
INTRODUCTION
Atorvastatin calcium (ATR) is one of the most favored statins, used to treat moderate-to-severe hypercholesterolemia. It leads to decrease in levels of low-density lipoproteins (LDL), i.e., bad cholesterol and triglycerides and simultaneously increases high-density lipoproteins level (HDL), which is the good cholesterol. This successful molecule was introduced by Pfizer Inc., USA as Lipitor®. However, the low aqueous solubility of drug (i.e., 0.1 mg/ml; BCS class II drug), incomplete intestinal absorption, hepatic first pass metabolism, and crystalline nature makes it poorly bioavailable (only 12%). ATR is unstable to light, heat, and moisture. Due to this, the hydroxyl acid form of ATR is changed to the lactone form. The hydroxyl acid form act as a free acid moiety and it exhibits 15 times more solubility than lactone form due to the ionization of hydroxyl acid. The difference in physicochemical properties (Log p value 1.53 for hydroxyl acid and 4.2 of lactone at pH of 7.4) of the hydroxyl acid and lactone of ATR affects their biologic and therapeutic performance. ATR can further be destabilized by excipients used in the development of formulation (Khan et al., 2011). Thus, this preferred statin has a good therapeutic value, but absorption is limited to biological system.
Poor performance of ATR in biological system leads to need of large doses resulting in liver abnormality, kidney failure, arthralgia, rhabdomyolysis, and other adverse effects (Choudhary et al., 2012). There is the unmet need to develop more bioavailable and stabilized formulation of poorly soluble drug ATR.
Enhancement of solubility, stability and vis-a-vis bioavailability (BA) of a poorly soluble drug is a challenging job for a drug formulation scientist. Most of the new chemical entities have good therapeutic impact, but their absorption is limited to the biological system due to poor solubility (Fazil et al., 2016).
Many approaches have been tried by the researchers to enhance BA of ATR, which includes solid dispersions (Rodde et al., 2014; Shaker, 2018), use of surfactants (Patel et al., 2014), complexation with cyclodextrins, polymorphism, salt formation, size reduction, emulsification (Yeom et al., 2016), gastroretentive formulation (Khan et al., 2011), and nanotechnology (Zidan et al., 2018) etc.
Choudhary et al. (2012) formulated solid dispersions of ATR employing skimmed milk as carrier, suggested as a promising approach for oral delivery of atorvastatin. To overcome the problem of unstability and BA, various experiments were performed, which included ATR-cyclodextrin complexation. Zidan et al. (2018) explored the complexation of ATR with β-cyclodextrin (β-CD) based nanosponge carriers with improved dissolution characteristics and eventually the enhanced oral BA of ATR.
Cyclodextrins (CD) lead to improved BA of poorly aqueous soluble moieties by stabilization of drug molecules at the biomembrane surface. Moreover, use of CD leads to reduced gastrointestinal drug irritation, conversion of liquid drugs into microcrystalline form or in amorphous state and also overcome drug-drug and drug-excipient interactions etc. Palem et al. (2009) observed increase in stability, solubility and in vitro dissolution characteristics of ATR through inclusion complexation with β-CD.
The natural CDs are generally proven to be non-toxic for oral administration. However, γ-cyclodextrin tends to aggregate in aqueous solutions and therefore not preferred in parenteral formulations. The reported nephrotoxicity of β-CD limit their use in parenteral products. Lipophilic cyclodextrin derivatives, i.e., methylated cyclodextrins are reported to be toxic in parenteral formulations. Modified cyclodextrins are helpful in better fitting and association between cyclodextrin and entrapped drug, with simultaneous stabilization of the entrapped entity thereby reducing drug mobility and reactivity and also help in attachment of specific (catalytic) groups to the binding site.
