
INTRODUCTION
Psoriasis is a chronic multisystem inflammatory disease with predominant skin and joint involvement. It occurs at various age levels: 16–22 years and 57–60 years affecting both the sexes. Psoriasis is multifactorial involving dysregulated inflammation and genetic association (Whan et al., 2017). Active inflammatory psoriasis is characterized by the Koebner phenomenon, in which new lesions arise at sites of trauma or pressure. Other types of psoriasis are site specific, such as flexural psoriasis, generalized pustular psoriasis, palmoplantar psoriasis, and psoriatic arthritis (Griffiths and Barker, 2007).
Clobetasol propionate (CP) is commercially available as cream, ointment, scalp solution, gel, foam, or emollient cream formulations that contain 0.05% w/w of the steroid. It is available in combination with calcipotriol 0.005% as ointment. Commercially available CP products are Dermovate®, Dovate®, and Xenovate®. The combination product of clobetasol propionate 0.05% w/w with calcipotriol 0.005% w/w is a popular topical product used for the treatment of psoriasis.
Clobetasol propionate is a Di-halogenated analog of prednisolone which is about 1,800 times more potent than the hydrocortisone. It is highly susceptible for hydrolysis with its corresponding acid and alcohol (Gordon, 1988). As per the official monographs of Clobetasol propionate drug substance available in United States pharmacopeia (USP), European pharmacopeia (EP), and Indian pharmacopeia, clobetasol propionate has a total of 13 related substances. The EP monograph (2018) recommends to quantify the impurities (A to E, L, and M), where L and M are structurally unknown impurities. However, there is no analytical method available for the estimation related substances of clobetasol propionate in its combination drug product with calcipotriol.
Control of impurities and potency is essential to ensure the safety of patients and efficacy of the product. As per the Food and Drug Administration (FDA) guidance for industry, each new drug application (NDA) and Abbreviated new drug application (ANDA) must include the analytical procedures necessary to ensure the identity, strength, quality, purity, and potency of the drug substance and drug product (FDA, 2015). Hence, it is required to assess the level of impurities present in the product.
The predominant analytical techniques used for the quantitative analysis of impurities in the pharmaceutical industry are high-performance liquid chromatography and ultra-high performance liquid chromatography. Several analytical methods have been developed for the quantitative estimation of clobetasol propionate drug substance alone by Thin layer chromatography (TLC) Densitometric (Bassuoni et al., 2016), HPLC (Angelo et al., 2017; Badilli et al.,2013; Bhuyian et al., 2015; Fauzee and Walker, 2013; Fontana et al., 2010; Gagliardi et al., 2000; Manoharan, 2017; Marika et al., 2008; Nidhi et al., 2016; Reepmeyer et al., 2008; Turabi and Khatatbeh, 2014), LC-MS (Nam et al., 2011; Sparidans et al., 2010; Van Velsen et al., 2012), UV Spectroscopy (Amr et al., 2010; Mostafa et al., 2002; Neelam et al., 2016), and High performance Thin layer chromatography (HPTLC) (Mrunali, 2010). Most recently an analytical method was developed for quantitation of ketoconazole and Clobetasol Propionate Cream by HPLC (Wenling, 2018). This paper emphasized the separation of only impurity-C and impurity-J with a high limit of quantitation of 0.5% [International Conference on Harmonisation (ICH) requirement is <0.1%]. This is could be due to challenge in extraction of desired impurities from the sample matrix. As per the ICH guidelines, all the impurities which are above 0.1% should be reported. Hence, the analytical method must have minimum LOQ of 0.1%. Thus, based on authors knowledge on entire literature search, there is only one method (Badilli et al., 2013) available for estimation clobetasol propionate and there is no method available for quantitative estimation impurities in its combination product with calcipotriol. Hence, there is an abrupt need in the pharmaceutical industry to have a sensitive, selective, precise, accurate, linear, and robust analytical method for the estimation of related substances of clobetasol propionate.
Thus, author took an opportunity to develop a rapid Ultra high performance liquid chromatography (UHPLC) analytical method with an efficient and simple sample extraction procedure for the determination available five related substances of clobetasol propionate (Fig. 1) in its topical combination dosage forms in a gradient elution. Method is validated as per ICH Q2 (R1) guideline (ICH, 2005) and found that the method is specific, sensitive, linear, precise, accurate, and robust to implement successfully in quality control and product development laboratories.
