t, respectively.
Co and C and the amount of DCN initially and at time t, respectively.
Ko, K1, and K2 are constants.
Statistical analysis
One-way analysis of variance single factor was used, p value of <0.05 will be considered statically significant (Elkhodairy et al., 2014).
RESULTS AND DISCUSSION
Wave length of maximum absorption
The wave length of maximum absorption of DCN in phosphate buffer pH 6.8 and 7.4 at 345 nm and in distilled water 344 nm.
Standard calibration curve of DCN
The calibration curves of DCN in distilled water, phosphate buffer pH 7.4 and pH 6.8 were constructed at ÊŽmax of 344 nm and 345 nm, respectively. The concentrations from 10 to 60 μg/ml were found to obey Beer's–Lambert law.
Drug content
The percentage drug content of the prepared SDs was in the acceptable range 93.65 and 97.85% w/w (El-Shenawy et al., 2017)
In vitro dissolution of DCN SDs and their corresponding physical mixtures
The in vitro dissolution of pure medicate was 52.39% after 45 minutes. Figure 2a and b shows the in vitro release of DCN with different PEG 4000 ratios (1:1, 1:2, and 1:3), The solid dispersion showed higher dissolution rate than comparing physical mixture and pure medicate. The medicate to polymer ratio (1:3) showed the highest percentage of dissolution after 45 minutes in both SDs and PMs (94.11% and 68.73%, respectively). The same result obtained with indomethacin (El-Badry et al., 2009) demonstrated that the ratio of indomethacin dissolved from PEG 400 solid dispersion was increased as the medicate to polymer ratio increased. Moreover, the dissolution rate of repaglinide-PEG 4000 SDs was increased by increasing the concentration of PEG4000 (Yang et al., 2016).
 | Figure 2. Dissolution profiles of (a) DCN SD and (b) PM with PEG4000 1:1, 1:2, and 1:3. [Click here to view] |
Figure 3a and b shows the in vitro release of DCN with different PVP K25 ratio (1:1, 1:2, and 1:3), The higher percentage of dissolution was found for SDs then PMs and pure drug. The ratio of 1:3 drug to polymer showed the best result after 45 minutes 90.59% and 66.47% for SDs and PMs, respectively. This data agreed with curcumin-PVP solid dispersion that showed an elevation in the dissolution rate of curcumin-PVP solid dispersion by increasing the concentration of PVP (Paradkar et al., 2004)
 | Figure 3. Dissolution profiles of (a) DCN SD and (b) PM with PVPK25 1:1, 1:2, and 1:3. [Click here to view] |
 | Figure 4. Dissolution profiles of (a) DCN SD and (b) PM with Sorbitol 1:1, 1:2, and 1:3. [Click here to view] |
Figure 4a and b showed the in vitro release of DCN with different Sorbitol ratios (1:1, 1:2, and 1:3), the higher dissolution rate was obtained SDs compared to PMs and pure drug. A 1:3 drug: polymer ratio had the best dissolution rate after 45 minutes (99.7% and 73.8% for SDs and PMs, respectively) (Garthe et al., 2013).
From the obtained results, it could be concluded that the dissolution rate of DCN from its solid dispersions and PMs were arranged in a descending order as follows: Sorbitol > PEG400 > PVPk25, this result may be due to the difference in the hydrophilic character of these polymers.
The higher dissolution rate of DCN from solid dispersions, in comparison to the immaculate drug, might be ascribed to improved wettability, dispersibility, local solubilization, medicate molecule size diminishment, or arrangement of high energy amorphous phase (El-Badry et al., 2009; Mohanty et al., 2010). The encompassing of the drug by the polymer diminishes the accumulation between the medicate particles which makes water to contact and wet the medicate particles and subsequently elevate the dissolution rate (Khadka et al., 2014).