Captisol, i.e., Sulphobutylether β-Cyclodextrin (SBEβ-CD) is an exclusively modified form of cyclodextrin. The chemical structure is logically designed for maximum safety and optimized interaction with drug molecules for improving the stability, solubility and BA along with decreasing the volatility, irritation, any odor or taste. The solubility of Captisol in water is 70 g in 100 ml which is remarkably higher than β-cyclodextrin (i.e., 1.85 g in 100 ml). No nephrotoxicity and cytotoxicity is reported with captisol (Lankalapalli et al., 2018).
Kulkarni et al. (2018) also demonstrated that Captisol inclusion complex is an effective strategy for solubility, stability and BA enhancement of asenapine. Ajmeera et al. (2015) indicated that captisol can be successfully employed as a potential carrier for the oral delivery of telmisartan with enhanced BA and industrial applicability in terms of scale up.
Based on this hypothesis, the intent of current study was to explore the use of modified β-cyclodextrin i.e. captisol, for enhancing the oral BA of ATR by formulating its inclusion complex with captisol.
MATERIALS AND METHODS
Materials
Atorvastatin was kindly gifted by Mylan Labs, Hyderabad. Captisol (Sulfobutyl ether β-CD) was kindly gifted by Cydex Pharmaceuticals, Kanas; β-CD was procured from Sigma-Aldrich, India. All the other chemicals used in the study were of AR grade.
Methods
Formulation of inclusion complexes using β-CD and SBE β-CD
In current study, the inclusion complexes of atorvastatin were developed by using two approaches.
Kneading method: Accurately weighed the drug atorvastatin and polymers β-cyclodextrin and SBE β-cyclodextrin were taken in a ratio 1:1, 1:3, and 1:5 in the mortar and kneaded with small quantity of the 70% ethanol. The kneaded mixture was placed in hot air oven till a constant weight of dried product was obtained and sieved through 80 (#) mesh. The resulting mixture was preserved in the air tight container at room temperature.
Lyophilization method: Inclusion complexes were prepared via solvent evaporation method. Pure drug atorvastatin and the polymers, i.e., β-cyclodextrin and SBE β-cyclodextrin, were mixed in the ratios of 1:1, 1:3, and 1:5 in the 25 ml of solvent (water) in the beaker. The mixture was kept on magnetic stirrer for 2 hours. The mixtures were removed and cooled at room temperature and later kept for pre-freezing at a temperature of −20ºC for 90 minutes. The pre-freezed sample was kept for freeze drying at a temperature of −70ºC for 48 hours resulting in formation of fine powdered inclusion complexes.
Table 1 shows the composition of various inclusion complexes.
 | Table 1. Composition of inclusion complexes. [Click here to view] |
Evaluation of inclusion complexes
Equilibrium solubility studies: The solubility of various inclusion complexes and ATR was determined in phosphate buffer of pH 6.8 at 37°C. The prepared formulations equivalent of 20 mg of drug was kept in covered conical flasks containing 25-ml phosphate buffer and further the dispersion was sonicated for 30 minutes at 37 ± 0.5°C. After sonication, the flasks were kept in orbital shaker incubator at a temperature of 37 ± 0.5°C for 24 hours. The resulting solution was filtered and analyzed for drug spectrophotometrically (Systronics, India) at 244 nm.
Fourier transform infrared spectroscopy (FTIR): FTIR spectra of the samples were determined via potassium bromide pellet method using FTIR spectrophotometer (Spectrum 400, Perkin Elmer). Transparent pellets obtained were scanned over frequency range from 400 to 4,000 cm−1.
X-Ray diffraction: Powder X-Ray Diffraction patterns were traced of the samples using Ni filtered Cu (K-α) radiations and at a voltage of 45 kV and a current of 40 mA using (X’Pert Pro). The samples were scanned over 2θ range of 0–50o with scan step size of 0.0170o (2θ) and scan step time of 25 seconds.
Differential scanning calorimetry: Thermal properties of atorvastatin calcium and inclusion complexes were studied via differential scanning calorimetric analysis (DSC 821e , Mettler Toledo, USA). The samples were sealed in aluminium pan and scanned in temperature range from 30 oC to 300°C and at a heating rate of 10°C/minute in nitrogen atmosphere.
