
INTRODUCTION
Controlled release dosage forms can provide many benefits over traditional ones, such as improve patient compliance and reduced fluctuation in plasma concentration (Niraj et al., 2013). The benefit will become even greater if the developed system was in the form liquid oral formulation which makes it easier for elder patient to take medication (Emami et al., 2008).
Many strategies have been developed to prepare oral liquid sustained release formulations. Preparing sparingly soluble salt of the drug in suspension form was early developed (Shah and Chafetz, 1994), latter, suspending controlled release microparticles in liquid vehicle was also explored (Ribeiro et al., 2005). Recently, developing of oral liquid in situ gelling system is considered as a promising approach (Nirmal et al., 2010). Theophylline was the first drug to be formulated as suspension in the presence of sodium alginate. The later reported to form gel when in contact with the acidic gastric environment (El Maghraby et al., 2012). To increase crosslinking, and consequently gelling ability, sodium alginate was combined with calcium in the form of calcium citrate complex. This complex preserves the liquid consistency of the system while in the container. After administration, the complex breaks immediately and liberated the divalent calcium ions become immediately available to interact with alginate chains as a cross-linker, forming the gel structure (Kubo et al., 2003). The resultant gel was found to have a pH-dependent solubility where they undergo immediate phase transition from gel to sol on increasing the pH value. Therefore, the gel structure was stable as long as it is in the gastric environment but dissolves rapidly after gastric emptying with the release of most of the drug payload. Consequently, the release pattern of the drug will vary in accordance to the gastric emptying rate that is highly unpredictable and variable. This would put the patient at high risk of dose dumping with possible complications. This problem was latter eliminated by the incorporation of chitosan with the in situ gelling formulation that reduced the gel breakdown following pH shifting to a higher value in the intestinal environment (El Maghraby et al., 2012; 2015).
Losartan potassium is used orally in treating hypertension. It acts as an angiotensin II receptor antagonist. It is a non-peptide specific receptor antagonist mainly blocking the AT1 receptors (Sarwar and Hossain, 2012). It is known to be readily absorbed from the gastrointestinal tract (GIT) following oral administration. Nonetheless, losartan potassium exhibits poor bioavailability (25%–35%) since it experiences an extensive first-pass metabolism, in addition to having a low elimination half-life. Owing to its narrow therapeutic index and short biological half-life (1.5–2 hours) (Vohra et al., 2012), losartan potassium showed low therapeutic effectiveness (Gollapudi et al., 2014). Additionally, losartan is subjected to hepatic first pass metabolism which liberates the active metabolite which is more powerful than the parent compound (Sica et al., 2005). Accordingly, controlled release is expected to deliver the drug gradually which ensure complete transformation to active drug.
The objective of this work was to develop candidate formulations for sustained delivery of losartan potassium from oral liquid formulation for elder patients with consistent drug release throughout the gastrointestinal tract. As losartan potassium is soluble, losartan particles were coated with polymer that is insoluble in the gastric pH to retard drug release in the gastric phase. Eudragit® L100 is soluble in pH of about 6.0–6.5; therefore, it was selected for this purpose (Cetin et al., 2010). The study combined the alginate-based in situ gelling system with a pH-dependent polymer. Eudragit® L100 was used at different concentrations to prepare solid dispersion (SD) microparticles of the drug that was suspended in the alginate solution. Additionally, chitosan was also added to alginate solution as an alternative strategy to control over drug release through the intestinal phase.
MATERIALS AND METHODS
Materials
Losartan potassium was obtained from Epico Pharmaceutical Industries, Cairo, Egypt. High molecular weight chitosan (approximately 310,000–>375,000) was purchased from NAIKAI International Trade Company, Shanghai, China. Sodium alginate (low viscosity grade with M/G ratio of 1.56) and trisodium citrate were a generous gift from SIGMA Pharmaceutical Industries, Quesna, Egypt. Eudragit (L100) was purchased from Sigma Aldrich, St. Louis, MO. Disodium hydrogen phosphate, ethanol, sodium hydroxide, and calcium chloride dihydrate were obtained from El Nasr Pharmaceutical Chemicals Company, Cairo, Egypt.