 | Figure 5. IR spectra of (a) DCN, (b) Sorbitol, (c) DCN-Sorbitol solid dispersion, and (d) DCN–Sorbitol physical mixture. [Click here to view] |
FT-IR spectroscopy analysis
FTIR spectroscopy of pure DCN, Sorbitol, solid dispersion, and physical mixture are shown in Figure 5. The elucidation of the peaks showed that the characteristic peaks of DCN at 1,079.06 cm−1 of C-O stretching of the ester, 1,678 cm−1 of C=O stretching of the ketene, 1,693.47 cm−1 of C=O stretching of the COOH, 1,768.47 of C=O stretching of the ester. Also showed the peaks of Sorbitol at 3,381.55 cm−1 of O-H stretching broad, 2,932.18 cm−1 of C-H stretching aliphatic, 1,307.74 cm−1 of in plane O-H bend, 1,095.65 cm−1 of C-O stretching. All these peaks were well observed with no critical alteration in both physical mixture and solid dispersion which confirm the absence of interaction between the drug and Sorbitol.
Powder XRPD
P-XRD was employed to investigate the physical form of DCN in the prepared solid dispersions and their corresponding PMs. P-XRD of DCN, sorbitol, their 1:3 w/w solid dispersion with DCN and corresponding PMs are presented in Figure 6. DCN showed a crystalline structure as demonstrated by sharp and intense diffraction peaks observed at 2θ of 5o, 10o, 17o, 25o, and 27o as shown in Figure 6a. The diffraction pattern of Sorbitol showed many sharp peaks indicating its crystalline nature as appeared in Figure 6b. The characteristic peaks corresponding to DCN in the PMs and SDs disappears except those at 5o and 17o which had a much-reduced intensity (Fig. 6c and d). These observations indicate that most of the drug was in the amorphous state.
Differential scanning calorimetry (DSC)
The DSC thermogram of DCN displayed sharp endothermic peak at 253.42oC demonstrating its melting point and explaining the crystallinity of DCN as shown in Figure 7a. The DSC thermogram of Sorbitol showed a sharp endothermic peak at 102.45oC corresponding to its melting point and the crystalline state as appeared in Figure 7b. The DSC thermograms of DCN-Sorbitol SDs and PMs, respectively, showed the absence of endothermic peak of medicate as illustrates in Figure 7c and d, the absence of drug endothermic peak in both SDs and PMs may be due to the solubility of drug in Sorbitol in the pan during analysis or may be due to the amorphous state of drug. This result was checked by XRPD studies that prove that the drug was present in the amorphous state.
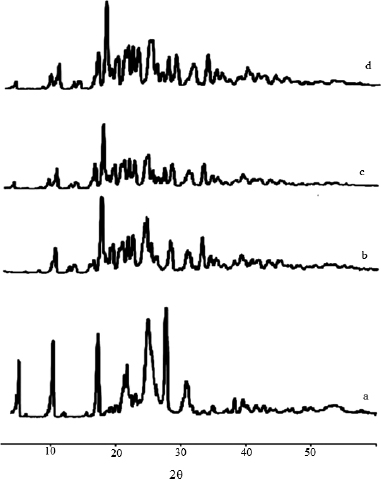 | Figure 6. XRPD pattern of (a) DCN, (b) Sorbitol, (c) DCN-Sorbitol solid dispersion, and (d) DCN–Sorbitol physical mixture. [Click here to view] |
 | Figure 7. DSC thermogram of (a) DCN, (b) Sorbitol, (c) DCN-Sorbitol solid dispersion, and (d) DCN–Sorbitol physical mixture. [Click here to view] |
 | Figure 8. SEM micrograph of (a) DCN, (b) Sorbitol, (c) DCN-Sorbitol solid dispersion, and (d) DCN–Sorbitol physical mixture. [Click here to view] |
Scanning electron microscopy (SEM)
The SEM image of DCN (Fig. 8a) demonstrates that it presents in crystal form and Sorbitol (Fig. 8b) as smooth surface ovoid particles. SDs (Fig. 8c) appeared relatively more unequal and rough surface with different length and surface, an increment in the surface area is available for DCN to attached itself to the surface of the solid dispersion, the presence of DCN is difficult to distinguish within the carrier as the morphology of the drug in the carrier is completely changed in the SDs due to the fine dispersion of medicate in the carrier. PMs (Fig. 8d) showed that the drug present in crystal form, it easy to distinguish the drug from polymer molecules. The contrast in the surface features between PMs and SDs is attributed to the arrangement of the molecules during preparation of the solid dispersion. SEM showed that Diacerein exists either in amorphous form or in very fine crystal form (Fig. 8c).