MATERIALS AND METHODS
Chemicals and reagents
HPLC grade methanol, n-heptane, and AR grade ammonium acetate was procured from Merck, Mumbai, India. Samples of Clobetasol propionate and all its impurities (A–D and J) were procured from analytica chemie Inc, Bangalore, India. High pure water was obtained using TKA water purification system. Ascentis Express C18 100 mm × 4.6 mm column with 2.7 μm particle size (cat no. #53827-U) was procured from Sigma Aldrich, Bangalore, India. Centrifuge model Heraeus Biofuge stratos was procured from Thermo Electron Corporation, supplied by Mas-tek instruments, Secunderabad, Telangana, India.
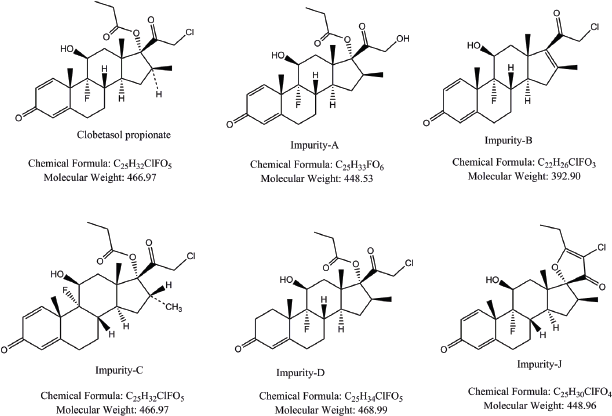 | Figure 1. Structures of clobetasol propionate and its impurities. [Click here to view] |
Instrumentation and software
Method development, forced degradation, and validation studies were performed on Acquity-UPLC system (Waters Corporation, USA) equipped with an LC pump (model ACQ-BSM, S. No: K10UPB207A), an online degasser, auto sampler (model ACQ-SM, S. No: K10UPA060M) with thermostat, and a photodiode array (PDA) (model ACQ-PDA, S. No: J10UPD130A) detector. The chromatographic data were acquired, monitored, and processed using Empower 3 software from Waters Corporation
Chromatographic conditions
The column oven temperature is set at 45°C. The injection volume of sample and standard was 10 µl. The stationary phase, Ascentis Express C-18, 100 mm × 4.6 mm column with 2.7 μm partial size (cat no. #53827-U) was employed in the method development and validation. Ammonium acetate in water (10 mM), as a mobile phase-A Mixture of methanol and water in the ratio of 90% and 10% v/v, is used as a mobile phase-B. The flow rate of the mobile phase is set at 1.5 ml minute−1. The gradient program time (in minute)/% mobile phase-B is set as 0/58, 11/70, 13/95, 17/95, 18/58, and 20/58. The mixture of methanol and water in the ratio of 80 and 20 is used as a diluent. The extraction of desired components achieved using the solvent extraction approach using n heptane and diluent. The chromatograms were acquired and monitored at wave length 240 nm.
Standard solutions
Standard for related substances analysis (diluted standard)
For standard preparation of related substances analysis, 10 mg of clobetasol propionate is transferred in 100-ml volumetric flask, added 60 ml of diluent and sonicated for 5 minutes to dissolve, and made up to the mark with diluent.
Transferred 2 ml of above solution (100 µg ml−1) in a 50-ml volumetric flask and diluted up to the mark with diluent to get 4 µg ml−1 solution.
Extraction procedure for Topical dermatologic formulations samples and related substances
Accurately transferred, 4,000 mg of clobetasol propionate and calcipotriol ointment in a 100-ml volumetric flak. To this, added 20 ml of n heptane and vortexed until a homogeneous dispersion is achieved. To this, 10 ml of diluent was added and vortexed again for 5 minutes with an intermittent hand shaking after every 1 minute. The whole solution was transferred into a separating funnel and allowed the solution for layer separation. Carefully collected the clear diluent layer in centrifuge tube. The solution was centrifuged at 10,000 rpm for 10 minutes. Collected the clear solution (some turbidity is expected on the surface of the solution due to traces of carryover of n heptane in the diluent) judiciously using the needle of syringe and transferred in the vial. Injected the clear solution in the UHPLC. Prepared the placebo same as the sample.