Methods
Construction of calibration curve
Standard curves of losartan were conducted at different pH values. The selected media were 0.1 HCl of pH 1.2 and phosphate buffer of pH values of 6.8 to simulate the gastric and intestinal pH, respectively. A standard stock solution containing 1.0 mg losartan per 1.0 ml distilled water was prepared. A series of concentrations in the range of 6–20 µg/ml were prepared for different media. The absorbance of each dilution was measured spectrophotometrically using UV Spectrophotometer (Thermo-Fisher Scientiï¬c, USA) at 234 nm. The absorbance values were plotted as a function of concentration, in order to produce two standard curves. The results revealed similar response in the tested pH values. The equations of the calibration curve for 0.1-N HCl system was: Y = 0.0437X − 0.0245, while for alkaline solution (pH 6.8) the equation was: Y = 0.0477X + 0.008.
Preparation of solid dispersions
As losartan potassium is freely soluble in acidic media, the aim of this step was to produce SD with polymeric carrier that has limited solubility in the acidic environment. Eudragit L100 is known to have a pH-dependent solubility, with alkaline environment is optimum for its dissolution. Therefore, it was selected to prepare SDs with losartan. SDs were prepared by solvent evaporation method (Essa et al., 2015). The composition of all formulations is shown in Table 1. Both losartan and Eudragit L100 were dissolved in ethanol that was then removed by gentle heating at 60°C. The co-evaporates were kept overnight at the ambient temperature till complete dryness. The resulting matrix was powdered and sieved to obtain a powder with particle size of 350 µm or below.
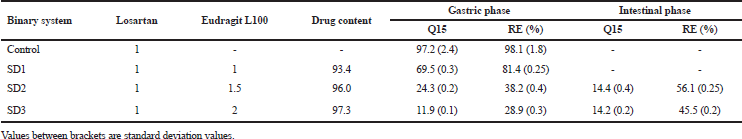 | Table 1. The composition of the prepared binary SDs systems of losartan potassium, together with drug content, amount released after 15 minutes (Q15) and RE in gastric (pH 1.2) and intestinal (pH 6.8) phases. |
Determination of drug content
The drug content of the prepared particles was determined for the prepared formulations. An amount equivalent to 20 mg of the drug from each formulation was dissolved in 100-ml dibasic sodium phosphate (pH 6.8) to dissolve both drug and polymer. The solution was suitably diluted and measured spectrophotometrically at 234 nm (Essa et al., 2017). All the experiments were performed in triplicates.
Characterization of solid dispersion
Differential scanning calorimetry
Physical state of unprocessed drug and that in SD form were analyzed using differential scanning calorimetric using differential scanning calorimetry (DSC)-60, Shimadzu, Japan. Approximately, 2 mg of the drug (or equivalent weight from each formulation) was loaded into aluminum pans and the lids were crimped using a Shimadzu crimper. The thermal behavior of each sample was monitored under nitrogen at a heating rate of 10°C/minute, covering temperature ranges of 25°C–350°C. Data analysis was conducted using the TA 60WS thermal analysis software.
Fourier transform infrared spectroscopy
The Fourier transform infrared (FTIR) spectra of Losartan, Eudragit L100, and their prepared SDs were recorded using FTIR-8400S spectophotometer, Shimadzu, Japan. The samples were mixed with potassium bromide (spectroscopic grade) and compressed into disks using a hydraulic press then scanned from 4,000 to 600 cm–1.
Preparation of in situ gelling system
The in situ gelling systems were prepared according to the method adopted by El Maghraby et al. (2012; 2015). Briefly, two-thirds of the required volume of distilled water was used to dissolve sodium alginate while heating at 60°C with continuous stirring using magnetic stirrer. Sodium citrate (0.45% w/v) and calcium chloride (0.15% w/v) were dissolved in the remaining one-third of water and added to the alginate solution after being cooled. Stirring was continued at the room temperature till homogeneity. The accurately weighed amount of crude losartan potassium or SD enough to produce 5 mg/ml, and chitosan (if present) were gently levigated with the alginate solution using mortar and pestle. The obtained fine dispersion was further mixed for 12 hours, using magnetic stirring, to achieve complete hydration.
To investigate the effect of alginate concentration on the drug release, a series of in situ gelling liquids were prepared with increasing concentrations of sodium alginate (Table 2). The alginate concentration capable of controlling the release of the drug and, at the same time, maintaining the fluidity of the final formulation will be selected to study the drug release from the prepared SD.