Characterization of FDTs
Pre-compression parameters of the powder blends
All formulations showed adequate to good flow properties as exhibited by the values of angle of repose (21.53°–25.8°). Hausner’s ratios (1–1.26) demonstrating good flowability. Carr’s index showed values between 16.47 and 20.65 denoting a good flowability exhibited by these formulations (Table 2).
Post-compression characterization of DCN FDTs
The prepared FDTs weights were in the range 445.34–448.93 mg. Their drug content was in the range 91.15%–101.27%. The friability values were in the range of 0.58%–0.81% indicating good mechanical characteristics. The tablets hardness was between 4.71 and 5.18 kg/cm2 while the thickness was between 4.19 and 4.93 mm.
All formulations disintegrated in a time period between 54 and 128.67 seconds. The wetting time values were between 61.67 and 137.67 seconds. The disintegration and wetting time decrease by increasing the concentration of CP from 10% to 30% w/w as shown in F2 and F4, respectively. In formulation F7 containing both superdisintegrant (CP) and subliming agent (camphor), the disintegration and wetting time decrease compared to F4 without camphor. The decrease in the disintegration and wetting time in all formulations may be due to the presence of CP which swells rapidly in water and produce rapid disintegration. Moreover, the presence of camphor in the formulation prepared by the combination of sublimation and superdisintegrant, resulted in development of porous tablet which assist the diffusion of wetting medium (Table 2).
In vitro release of DCN from the prepared FDTs
The in vitro release rate after 15 minutes was used for comparison between the formulations according to Elkhodairy et al. (2014).
The in vitro release study of the prepared FDTs revealed that F7 released the highest amount of the drug (89.84 ± 1.45 %) at 15 minutes. On the other hand, control tablets (F1) showed the lowest amount of drug released at the same time period (59.84 ± 1.25 %) (Fig. 9 and Table 3).
The dissolution rate after 15 minutes of F1 (as control) and F2, F3, and F4 showed that adding an increasing concentration of CP (F2, F3, and F4) results in a significant increase in the dissolution rate as compared to F1 (control) (p = 0.0267). Formulations containing a combination of superdisintegrant CP and subliming agent (camphor) F5, F6, and F7 showed a significant increase in the dissolution rate after 15 minutes (p = 0.00097) as compared to F4 containing superdisintegrant only and control (F1).
CP acts by both wicking and swelling action (Swamy et al., 2016), Due to its high cross-linking density, CP swell rapidly in water without gelling. Its particles are found to be granular and highly porous which facilitate wicking of liquid into tablets and particles to generate rapid disintegration.
 | Figure 9. In vitro release profiles of DCN from the prepared FDTs formulations. [Click here to view] |
 | Table 2. Different properties of DCN fast dissolving tablets. [Click here to view] |
From Figure 9, it showed that the in vitro release of DCN from FDTs was higher than control tablets, due to the presence of superdisintegrant in the FDTs, which accelerates the disintegration of tablets, due to their ability to absorb large quantity of water when exposed to the aqueous environment (Moghal et al., 2016). By increasing CP concentration in the prepared formulations, the dissolution rate increased. This result agreed with the previous work on FDTs containing CP as superdisintegrant (Mettu and Veerareddy, 2013; Moghal et al., 2016; Parhi, 2014; Tanuwijaya, 2013).
The incorporation of camphor as a subliming agent together with superdisintegrant result in a decrease in the disintegration time and faster dissolution rate, due to the porous structure produced by sublimation of the camphor which accelerates the rate of medium penetration, thus fasten the action of superdisintegrant (Elkhodairy et al., 2014; Furtado et al., 2008; Sammour et al., 2006).
The result also showed that by increasing camphor concentration from 3.75% (F5) to 11.25% (F7), the disintegration time decreased (54 seconds) and dissolution rate increased (89.84% after 15 minutes) as shown in Table 2 and Figure 9, respectively (El Rasoul and Shazly, 2017; Gohel et al., 2004; Sammour et al., 2006). This data in good agreement with Furtado et al. (2008) that proved that the disintegration time of Famotidine orodispersible decreased from 22 to 14 seconds by increasing camphor concentration from 10% to 20%.