Procedure and calculation of impurity content in the formulation samples
Injected blank, standard solution, placebo, and formulation samples with the chromatographic conditions mentioned in the Section 2.3 for the analysis.
The estimation of Clobetasol propionate related compounds is calculated using Eq. 1.
where, AT is the peak area due to Known/Unknown impurity in the sample preparation, AS is the Average peak area due to Clobetasol propionate in the standard, WS is the weight of the standard in mg, WT is the weight of formulation sample in mg, P is the potency of the standard in %, and LC is the label claim of clobetasol propionate in the formulation (%w/w). RRF is the relative response factor. The relative response factors of impurity-A, B, C, D, and J are 1.45, 0.91, 1.03, 1.17, and 1.16, respectively. The relative retention times of impurity-A, B, C, D, and J are 0.67, 0.62, 0.85, 1.16, and 1.20, respectively.
RESULTS
Method development
Analytical method development, validation, and transfer play an important role in the identification and quantification of impurities in pharmaceutical products. Since the industry is now looking for reducing the cost of processes involved in the making the products, it is imperative to minimize the cost of analysis. The use UHPLC not only reduces the cost of solvents but also provides advantages, such as increased speed, sensitivity, and resolution, compared to conventional HPLC (Michael, 2005). Thus, UHPLC instrument was selected for the method development.
Selection of stationary phase
The molecular weight of Clobetasol propionate and its impurities suggests that C18 column is apt for the separation of impurities and Clobetasol propionate. Waters BEH C18 column was evaluated initially but fronting in the peak shape was observed. Fused core particle technology column, Ascentis Express C18, 100 mm × 4.6 mm column with 2.7 μm was given the next preference considering the advantages with respect to its performance in the separation of impurities as well as its flexibility to operate in UPLC, UHPLC, and HPLC (Joseph et al., 2013). The peak shape are found be acceptable but selectivity was not achieved among the impurities. To achieve the separation/selectivity, extensive work was done in mobile phase optimization.
Selection of mobile phase
As one of the objectives of the method is to develop a method with mass compatible mobile phase, Formic acid, Glacial acetic acid, and ammonium acetate were used to screen the pH for the selectivity. Initially, it was thought that pH will not have any impact as molecule under the investigation is a steroid. But, the pattern of placebo has the significant impact on the mobile phase pH/selection buffer. When formic acid (0.1%, v/v) and glacial acetic acid (0.1%, v/v) is used as a mobile phase, the placebo peaks were interfered with the peaks of interest. When the pH of mobile phase is shifted toward neutral, i.e., when we use ammonium acetate (10 mM) as a mobile phase, a clean base line was observed at the peaks of interest and all the placebo peaks were eluted near to dead volume. Thus, mobile phase-A is selected as 10 mM ammonium acetate. Acetonitrile was initially used as the mobile phase-B. All the impurities of clobetasol propionate were very well separated but calcipotriol impurities were co-eluted with them. To reduce the elution strength of mobile phase-B introduced 50% of methanol in the Acetonitrile. The calcipotriol related impurities were retained more compared to clobetasol propionate impurities but enough resolution was not achieved. When methanol alone was tried to verify the selectivity, the clobetasol propionate and calcipotriol impurities were completely separated as two groups but poor resolution was observed between clobetasol impurity-A and B pair as well as clobetasol impurity-D and J pair. An introduction of 10% of water in the methanol has provided very good selectivity among all the desired peaks. Thus, mobile phase-B was selected as a mixture of water and methanol in the ratio of 10% and 90% v/v.
Selection of diluent
Extensive work has been conducted in selecting the diluent for this product. Due to complex excipient involved in the formulation, it is not possible to achieve the extraction drug and impurities. The approach followed in this method can provide the attention to all the researchers who are working on topical formulations with low drug strength. Sample preparation plays a key role in achieving the extraction/recovery, solution stability, as well as the sensitivity. The detailed experimental trials conducted for the selection of diluent was tabulated in Table 1. Based on the experiments, n heptane is used as the dispersion solvent and mixture of methanol and water in the ration of 80 and 20 v/v is selected as a diluent.