Evaluation of the gel-forming capacity
This study was conducted to measure the in situ gelling capacity of the tested formulations. Three different formulations were prepared to contain increasing concentrations of sodium alginate. Cellulose bags containing 10 ml of each system were immersed separately in 100 ml of 0.1-N HCl kept at 37°C for 24 hours. Afterward, each cellulose bag was separated from the immersing media and the weight of the gel produced was determined. The gel was visually inspected and the relative weight of the gel was taken as a measure for the gel strength (Itoh et al., 2006).
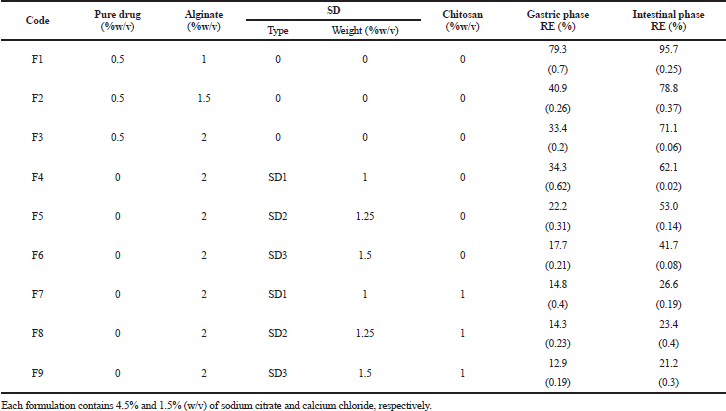 | Table 2. The composition of the in situ gel forming systems with the drug RE of losartan potassium in simulated gastric and intestinal phases. |
In vitro drug release studies
The release study employed the continuous pH variation method with modification (El Maghraby et al., 2012; 2015). This method employed two phases of dissolution representing the variation in pH values through the gastrointestinal tract. Phase I, in which the drug release was monitored at pH 1.2 for 2 hours. Then, pH was changed to 6.8 for 4 hours (Phase II). This design allowed monitoring drug release after in situ gelation in the stomach condition with an extension to monitor the release behavior after gastric emptying. The experiments employed a United state pharmacopoeia (USP) dissolution apparatus (Paddle type, Copley, England) where the paddle speed was maintained at 50 rpm. For Phase I, the release medium was 500 ml of 0.1-N HCl (pH 1.2) simulating gastric fluid without enzymes maintained at 37°C ± 0.2°C. For SDs, an amount equivalent to 50 mg of losartan was loaded in the dissolution vessel, using unprocessed drug as a control. For in situ gelling systems, 10 ml of each system was placed in a petri dish of 2.7 cm internal diameter, which was then immersed carefully into the bottom of dissolution vessel containing the release medium. Samples (5 ml) were collected at predetermined time intervals, then ï¬ltered instantaneously through a 0.45-mm Millipore ï¬lter. The release medium was replenished with the fresh media. The pH of the release medium was then adjusted to 6.8 by adding 200 ml of 0.3-M dibasic sodium hydrogen phosphate and 25 ml of 1-N sodium hydroxide, in order to simulate the intestinal conditions. Sample collection was continued for another 4 hours. The drug concentration in each sample was determined spectrophotometrically.
 | Figure 1. Photographs of the gel formed after immersing different gelling systems in 0.1 N Hcl at 37°C for 24 hours for formulations F1 (A), F2 (B), and F3 (C). Detailed formulations are shown in Table 2. |
The cumulative amounts of the drug released (expressed as % of the total drug added) were plotted as a function of time, so as to produce the drug release proï¬les. The release efï¬ciency (RE) was then calculated from the area under the release curve at time t and expressed as a percentage of the area of the rectangle described by 100% release in the same time (Khan, 1975). In addition, the release data for each phase were ï¬tted to different kinetic models to determine the release kinetics.
Statistical analysis
All experiments were conducted in triplicates and statistical analysis employed Student t-test. Results were quoted as significant where p < 0.05.
RESULTS AND DISCUSSION
Characterization of the gel-forming systems
Development of liquid oral in situ gelling system, it was important to prepare liquid formulation of good fluidity and at the same time, ensure formation of firm gel mass after administration Accordingly, viscosity and gel-forming capacity were used to evaluate the physical properties of the in situ gelling systems.