Kinetic study
The kinetic studies of the in vitro release studies showed that F7 (composed of 120 mg CP, 45 mg camphor, 200 mg SD containing 50 mg DCN, 7.5 mg aspartame, 2.5 mg menthol, 2.5 mg Magnesium stearate, and 22.5 mg lactose) followed first-order kinetics based on the calculated correlation coefficient (Table 4).
Stability study
The results of the stability study showed that there is no significant difference between the recent and the stored F7 (composed of 120 mg CP, 45 mg camphor, 200 mg SD containing 50 mg DCN, 7.5 mg aspartame, 2.5 mg menthol, 2.5 mg Magnesium stearate, and 22.5 mg lactose) in the drug content, weight variation, percentage friability, hardness, wetting time, in vitro disintegration time, and amount of DCN released at 15 minutes after storage for 12 weeks at 30oC/75% RH and 40oC/75% RH as shown in Table 5.
 | Table 3. Dissolution parameters of DCN-FDTs. [Click here to view] |
 | Table 4. The calculated correlation coefficient for the in-vitro release of F7 employing different kinetic order or systems. [Click here to view] |
 | Table 5. Characterization properties of F7 after storage under different conditions for 12 weeks compared with the recently prepared tablets. [Click here to view] |
CONCLUSION
Diacerein solid dispersions were prepared using PEG4000, PVP K25 and Sorbitol as water-soluble carriers; Comprehensive characterization of SDs with SEM, XRD, and DSC studied indicated that Diacerein existed in an amorphous state or in the very fine crystalline state. FTIR studies showed that there were no drug carrier interactions. SDs have better dissolution profile as compared to PMs and pure drug, The DCN-Sorbitol solid dispersion has shown comparatively better physicochemical characteristic among all SDs. SD with Sorbitol was selected for the preparation of fast dissolving tablet with subsequent rapid dissolution. The obtained data demonstrated that the amount of both CP and camphor significantly influence the disintegration time, and hence dissolution profile of drug from FDTs. The use of superdisintegrant in addition to subliming agent in the formulation results in tablets with good properties. Therefore, F7 (composed of 120 mg CP, 45 mg camphor, 200 mg SD containing 50 mg DCN, 7.5 mg aspartame, 2.5 mg menthol, 2.5 mg Magnesium stearate and 22.5 mg lactose) was selected as an optimized FDTs formula as it showed the shortest disintegration time (54 seconds) and the highest dissolution rate. Storage of F7 at 30oC/75% RH and 40oC/75% RH for 12 weeks showed no significant difference between the recent and the stored one in the drug content, weight variation, percentage friability, hardness, wetting time, in vitro disintegration time, and amount of DCN released at 15 minutes, demonstrating a good stability of the this formulation.
ACKNOWLEDGMENTS
The authors are grateful to Faculty of Pharmacy Zagazig University for helpful facilities during this work.
CONFLICT OF INTEREST
The authors declared that they have no conflicts of interest.
REFERENCES
Auda SH, Fathalla D, Fetih G, El-Badry M, Shakeel F. Niosomes as transdermal drug delivery system for celecoxib: in vitro and in vivo studies. Polym Bull, 2016; 73(5):1229–45. CrossRef
Aulton ME. Pharmaceutics: the science of dosage form design: powders and granules. 2nd edition, Churchill Livingstone, Spain, pp 360, 2002.
Bhardwaj V, Bansal M, Sharma P. Formulation and evaluation of fast dissolving tablets of amlodipine besylate using different super disintegrants and camphor as sublimating agent. Am Euras J Sci Res, 2010; 5(4):264–9.
Bhirud YD, Phalak HM. Advances in solid dispersion technology and its applications in the development of solid dosage forms. J Drug Deliv Ther, 2016; 6(6):40–7. CrossRef
Borgmann S, Parcianello L, Arend M, Bajerski L, Cardoso S. Development and validation of a dissolution method with spectrophotometric analysis for diacerhein capsules. Sci Pharm, 2008; 76(3):541–54. CrossRef
Bradoo R, Shahani S, Deewan B, Sudarshan S. Fast dissolving drug delivery system. JAMA, 2001; 4:27–31.