Scanning electron microscopy: External morphology of various samples was studied scanning electron microscopy (JSM 6100, JEOL). The samples were mounted onto the stubs using double-sided adhesive tape and which was coated with gold palladium alloy (150–200 Aº) using fine coat ion sputter.
In vitro dissolution studies: The in vitro release of ATR from various inclusion complexes was determined in 900-ml phosphate buffer of pH 6.8, kept at 37 ± 0.5°C and at 100 rpm using USP (Type II) dissolution apparatus (Electro Lab, India). 5 ml aliquots were withdrawn at specified intervals (5, 15, 30, 45, 60, 90, and 120 minutes), suitably diluted and analyzed spectrophotometrically for drug at 244 nm.
In vivo pharmacodynamics studies
Animals
Wistar male rats were employed in current study. The experimental protocol was suitably approved by Institutional Animal Ethics Committee (1181/PO/ReBi/S/08/CPCSEA). Animals (rats) were kept according to guidelines of CPCSEA, i.e., Committee for the Purpose of Control and Supervision of Experiments on Animals in Chitkara College of Pharmacy, Chitkara University Punjab, India. Rats were fed with normal chow diet (i.e., commercial rat pellets from Kisan Feeds Ltd. Mumbai) and ad libitum under controlled temperature and relative humidity (RH) of (24â—¦C–28â—¦C and 60%–70 %, respectively) and natural light/dark cycle (12:12) animals.
High fat diet (HFD) induced obesity: Rats were kept on prepared HFD and water ad libitum for the 8 weeks time period. The experimental diet (g/kg) contents were as per the formula of Srinivasan et al. (2005).
Experimental protocol: 24 rats were used and divided into four groups of 06 rats each. Group I rats were maintained on standard chow diet for 08 weeks and water ad libitum, with no treatment. Group II rats were maintained on HFD for 8 weeks to persuade obesity. Group III rats received pure drug atorvastatin (10 mg/kg/day, p.o) for 4 weeks, started at the end of 4th week. Group IV rats received optimized test formulation (10 mg/kg/day, p.o) for 4 weeks, started at the end of 4th week.
Morphological parameters for obesity
Body weights of rats were measured once in a week.
Body mass index (BMI) was calculated from formula (Friedewald et al., 1972; Krachler et al., 2013):
BMI = body weight (g)/length2 (cm2).
Sample collection: Blood samples from overnight fasted rats were collected at the end of 8th week by retro-orbital puncture under ether anesthesia. The separated serum samples were stored at −20°C until used for further calculation of various biochemical parameters by enzymatic colorimetric methods using commercial kits.
Biochemical estimation
Estimation of HDL, LDL and total cholesterol (TC) was done by autoanalyzer using Bayer Diagnostic kit and triglycerides estimation was done by using Erba Diagnostics kit. Glucose-peroxidase method was used to calculate serum glucose levels.
Histopathological analysis: Separated liver samples from rats were fixed in buffered formalin (10% neutral), embedded in paraffin for histological examination. Standard liver tissue sections of 5 mm thickness were cut; stained with eosin and haematoxylin, and then examined under light microscope.
Statistical analysis: Results were expressed as mean ± standard error of mean (SEM.). Data were analyzed using one-way analysis of varience followed by Bonferroni multiple comparison test as post-hoc analysis. A value of p < 0.05 was considered to be statistically significant.
RESULTS AND DISCUSSION
Equilibrium solubility studies
The solubility of the ATR in pH 6.8 phosphate buffer was found to be 54.1212 μg/ml. Addition of cyclodextrins (β-CD and SBE β-CD) leads to increase in equilibrium solubility of drug. Captisol (i.e., SBE β-CD, modified form of cyclodextrins) based inclusion complexes showed significant increase in solubility of drug in comparison to inclusion complexes of drug using β-CD.