The viscosity of the formulation depended on the concentration of sodium alginate where the viscosity increases with increasing concentration of alginate. This can be attributed to greater chance for chain entanglement in the presence of high polymer concentration (Miyazaki et al., 2000). The flow behavior of the systems followed a shear thinning non-Newtonian flow characteristics. This indicates that the formulations will become more fluid upon shaking of the container to allow easy withdrawal of the required volume. Similar flow pattern was reported for the same system (El Maghraby et al., 2012; 2015).
The gel-forming property of the formulation is another important factor that can determine the rate of the drug release from in situ gelling systems. This parameter was previously used as a measurement for the ability of liquid system to convert to gel form (El Maghraby et al., 2012). The gel-forming property was investigated by packing the liquid formulation into cellulose bags and incubation in 0.1-N HCl at 37°C. Gel formation was visually inspected and the weight of the obtained gel phase was recorded and used to compare between different formulations. Larger weight is usually taken as an indication for the strength of the gel (Itoh et al., 2006; El Maghraby et al., 2012; 2015). Figure 1 shows photographs of the gels which were formed in acidic medium. The photographs showed different gel consistency for different formulations reflecting the dependence of the in situ gelling capacity on the composition of the system. Liquid comprising 1% alginate produced soft gel. The gel became relatively more firm on increasing alginate concentration.
Effect of alginate concentration on the release of pure losartan
Release profiles of the drug from different in situ gelling liquids are presented as cumulative amount released as a function of time (Fig. 2A). Calculated release efficiency (RE) values in simulated gastric and intestinal media are presented in Table 1.
The drug release pattern indicating dependency of release rate on the concentration of sodium alginate. For formula F1 (containing alginate at 1% w/w), there was a rapid drug release as reflected by the liberation of about 60% of the loaded dose after 15 minutes Almost all the drugs were released at the end of the gastric phase. This behavior can be attributed to the weak gel consistency at such low polymeric concentration. The RE in gastric phase was about 79% with overall efficiency of 96% after shifting to basic phase. For F2 and F3 (containing 1.5 and 2% w/w alginate, respectively), there was a significant reduction (p <0.05) in the initial drug released in gastric phase compared to F1 with a total release of 41% and 33% of loaded drug at the end of gastric phase for F2 and F3, respectively. This indicates that increased alginate concentration can retard drug release most probably due to increased gel barrier formed from the gelation of alginate. Both systems showed prompt drug release shortly after shifting to the basic phase followed by gradual release to reach 100% at the end of this phase (Fig. 2A). The RE in gastric and intestinal phases were significantly lower than that for F1 (Table 1).The release kinetics pattern in gastric phases were best fit to Higuchi equation for the three formulations (Table 3).
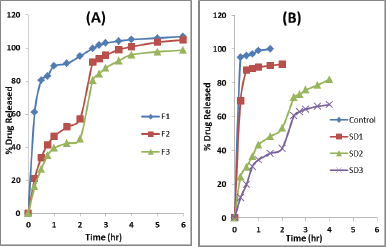 | Figure 2. In vitro release profiles of losartan potassium from in situ gelling systems (A) and from SD formulations (B) using the pH change protocol. For detailed formulations refer to Tables 1 and 2.ons are shown in Table 2. |
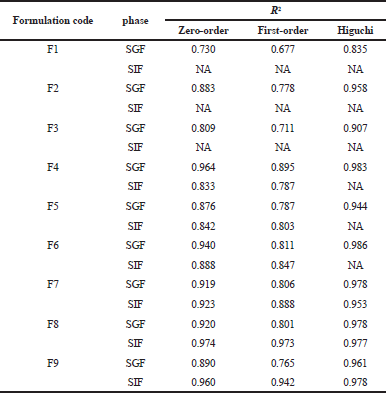 | Table 3. The coefficient of variation (R2) obtained after fitting the drug release data to different kinetic models in simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) phases. |
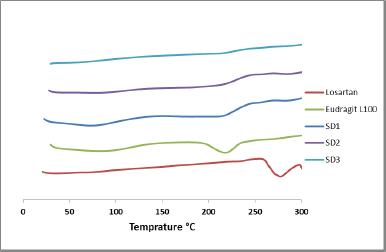 | Figure 3. The DSC thermograms of losartan potassium, Eudragit L100 and their SD formulation. |
The results of release profile and the gel forming property study revealed that 2% w/w alginate is the suitable concentration to sustain drug release without compromising the fluidity of the final formulation. Therefore, it was selected as a standard vehicle for all formulations in this work. Nevertheless, the liberated drug in the gastric phase is more than expected. This may be due to the hydrophilic nature the losartan potassium. Therefore, SDs of the drug in Eudragit® L100 were prepared to reduce the liberated drug during phase I.