Selection of detector wavelength
Selection of wave length plays a key role in the detection of various impurities. In the case of Clobetasol propionate, based on the UV spectra of all the impurities and clobetasol propionate, 240 nm is selected as the wavelength for the detection.
Selection of column temperature
Steroids show temperature-dependent separation. Clobetasol propionate also showed similar observation proving that it has no exemption. The separation was screened from 35°C to 50°C of column temperature. From 40°C column temperature, superior peaks separation was observed. Keeping the robustness of the method in the mind, 45°C is finalized as the column oven temperature.
Selection of flow rate
The separation was screened from 1.0 to 1.7 ml/minute of flow rate. Keeping the robustness of the method in the mind, 1.5 ml/minute is finalized as the flow rate of the method.
Method validation
The method is validated in accordance with ICH Q2 (R1) guideline.
System suitability
The system suitability parameters were evaluated for Clobetasol propionate and its five impurities (Fig. 1) in the above optimized UHPLC conditions. The critical system suitability parameters, such as tailing factor and resolutions, for all the impurities were evaluated. The tailing factor is less than 1.5 for all the peaks of interest and resolution between any pair of components that are eluting closely was more than 2.0. The results are summarized in Table 2.
Specificity
The selectivity and specificity of the method was verified for interference due to placebo, known impurities and all the degradation impurities. The peak homogeneity was determined for all the desired peaks using photo diode array detector. Blank, placebo matrix, and individual impurities were prepared and injected in UHPLC. The interference due to placebo, blank was verified at the retention times of all the impurities and Clobetasol propionate. All the impurities are separated from each other and there is no interference at any of these impurities and Clobetasol propionate.
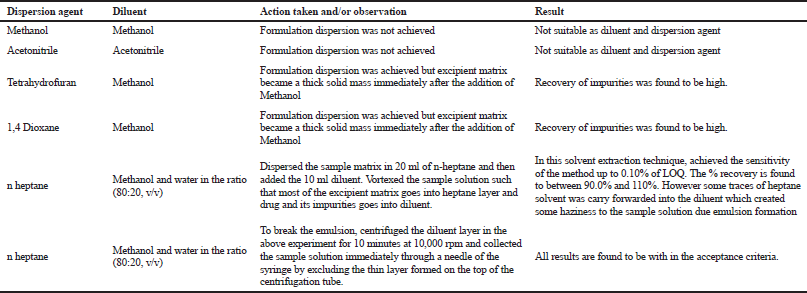 | Table 1. Optimization trials conducted for selection of diluent. [Click here to view] |
Linearity
For determining the linearity of impurities, a series of different solutions with concentrations ranging from 0.2 to 6.0 µg ml−1 (i.e., 0.1%–3.0%) were prepared and injected in the UHPLC where 0.2 µg ml−1 is LOQ for all the impurities and Clobetasol propionate. A plot is drawn by taking the concentration of impurity (µg ml−1) in on x-axis and absorbance/area on y-axis. Correlation coefficient, % Bias, and slope were calculated from plot. The results showed that the method is linear in the proposed range. The relative response factors for all the impurities were calculated using the slope ratio method. The results are summarized in Table 3.
Accuracy
For the determination of accuracy of related substances method, recovery study was carried out by spiking known amounts of impurities in the placebo matrix at three different levels, 50%, 100%, and 150% of specification. The recovery of each impurity was calculated using diluted standard area of Clobetasol propionate and relative response factor of each impurity. The results are summarized in Table 3. From the data, it is evident that the accuracy is ranged from 94.4% to 108.0%. This study helped in concluding that all the desired components are extracted in the selected diluent for the accurate determination impurities of Clobetasol propionate.
Precision
The precision of the method was established by conducting system precision and method precision experiments.
System precision: Precision of the system was determined by injecting six replicate injections of standard solution of clobetasol propionate prepared at 4 µg ml−1 in a short interval of time. In the UHPC, as per the procedure mentioned in the Section 2.3, precision of the system was measured using statistical tool relative standard deviation. The %RSD of area standard was found to be 0.36.