The effect of method of formation of inclusion complexes was also observed, i.e., freeze dried or lyophilized formulations showed dramatic increase in solubility of the drug in comparison to that of kneading method. Moreover, the yield of final formulations is less in kneading method and lyophilized batches yield almost 95%–100% final product. The results revealed that Captisol inclusion complexes of drug prepared by freeze drying method lead to maximum solubility of drug in 1:3 ratio of drug to Captisol. The comparative solubility data is shown in Figure 1.
Fourier transform infrared spectroscopy
FTIR spectrum of ATR showed characteristic peaks at 1,651 cm−1 (C=O stretch), 3,365 cm−1 (O-H stretch), 1,435 cm−1 (C-F stretch), 1,317 cm−1 (C-O stretch), 1,216 cm−1 (C-N stretch), and signal at 691.54 cm−1 is due to aromatic out of plane bend. FTIR spectrum of β-CD showed C-H stretching at 2,925 cm−1, N-H stretching at 3,307 cm−1, C-H bending at 1,417 cm−1, and C-Br stretching at 609 cm−1. FTIR spectrum of SBE β-CD showed C-H stretching at 2,938 cm−1, C-O stretching at 1,412 cm−1 and C-N vibrations at 1,043 cm−1. FTIR spectra of various inclusion complexes (KN5β, KN5C, FD5β, and FD5C) showed that most of the characteristic peaks of drug are missing (signals at 3,365, 1,317, 1,216 cm−1) and some signals have shifted (e.g., 1,435 to 1,420 cm−1) which is indicative of interaction of drug with both β-cyclodextrins (Figs. 2 and 3).
X-Ray diffraction
Atorvastatin calcium showed sharp peaks of the diffraction angle of 2θ at 9.2367, 10.3544, 11.9284, 17.0248, and 21.5218. Diffraction patterns of β-CD showed various peaks at diffraction angle of 2θ at 12.4921, 19.5868, 22.7177, 27.0674, and 39.9618, suggested crystalline nature of β-CD. In β-CD inclusion complexes (i.e., KN5β and FD5β), some of the peaks of drug are present but with reduced area and intensity and in addition peaks of β-CD was also observed (Fig. 4). The diffraction pattern of SBE β-CD with no sharp peaks, suggests amorphous nature of polymer. The absence of main peaks of drug or decreased area of some characteristic peaks of drug in inclusion complexes prepared by freeze drying and kneading (KN5β; FD5β and KN5C; FD5C) indicated strong interaction of drug with β-CD and SBE β-CD and suggested formation of single amorphous complex (Fig. 5).
 | Figure 1. Equilibrium solubility studies of Atorvastatin Calcium inclusion complexes with β-CD and SBEβ-CD. [Click here to view] |
 | Figure 2. Overlay diagram of FTIR of ATR, β-CD, FD5β, and KN3β. [Click here to view] |
 | Figure 3. Overlay diagram of FTIR of KN5C, SBEβ-CD, ATR, and FD5C. [Click here to view] |
 | Figure 4. Overlay diagram of XRD of ATR, β-CD, PM5β, KN5β, and FD5β. [Click here to view] |
 | Figure 5. Overlay diagram of XRD of ATR, SBEβ-CD, PM5C, KN5C, and FD5C. [Click here to view] |
Scanning electron microscopy
Scanning electron micrographs of pure drug ATR, SBEβCD and inclusion complex (FD5C and KN5C), are shown in Figure 6. The drug appeared as discrete particles rectangular needle shaped indicating its crystalline nature and the polymer (SBE β-CD) appeared as round, oblong in shape. SEM images of FD5C and KN5C inclusion complexes indicated changes in topological characteristics of drug particles aggregated with each other, suggesting the amorphous nature of the product and existence of one component suggesting the maximum complexation.