Characterization of solid dispersions
Eudragit L100 is an anionic copolymer based on methacrylic acid and methyl methacrylate. It has a pH-dependent solubility in the basic media (Cetin et al., 2010). Therefore, it was selected a matrix former to reduce drug solubility in the simulated gastric fluid (Phase I). Both drug and polymer were co-evaporation from their organic solvent and the obtained solid matrix was size-reduced and sieved. Three binary co-precipitates were prepared at drug:polymer concentration of 1:1, 1:1.5, and 1:2 w/w weight ratios. The drug content of the prepared formulations was in the acceptable limit (Table 2). It ranged from 93% to 97% w/w, indicating good recovery and excluding any segregation of the drug or polymer during formation.
Differential scanning calorimetry
Thermograms of pure losartan potassium, pure Eduragit L100, and different binary SDs are shown in Figure 3. For pure drug, the thermogram showed a single broad endothermic peak with onset of 254°C, endset of 292°C, and transition temperature (Tm) of 271.3°C corresponding to drug melting transition. This thermal pattern is close to reported data and reflects the crystalline nature of drug (Khattab et al., 2014; Velmurufan and Srinivas, 2013).
The thermogram of Eudragit L100 showed two endothermic peaks. The first is a broad peak between 58°C and 109°C, which can be explained by the evaporation of the absorbed moisture. The other peak appeared at higher temperature with onset 194°C, endset of 236°C, and Tm of 218°C. The value of this transition correlates well with the published data (Sharma et al., 2011). For the prepared drug/polymer co-evaporate particles, the thermograms showed initial reduction in Tm of the endothermic peak of the drug in case of SD1 compared to unprocessed drug. Further increase in polymer concentration in the particles (SD2 and SD3) resulted in complete disappearance of the endothermic peak of the drug. This could indicate loss of drug crystallinity and formation of amorphous form (Kim et al., 1985).
Fourier transform infrared spectroscopy
The infrared study was performed to examine any possible interaction between pure drug and additives. Figure 4 shows the FTIR spectra of losartan potassium, Eduragit L100, and sample SD (formula SD3).
The FTIR spectrum of losartan potassium (Fig. 4A) showed a broad absorption band at 3,197.9 cm−1 and twin bands at 995.27 and 1,008.7 cm−1 due to tetrazole ring and a sharp band at 1,458.18 cm−1 due to imidazole ring. The characteristic bands due to O-H and C-O stretch appear at 933.5 and 1,072.4 cm−1. N-H stretch gives a band at 2,954 cm−1 and C=N stretch gives another one at 1,423.47 cm−1. Tertiary amine band (C-N stretch) at 1,260 cm−1. Similar findings were reported previously (Khattab et al., 2014).
The FTIR spectrum of Eudragit L100 (Fig. 4B) showed the characteristic bands of the c=o vibrations of the carboxyl group at 1,730 cm−1 and the esterified carboxyl groups at 1,730 cm−1. The O-H band which appears between 2,500 and 3,500 cm−1 is superimposed by CH vibrations at 2,900–3,000 cm−1. The spectrum also shows ester vibrations at 1,157, 1,190, and 1,265.3 cm−1. This spectrum correlates with the supplier monograph and published data (Sharma et al., 2011).