Chaulang G, Patel P, Hardikar S, Kelkar M, Bhosale A, Bhise S. Formulation and evaluation of solid dispersions of furosemide in sodium starch glycolate. Trop J Pharm Res, 2009; 8(1):43–51. CrossRef
Dalvi P, Gerange A, Ingale P. Solid dispersion: strategy to enhance solubility. JDDT, 2015; 5(2):20–8. CrossRef
Damodar R, Movva B, Mallikarjun P, Pasumarthy C, Kona N, Varsha P. Formulation and evaluation of fast dissolving tablets of diclofenac sodium by novel hole technology. J Mol Pharm Org Process Res, 2014; 2:1–6. CrossRef
Delahaye N, Duclos R, Saiter J, Varnier S. Characterization of solid dispersion phase transitions using a new optical thermal analyzer. Drug Dev Ind Pharm, 1997; 23(3):293–303. CrossRef
Deshmukh D, Gaikwad P, Bankar V, Pawar S. Dissolution enhancement of poorly water soluble diacerein by solid dispersion technique. J Pharm Sci Res, 2010; 2(11):734–9.
El Maghraby GM, Elsergany RN. Fast disintegrating tablets of nisoldipine for intra-oral administration. Pharm Dev Technol, 2014; 19(6):641–50. CrossRef
El Rasoul SA, Shazly GA. Propafenone hcl fast dissolving tablets containing subliming agent prepared by direct compression method. Saudi Pharm J, 2017; 25(7):1086–92. CrossRef
El-Badry M, Fetih G, Fathy M. Improvement of solubility and dissolution rate of indomethacin by solid dispersions in gelucire 50/13 and peg4000. Saudi pharm J, 2009; 17(3):217–25. CrossRef
Elkhodairy KA, Hassan MA, Afifi SA. Formulation and optimization of orodispersible tablets of flutamide. Saudi Pharm J, 2014; 22(1):53–61. CrossRef
El-Shenawy AA, Ahmed MM, Mansour HF, El Rasoul SA. Torsemide fast dissolving tablets: development, optimization using box–bhenken design and response surface methodology, in vitro characterization, and pharmacokinetic assessment. AAPS PharmSciTech, 2017; 18(6):2168–79. CrossRef
Fu Y, Yang S, Jeong SH, Kimura S, Park KJ. Orally fast disintegrating tablets: Developments, technologies, taste-masking and clinical studies. Crit Rev Ther Drug, 2004; 21(6):433–75. CrossRef
Furtado S, Deveswaran R, Bharath S, Basavaraj B, Abraham S, Madhavan V. Development and characterization of orodispersible tablets of famotidine containing a subliming agent. Trop J Pharm Res, 2008; 7(4):1185–9. CrossRef
Garthe OM, Kothawade PS, Mahajan VR. Solubility enhancement of diacerein by solid dispersion technique. Int J Pharm Res Allied Sci, 2013; 2(2):47–55.
Gohel M, Patel M, Amin A, Agrawal R, Dave R, Bariya N. Formulation design and optimization of mouth dissolve tablets of nimesulide using vacuum drying technique. AAPs PharmSciTech, 2004; 5(3):10–5. CrossRef
Gupta K, Singhvi I. Formulation and characterization of mouth dissolving tablets of ezetimibe by frosta technique using ezetimibe: hydroxypropyl-β-cyclodextrin solid dispersion. Int J Pharm Erud, 2015; 5:18–28.
Higuchi T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci, 1963; 52(12):1145–9. CrossRef
Khadka P, Ro J, Kim H, Kim I, Kim JT, Kim H, Cho JM, Yun G, Lee J. Pharmaceutical particle technologies: an approach to improve drug solubility, dissolution and bioavailability. Asian J pharm Sci, 2014; 9(6):304–16. CrossRef
Luca D. Fast melting tablets development and technologies. Pharm Tech, 2002; 2:42–4.
Maski N, Kumaran A, Girhepunje K, Ghode P, Randive S, Pal R. Studies on the preparation, characterization and solubility of β-cyclodextrin-diacerein inclusion complexes. Int J Pharm Pharm Sci, 2009; 1(2):121–35.