Differential scanning calorimetry
DSC thermogram of ATR showed sharp endothermic peak at 165.41°C with enthalpy of fusion 80.873 J/g relating to its melting point (Fig. 7). DSC thermogram of SBE β-CD showed (Fig. 8) sharp endothermic peak at 272.22 °C and broad endotherm at 96.03°C with enthalpy of fusion 49.733 J/g and 214.70 J/g, respectively, and subsequent to its melting point. DSC thermogram of FD5C showed two endothermic peaks (related to SBE β-CD) at 99.00°C and 240.06°C with enthalpy of fusion 113.56 J/g and 41.485 J/g (Fig. 9). A slight shift and very small peak of drug (at 163.03ºC) in DSC thermogram of FD5C suggested strong interaction of drug with SBEβ-CD. This is also supported by a shift in the endothermic peak of SBE β-CD at 272.22 ºC to 240.06°C in FD5C which indicated formation of stable complex.
 | Figure 6. Scanning electron micrograph of ATR, SBEβ-CD, FD5C, and KN5C. [Click here to view] |
In vitro dissolution studies
In vitro drug release profile of pure ATR and various β-CD inclusion complexes and SBEβ-CD inclusion complexes is shown in Figures 10 and 11, respectively. The lyophilized ATR- SBEβ-CD inclusion complex (FD3C) exhibited 100% drug release in 2 hours while it is only 65% in pure drug. The drug release from lyophilized β-CD inclusion complex (FD5 β) showed 96% drug release in 2 hours. The kneading method is not supported as per the results of solubility studies and dissolution as well. The inclusion complexes formed by freeze drying/lyophilization exhibited significantly higher drug release in comparison to that formed by kneading method. 1:3 ratio of drug to cyclodextrins proved optimum (formation of stable inclusion complex), and it is also supported by dissolution results. The dissolution rate was increased with increase in concentration of β-CD (Figs. 10 and 11). The general solubility of chemically modified SBEβ-CD is 70 g/100 ml where as in case of naturally occurring β-CD it is just 1.85 g/100 ml which clearly shows that SBEβ-CD has more than 30 times greater solubility than β-CD; and the advantage of being amorphous in nature provides an opportunity to form better inclusion complex with higher dissolution rate (Devasari et al., 2015; Lankalapalli et al., 2018; Shiralashetti and Patil, 2014). On the basis of results of in vitro dissolution studies, the lyophilized batch FD3C will be utilized further for in vivo studies.
 | Figure 7. [Click here to view] |
 | Figure 8. DSC thermogram of SBEβ-CD. [Click here to view] |
 | Figure 9. DSC thermogram of FD5C. [Click here to view] |
In vivo pharmacodynamics studies
Effect of optimized formulation of Atorvastatin on body weight gain and body mass index of rats
Obesity was induced in rats by feeding a high fat diet for 08 weeks. The mean body weights of the four experimental groups were same with minimum variability at the start of the study. A significant increase in body weight and BMI was observed in rats of HFD control group after 08 weeks, as compared to normal control group. Furthermore, the once daily per oral treatment with Atorvastatin (Pure drug, 10 mg/kg, p.o.) for 4 weeks significantly decreased the body weight and BMI as compared to HFD control group (p < 0.01). Also, the treatment of animals with optimized test formulation of Atorvastatin (FD3C) (10 mg/kg, p.o.) for four weeks shows significant (p < 0.001) difference in the decreased body weight and BMI, as compared to HFD control rats (Figs. 12 and 13).
Effect of optimized formulation of Atorvastatin on high fat diet induced changes in blood glucose level of animals
Random blood glucose levels were measured at the end of study (08 weeks). Feeding with high fat diet for 12 weeks significantly increased the blood glucose level in HFD control group as compared to normal control group. Furthermore, the once daily per oral treatment with Atorvastatin (Pure drug, 10 mg/kg, p.o.) for 4 weeks significantly decreased blood glucose level as compared to HFD control group (p < 0.01). Also, the treatment of animals with optimized test formulation of Atorvastatin (FD3C) (10 mg/kg, p.o.), for 4 weeks shows significant (p < 0.001) difference in blood glucose level, as compared to HFD control rats (Fig. 14).