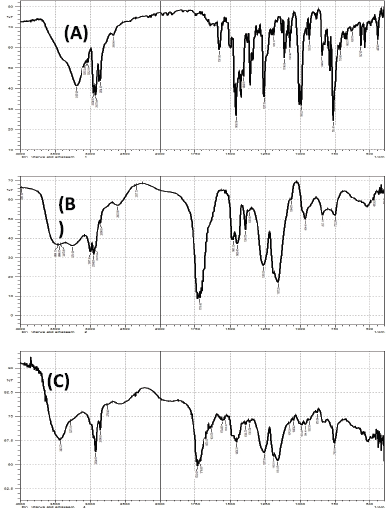 | Figure 4. FTIR spectrum of pure losartan potassium, Eudragit L100 and SD3. |
FTIR spectrums of tested SD formulation (Fig. 4C) showed the main absorption bands of losartan with no significant changes compared with the spectrum of unprocessed drug. This suggests absence of any interaction between the polymers and losartan potassium losartan potassium.
In vitro drug release of Losartan from co-evaporated particles
The dissolution profiles of losartan from the unprocessed powder and different SDs prepared by using Eudragit L100 are shown Figure 2B. The dissolution protocol involved two phases, Phase I comprises 2 hours dissolution in pH 1.2 followed by Phase II (2 hours in pH 6.8). The dissolution parameters represented as the percentage drug released after 15 minutes (Q15) and the percentage RE in each phase were measured and compared. These parameters are presented in Table 1.
Unprocessed drug showed burst dissolution with Q15 of 95% in Phase I. This could be attributed to salt form of the drug. SDs formula SD1 showed rapid drug release with most of the drug release at the end of Phase I with RE of >80%. This may be explained by the low concentration of the polymer with little enteric capacity. Increasing Eudragit L100 concentration increased the enteric power as reflected by reduction in drug release in both acid and alkaline phases. This is reflected by the reduced Q15 in Phase I. The extent of reduction in RE increased with increasing the Eudragit L100 concentration in the particles. This significant (p < 0.05) decrease can be attributed to the proposed hydrogen bonding that occurred between the amine group of losartan and the C=O of the carboxylic groups of Eudragit® L100 (El Maghraby et al., 2014). Shifting to basic media (Phase II), there was a rapid release of appreciable amount of the drug followed by gradual pattern. This could be due to the dissociation of the anionic polymer with chain opening and release of the drug from particles surface, followed by slow diffusion from the particular core.
In vitro drug release of Losartan- eudragit L100 co-evaporate from in situ gelling systems
The release studies employed formulations containing drug: Eudragit® L100 co-precipitate at increasing concentration of the polymer. Formula F4, F5, and F6 represents in situ gelling liquids containing SD1, SD2, and SD3, respectively. The prepared co-precipitate microparticles were dispersed in 2% alginate solution. Drug release was investigated in presence and absence of chitosan the in situ gel forming liquid at fixed concentration of 1%. This concentration was selected based on the previously published work, where 1% chitosan was optimum concentration to control nateglinide release from similar system (El Maghraby et al., 2015). The release profiles are shown in Figure 5, and calculated REs are present in Table 2.
The release pattern indicated reduction in drug release after incorporation of the Eudragit® L100 coated drug particles in alginate solution compared to the same formulations containing the drug in absence of Eudragit L100. The drug release decreases by increasing the Eudragit® L100 concentration in the co-precipitated microparticles (Table 2). This was reflected by decreased RE in both phases. Formula F6 (containing SD3) showed the highest control over drug liberation with RE of 17% in gastric phase, compared to 33% in the free drug loaded system. Alkaline phase was similarly reduced (Table 2). This signifies the ability of SD to slow drug liberation. Alginate gelation can provide additional contributing factor to reduced release by forming a gelatinous barrier layer through which drug should diffuse, thus slowing release from SD particle compared to drug release from microparticle alone.
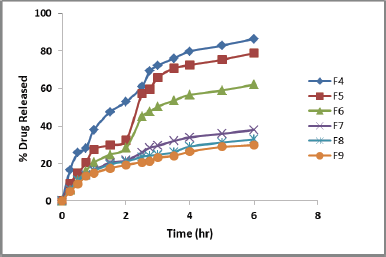 | Figure 5. In vitro release profiles of losartan Potassium from solid dispersion-containing in situ gelling alginate formulation in absence (F4–F6) and presence (F7–F9) of chitosan. For detailed formulations refer to Table 2. |
The release kinetic modeling is presented in Table 3. According to the values of the correlation coefficient (R2), the release kinetics was best fit to Higuchi's model in gastric phase. For intestinal phase, zero-order model was applied. The latter could be attributed to chain opening and biodegradation of alginate polymer (El Maghraby et al., 2014).