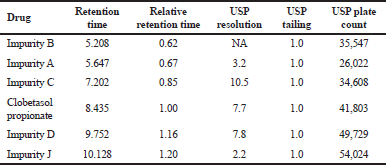 | Table 2. Results of system suitability. [Click here to view] |
Method precision: Method precision for related substance was demonstrated by preparing six samples of Clobetasol propionate and calcipotriol formulation by spiking the impurities at specification level. The samples were analyzed as per the method and calculated the % of impurity content. The relative standard deviation was calculated for the results obtained from all the six preparations. The results are summarized the data in the Table 3. The overlay of chromatograms of method precision is depicted in Figure 2.
Limit of detection and limit quantification
The limit of detection (LOD) and limit of quantitation (LOQ) are established for Clobetasol propionate and its related substance by diluting the standard stock solution of each impurity and Clobetasol propionate. Even though ICH guideline suggests having a signal to noise around 10, the method was developed such that the signal to noise for least absorbing component is above 15 at the LOQ concentration to ensure the suitability of the method even with aged detectors where relatively more noise base line is expected. At concentration 0.03 and 0.10 µg ml−1, the signal-to-noise ratios were found to be more than 6.5 and 15.4 for any impurity. Thus, these concentrations were finalized as LOD and LOQ concentrations. Further precision and accuracy for all the impurities at LOQ level was established. The precision was found to be less than 5.1% RSD and the average recoveries at LOQ level were found to be in the range of 94.4%–107.9%. The results are summarized in Table 3. The overlay of chromatograms of limit of quantitation for Clobetasol propionate and all other impurities is depicted in Figure 3.
Robustness
Robustness study is performed for ensuring the suitability of method with small and deliberate changes to method parameters. The critical method parameters which are expected to have a slight change in the method parameters are Flow rate, column oven temperature, and aqueous portion in the mobile phase-B. Hence, the robustness of these parameters was verified from 1.3 to 1.7 ml min−1 of flow rate, 40°C to 50°C of column temperature and 8%–12% of water concentration in mobile phase-B. The results are captured in Table 4. Based on the results, it is concluded that the method is robust and it can be easily implemented in quality control and product development laboratories for the regular analysis and formulation development of Clobetasol propionate and its combination product.
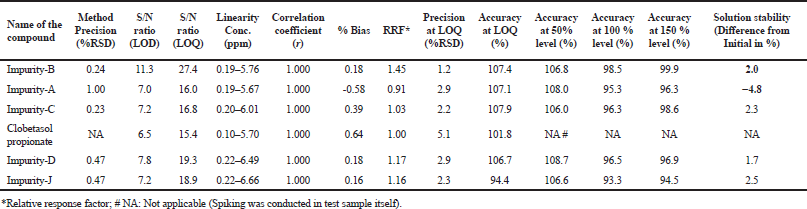 | Table 3. Results of LOD, LOQ, precision, linearity, RRF, solution stability, and accuracy. [Click here to view] |
Stability of solutions
The solution stability was conducted on a spiked sample of formulation. The study was conducted up to 2 days by storing the sample on bench top. Compared the data obtained on day-0 and on day-2. The results are tabulated in Table 3. Based on the data, it can be concluded that the solution are stable at least for 2 days.
DISCUSSION
The purpose of present work is to develop a sensitive, simple, short, robust, specific, and LC-MS compatible UHPLC method which can accurately and precisely quantitate related substances of Clobetasol propionate in its combination drug product with calcipotriol. After an extensive literature search on the availability of the method, it was understood that there is no single analytical method available which can able to separate and accurately quantitate all the five commercially available related substances of Clobetasol propionate.