 | Figure 10. In vitro release profile of inclusion complexes of ATR and β-CD. [Click here to view] |
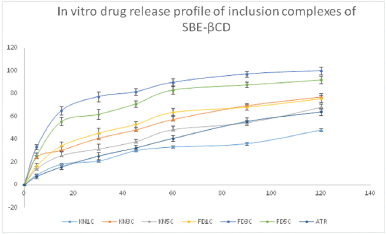 | Figure 11. In vitro release profile of inclusion complexes of ATR and SBEβ-CD [Click here to view] |
Effect of optimized formulation of Atorvastatin on high fat diet induced changes in Lipid Profile of animals
The evaluation of serum lipid profile of experimental animals was carried out for all groups. There was statistically significant (p < 0.01) increase in TC, triglycerides (TG), Low density lipoprotein (LDL) along with decreased high density lipoprotein (HDL) in HFD control group, as compared to normal control group. Further, the once daily per oral treatment with Atorvastatin (Pure drug, 10 mg/kg, p.o.) for four weeks significantly decreased the levels of TC, TG, LDL with increase in HDL as compared to HFD control group (p < 0.01).
 | Figure 12. Effect of optimized formulation of ATR on % body weight gain of rats. [Click here to view] |
 | Figure 13. Effect of optimized formulation of Atorvastatin calcium on BMI of rats [Click here to view] |
 | Figure 14. Effect of optimized formulation of Atorvastatin calcium on random blood glucose level of rats. [Click here to view] |
Also, the treatment of animals with optimized test formulation of Atorvastatin (FD3C) (10 mg/kg, p.o.), for 4 weeks significantly (p < 0.001) modulates the levels of TC, TG, LDL with increase in HDL as compared to HFD control rats (Table 2).
Effect of optimized formulation of Atorvastatin on histopathology
HFD consumption resulted in marked enlargement of adipocytes in highly fat diet control groups. More fat accumulation and consequently marked expansion of adipocytes size is seen in liver tissues. Distended adipocytes and extra fat deposits can be noticed in HFD groups when compared to normal control group. Administration of optimized test formulation of Atorvastatin (FD5C) (10 mg/kg, p.o.) has considerably reduced the size of adipocytes, when compared to disease control group. The comparative microscopic representation of histologically examined livers is shown in Figure 15.
 | Table 2. Effect of various treatments on high fat diet induced changes in lipid profile of rats. [Click here to view] |
 | Figure 15. Gp 1- Normal hepatic adipocytes; Gp 2- Distended adipocytes showing extra fat deposits; Gp 3- Pure drug Atorvastatin treatment (comparatively large size of adipocytes); Gp 4- FD5C inclusion complex treatment (reduced size of adipocytes). [Click here to view] |
CONCLUSION
The modified form of β-CD proved as a prospective carrier system for poorly soluble Atorvastatin calcium. The freeze drying method for preparation of inclusion complexes proved better in terms of yield and dissolution as well. The modified form of β-CD, being amorphous and stable in comparison to pure β-CD, yielded better dissolution characteristics. The lyophilized inclusion complex FD3C (of ATR and SBEβ-CD) lead to better hypolipidemic activity as seen in pharmacodynamic studies in comparison to ATR alone studies. Therefore CAPTISOL, i.e., SBE β-CD-based inclusion complexes can be potential carrier system for poorly soluble drugs and for also useful for oral drug delivery.
ACKNOWLEDGMENTS
The authors would like to thank Dr. Madhu Chitkara, Vice Chancellor, Chitkara University; Dr. Ashok Chitkara, Chancellor, Chitkara University; and Dr. Sandeep Arora, Director, Chitkara College of Pharmacy for providing necessary facilities and support.
CONFLICTS OF INTEREST
The authors declared that they have no conflicts of interest.
REFERENCES
Ajmeera T, Gera S, Pailla SR, Mallika A, Dodoala S, Sampathi S. Enhancement of dissolution and bioavailability of poorly soluble Telmisartan: designing modified cyclodextrin inclusion complexes. J Chem Pharm Res, 2015; 7(10):164–74.