For chitosan containing systems, a significant reduction in drug release was observed compared to same formulations in the absence of chitosan. Release of the drug from these systems was characterized by an initial retarded release in gastric phase that became higher on shifting to the intestinal phase. In gastric phase, there was a similar RE for all in situ gelling liquids containing chitosan, regardless of the SD concentration (p > 0.05).
The effect of chitosan was more clear in the intestinal phase in which the release rate was greatly reduced compared to chitosan-free systems. Formula F9 (containing SD3 microparticles) showed RE of about 21%, compared to 42% for the same system in absence of chitosan (Table 2). These results reflected the synergistic effect between chitosan and alginate with the formation of a firmer gel (El Maghraby et al., 2014; 2015). This firm structure can be explained on the bases of formation of strong polyelectrolyte complex arising from electrostatic forces of attraction between the amino groups of chitosan and carboxylic groups of alginate, with increased crosslinking intensity (Becherán-Marón et al., 2004; Murata et al., 1996; Wang et al., 2001).
The release kinetics modeling is presented in Table 3. The correlation coefficient (R2) suggested the Higuchi model for drug release in both gastric and intestinal phase. A condition that ensure the release of consistent amount of the drug throughout the gastrointestinal tract (Lee et al., 1995). In another word, chitosan containing formulations provide oral liquid in situ gel forming system capable of controlling drug release after gastric emptying, with no risk for dose dumping.
CONCLUSION
Combination of the enteric coating technique with the in situ gelling system is a promising strategy to develop a liquid oral sustained release system with close control of the release rate both in the gastric and intestinal conditions. Preparation of sodium alginate in situ gelling systems with unprocessed drug reduced drug release in gastric phase only at high alginate concentration; however, it showed burst release in intestinal phase with possible dose dumping. Preparation of drug-eudragt® L100 SD microparticles reduced drug release both in gastric and intestinal phase. Incorporation of drug microparticles in alginate solution reduced drug release. At 2% w/v alginate solution, increasing eudragit concentration decreased the liberated drug with total RE values of 62.1%, 53.0%, and 41.7% for 1:1, 1:1.5, and 1:2 drug:eudragit ratios, respectively. Presence of chitosan decrease further drug release, especially in the intestinal phase. These results indicate that manipulation of the in situ gelling system can provide formulations with improved therapeutic benefits of losartan potassium by controlling the onset and duration of action. Being oral liquid, the benefit becomes even greater by improving patient acceptability, especially among geriatric patient.
CONFLICT OF INTEREST
Authors declare that there is no conflict of interest.
FINANCIAL SUPPORT
None.
REFERENCES
Becherán-Marón L, Peniche C, Argüelles-Monal W. Study of the interpolyelectrolyte reaction between chitosan and alginate: influence of alginate composition and chitosan molecular weight. Int J Biol Macromol, 2004; 34(1–2):127–33. CrossRef
Cetin M, Atila A, Kadioglu Y. Formulation and in vitro characterization of Eudragit® L100 and Eudragit® L100-PLGA nanoparticles containing diclofenac sodium. AAPS PharmSciTech, 2010; 11(3):1250–6. CrossRef
El Maghraby GM, Elsisi AE, Elmeshad GA. Development of liquid oral sustained release formulations of nateglinide: in vitro and in vivo evaluation. J Drug Deliv Sci Technol, 2015; 29:70–7. CrossRef
El Maghraby GM, Elzayat EM, Alanazi FK. Investigation of in situ gelling alginate formulations as a sustained release vehicle for co-precipitates of dextromethrophan and Eudragit S 100. Acta Pharm, 2014; 64(1):29–44. CrossRef
El Maghraby GM, Elzayat EM, Alanazi FK. Development of modified in situ gelling oral liquid sustained release formulation of dextromethorphan and eudrgit S100. Drug Dev Ind Pharm, 2012; 38(8):971–8. CrossRef
Emami J, Varshosaz J, Ahmadi F. Preparation and evaluation of a liquid sustained-release drug delivery system for theophylline using spray-drying technique. Res Pharm Sci, 2008; 2(1):1–11.