Mettu SR, Veerareddy PR. Formulation, evaluation and pharmacokinetics of flurbiprofen fast dissolving tablets. Br J Pharm Res, 2013; 3(4):617–31. CrossRef
Moghal MM, Mazumder SC, Lira DN, Rouf ASS. Fabrication and in vitro evaluation of allopurinol fast dissolving tablets using croscarmellose sodium, sodium starch glycolate and crospovidone as superdisintegrants. Dhaka Univ J Pharm Sci, 2016; 15(1):73–81. CrossRef
Mohanty S, Biswal S, Biswal S, Sahoo J, Mahapatra A, Murthy P. Enhancement of dissolution rate of glimepiride using solid dispersions with polyvinylpyrrolidone k 90. Indian J Pharm Educ Res, 2010; 44(1):71–7.
Nayak S, Gopalkumar P. Design and optimization of fast dissolving tablets for promethazine theoclate. Indian Drugs, 2004; 41:554–6.
Paradkar A, Ambike AA, Jadhav BK, Mahadik K. Characterization of curcumin–pvp solid dispersion obtained by spray drying. Int J Pharm, 2004; 271:281–6. CrossRef
Parhi RJ. Improvment of dissolution rate of indomethacin from fast dissolving tablets. Indonesian J Pharm, 2014; 25(3):189–98. CrossRef
Patil SB, Shete DK, Narade SB, Surve SS, Khan ZK, Bhise SB, Pore YV. Improvement in the dissolution profile of diacerein using a surfactant-based solid dispersion technique. Drug Discov Ther, 2010; 4(6):435–41.
Pavelka K, Bruyere O, Cooper C, Kanis JA, Leeb BF, Maheu E, Martel-Pelletier J, Monfort J, Pelletier JP, Rizzoli R. Diacerein: Benefits, risks and place in the management of osteoarthritis. An opinion-based report from the esceo. Drug Aging, 2016; 33(2):75–85. CrossRef
Porter SC. Novel drug delivery: Review of recent trends with oral solid dosage forms. Am Pharm Rev, 2001; 4:28–36.
Sahitya G, Krishnamoorthy B, Muthukumaran M, Kishore G. Formulation and evaluation of fast dissolving tablets using solid dispersion of clopidogrel bisulphate. Int J Adv Pharm Gen Res, 2014; 2(1):15–23.
Sammour OA, Hammad MA, Megrab NA, Zidan AS. Formulation and optimization of mouth dissolve tablets containing rofecoxib solid dispersion. AAPS PharmSciTech, 2006; 7(2):E167–75. CrossRef
Seager H. Drug-delivery products and the zydis fast-dissolving dosage form. J Pharm Pharmacol, 1998; 50(4):375–82. CrossRef
Solignac M. Mechanisms of action of diacerein, the first inhibitor of interleukin-1 in osteoarthritis. Presse medicale, Paris, France, pp S10–12, 2004, 33(9 Pt 2). CrossRef
Swamy SK, Arun G, Srinivas B, Goud AB. Effect of various super disintegrants on the drug release profile of orally disintegrating tablets. Asian J Pharm Tech, 2016; 6(2):99–105. CrossRef
Tanuwijaya J. The effects of crospovidone and croscarmellose sodium as superdisintegrants on the characteristics of piroxicam nanoparticles ODT (orally disintegrating tablet). Int J pharm Tech Res, 2013; 5(4):1590–7.
Thenmozhi K, Yoo Y. Enhanced solubility of piperine using hydrophilic carrier-based potent solid dispersion systems. Drug Dev Ind Pharm, 2017; 43(9):1501–9. CrossRef
Varshosaz J, Tabbakhian M, Salmani Z. Designing of a thermosensitive chitosan/poloxamer in situ gel for ocular delivery of ciprofloxacin. Open Drug Delivery J, 2008; 2(1):61–70. CrossRef
Xie Y, Xie P, Song X, Tang X, Song H. Preparation of esomeprazole zinc solid dispersion and study on its pharmacokinetics. Int J Pharm, 2008; 360(1–2):53–7. CrossRef
Yang XD, Li WS, Tian YJ, Liu CG, Gao DH, Ma HL. Dissolution rate enhancement of repaglinide by solid dispersion. Trop J Pharm Res, 2016; 15(6):1123–8. CrossRef