Choudhary A, Rana AC, Aggarwal G, Kumar V, Zakir F. Development and characterization of an atorvastatin solid dispersion formulation using skimmed milk for improved oral bioavailability. Acta Pharma Sin B, 2012; 2(4):421–8. CrossRef
Devasari N, Dora CP, Singh C, Paidi SR, Kumar V, Sobhia ME, Suresh S. Inclusion complex of erlotinib with sulfobutyl ether-β-cyclodextrin: preparation, characterization, in silico, in vitro and in vivo evaluation. Carbohydr Polym, 2015; 134:547–56. CrossRef
Fazil SM, Ansari SH, Ali J. Atorvastatin solid dispersion for bioavailability enhancement. J Adv Pharm Technol Res, 2016; 7(1):22–6. CrossRef
Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem, 1972; 18(6):499–502.
Khan FN, Dehghan MHG. Enhanced bioavailability of atorvastatin calcium from stabilized gastric resident formulation. AAPS PharmSciTech, 2011; 12(4):1077–86. CrossRef
Krachler B, Völgyi E, Savonen K, Tylavsky F, Alén M, Cheng S. BMI and an anthropometry-based estimate of fat mass percentage are both valid discriminators of cardiometabolic risk: a comparison with DXA and bioimpedance. J Obes, 2013; 2013:862514; doi:10.1155/2013/862514 CrossRef
Kulkarni JA, Avachat AM, Avachat CM, Pradhan R, Suryawanshi TS, Khan EM, Martis EAF, Coutinho EC, Padhye S. Preferential formulation of second generation antipsychotic asenapine as inclusion complex with sulphobutylether-βCD (Captisol): in vitro and In vivo evaluation. Curr Drug Deliv, 2018; 15(4):520–31. CrossRef
Lankalapalli S, Beeraka NMR , Bulusu BT. Studies on oral bioavailability enhancement of Itraconazole salts by complexation with Sulfo-butyl7 ether β cyclodextrin. IJRPC, 2018; 8(1):131–43.
Palem CR, Patel S, Pokharkar VB. Solubility and stability enhancement of atorvastatin by cyclodextrin complexation. PDA J Pharm Sci Technol, 2009; 63(3):217–25.
Patel VF, Sarai J. Synergistic effect of hydrotrope and surfactant on solubility and dissolution of atorvastatin calcium: screening factorial design followed by ratio optimization. Indian J Pharm Sci, 2014; 76(6):483–94.
Rodde MS, Divase GT, Devkar TB, Tekade AR. Solubility and bioavailability enhancement of poorly aqueous soluble atorvastatin: in vitro, ex vivo, and in vivo studies. BioMed Res Int, 2014; Article ID 463895:10. CrossRef
Shaker MA. Dissolution and bioavailability enhancement of Atorvastatin: gelucire semisolid binary system. J Drug Deliv Sci Technol, 2018; 43:178–84. CrossRef
Shiralashetti SS, Patil JS. Design charecterization and evaluation of inclusion complexes of poorly soluble atorvastatin. Unique J Pharm Biosci, 2014; 2(2):88–96.
Srinivasan K, Viswanad B, Asrat L, Kaul CL, Ramarao P. Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: a model for type 2 diabetes and pharmacological screening. Pharmacol Res, 2005; 52(4):313–20. CrossRef
Yeom DW, Son HY, Kim JH, Kim SR, Lee SG, Song SH, Chae BR, Choi YW. Development of a solidified self-microemulsifying drug delivery system (S-SMEDDS) for atorvastatin calcium with improved dissolution and bioavailability. Int J Pharm, 2016; 506(1–2):302–11. CrossRef
Zidan MF, Ibrahim HM, Afouna MI, Ibrahim EA. In vitro and in vivo evaluation of cyclodextrin-based nanosponges for enhancing oral bioavailability of atorvastatin calcium. Drug Dev Ind Pharm, 2018; 44(8):1243–53. CrossRef