Essa EA, Diwakat M. Enhancement of simvastatin dissolution via surface solid dispersion; effect of carriers and wetting agents. J Appl Pharm Sci, 2015; 5(1):054–3.
Essa E, Negm M, Zin Eldin E, El Maghraby G. Fast disintegrating tablets of amiodarone for intra-oral administration. J Appl Pharm Sci, 2017; 7(01):064–72. CrossRef
Gollapudi R, Javvaji H, Tadikonda RR, Arpineni V. Formulation and evaluation of sustained release matrix tablets of Losartan potassium. Res J Pharm Biol Chem Sci, 2014; 3(2):526–34.
Itoh K, Kubo W, Fujiwara M, Watanabe H, Miyazaki S, Attwood D. The influence of gastric acidity and taste masking agent on in situ gelling pectin formulations for oral sustained delivery of acetaminophen. Biol Pharm Bull, 2006; 29(2):343–7. CrossRef
Khan KA. The concept of dissolution efficiency. J Pharm Pharmacol, 1975; 27(1):48–9. CrossRef
Khattab WM, Gad S, El-Sayed MM, Ghorab MM. Simple controlled release delivery system for an anti-hypertensive drug via buccal route. Br J Pharm Res, 2014; 4(10):1174–95. CrossRef
Kim KH, Frank MJ, Henderson NL. Application of differential scanning calorimetry to the study of solid drug dispersions. J Pharm Sci, 1985; 74(3):283–9. CrossRef
Kubo W, Miyazaki S, Attwood D. Oral sustained delivery of paracetamol from in situ-gelling gellan and sodium alginate formulations. Int J Pharm, 2003; 258(1–2):55–64. CrossRef
Lee BJ, Cui JH, Kim TW, Heo MY, Kim CK. Biphasic release characteristics of dual drug-loaded alginate beads. Arch Pharm Res, 1998; 21:645–50. CrossRef
Miyazaki S, Kubo W, Attwood D. Oral sustained delivery of theophylline using in-situ gelation of sodium alginate. J Control Release, 2000; 67(2–3):275–80. CrossRef
Murata Y, Miyamoto E, Kawashima S. Additive effect of chondroitin sulfate and chitosan on drug release from calcium-induced alginate gel beads. J Control Release, 1996; 38(2–3):101–8. CrossRef
Niraj VK, Srivastava N, Singh T, Gupta U. Sustained and controlled drug delivery system—As a part of modified release dosage form. Int J Pharma Sci, 2013; 2(5):586–601.
Nirmal HB, Bakliwal SR, Pawar SP. In-situ gel: new trends in controlled and sustained drug delivery system. Int J PharmTech Res, 2010; 2(2):1398–408.
Ribeiro AJ, Silva C, Ferreira D, Veiga F. Chitosan-reinforced alginate microspheres obtained through the emulsification/internal gelation technique. Eur J Pharm Sci, 2005; 25(1):31–40. CrossRef
Sarwar S, Hossain MS. Development and evaluation of sustained release losartan potassium matrix tablet using kollidon SR as release retardant. Brazilian J Pharm Sci, 2012; 48(4):621–8. CrossRef
Shah KP, Chafetz L. Use of sparingly soluble salts to prepare oral sustained release suspensions. Int J Pharm, 1994; 109(3):271–81. CrossRef
Sharma M, Sharma V, Panda AK, Majumdar DK. Development of enteric submicron particle formulation of papain for oral delivery. Int J Nanomed, 2011; 6:2097–111. CrossRef
Sica DA, Gehr TW, Ghosh S. Clinical pharmacokinetics of losartan. Clin Pharmacokinet, 2005; 44(8):797–814. CrossRef
Velmurufan S, Srinivas P. Formulation and in vitro evaluation of losartan potassium mucoadhesive tablets. Asian J Pharm Clin Res, 2013; 6(3):125–30.
Vohra DD, Pagi KS, Rajesh KS. Losartan potassium loaded sustained release matrix tablets: influence of various hydrophilic and hydrophobic polymers on drug release behaviour. J Pharm Bioallied Sci, 2012;4(5):S79–80. CrossRef
Wang L, Khor E, Lim LY. Chitosan-alginate-CaCl(2) system for membrane coat application. J Pharm Sci, 2001; 90(8):1134–42. CrossRef