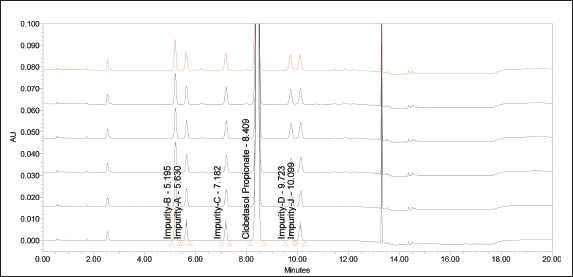 | Figure 2. Overlay chromatograms of method precision. [Click here to view] |
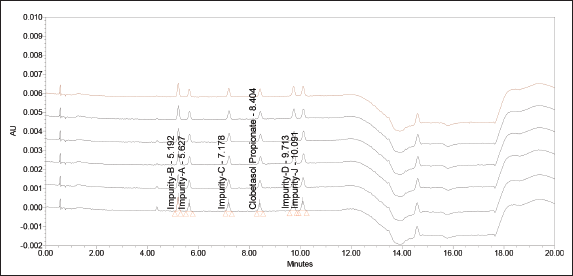 | Figure 3. Overlay chromatograms of limit of quantitation. [Click here to view] |
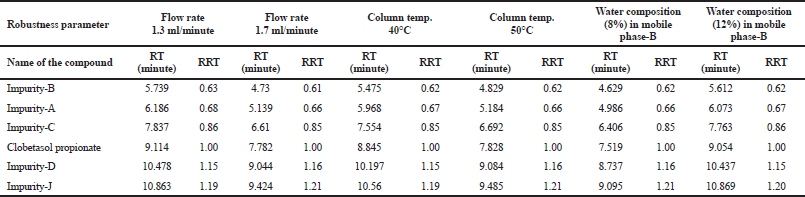 | Table 4. Results of robustness study. [Click here to view] |
The method development was initiated with a fused core particle column with the intention to utilize its flexibility to operate at higher flow rates. The mobile phase-A selection played a crucial role in separation of placebo component from the peaks of interest. The use of acetonitrile as organic modifier has made a very good separation among the impurities of clobetasol propionate but unfortunately calcipotriol impurities co-eluted. A mixture of water and methanol in the ratio 10 and 90 had provided a good separation among all the impurities. All the impurities and clobetasol propionate had wavelength maxima at 240 nm. Hence, the same is used to detect the impurities. At this wavelength, the method can able to detect up to 0.03% of impurity and accurately quantitate from 0.1% to at least 3%. For selection of diluent, significant efforts were made to achieve the complete extraction of compounds of interest. The details of the same are tabulated in the Table 1. The other chromatographic parameters, such as flow rate, column temperature, are optimized such the method is robust for at least 10% of their actual method conditions. The method is much superior over the available method in terms of time cost (low solvent consumption, and high throughput) and selectivity (able to estimate five impurities of Clobetasol propionate) and its applicability to clobetasol propionate drug product and its combination products with calcipotriol.
Application to pharmaceutical industry
As per the FDA guidance for industry, each NDA and ANDA must have an analytical method to ensure the identity, strength, quality, purity, and potency of the drug substance and drug product. The stability and product development data must be available at the submission to regulatory. The method used for the qualifying the product must have standards of accuracy, linear response, sensitivity to detect, specificity to separate all the impurities, and reproducibility. The method described in Section 2 can be successfully implemented in the product development labs for performing the drug and excipient compatibility studies to select the excipients, such as surfactants, gelling agents, skin permeation enhancers, solubilizes, preservatives emollients, and anti-oxidants. During the screening of these excipients for their suitability to formulation, a lot of time would be consumed but the proposed method being a short run time of 20 minutes, the industries can made conclusions in quick turnaround time. Further since method is validated considering ICH Q2 guideline recommendation, it can be implemented in all the phases of product development, i.e., from exploratory stage of product development to commercialization stage. This is the first reported analytical method which can accurately quantitate five related substances of clobetasol propionate with the highest sensitivity. Hence, the method finds a greater application in the pharmaceutical industry.
CONCLUSION
A reverse phase UHPLC method for identification and quantitation of five related substances of Clobetasol propionate, a super potent steroid was developed using a mass compatible mobile phase and fused core particle technology stationary phase. The proposed method is compatible with LC-MS. Hence, this method can be used for the identification of molecular weight of any unknown impurity generated during the stability studies and in the synthesis of Clobetasol propionate. Proposed method is proved to be robust by performing a deliberate change to the method parameter. This method is the highest sensitive method developed so far in the pharmaceutical industry for impurities estimation OF clobetasol propionate drug products with 0.03% LOD and 0.1% LOQ by UHPLC. This method can successfully be implemented in the quality control lab for the routine analysis of this product. Further this UHPLC method was successfully validated as per ICH Q2(R1) guideline and proved to be precise, linear, sensitive, accurate, and robust. This method is being short (<20 minutes), high throughput can be expected. As the solvent consumption is less due to the use of UHPLC, implementation of this method will be environment friendly as well as economical. This is the first reported RP-UHPLC method for estimation of impurities of clobetasol propionate in its combination product with calcipotriol.
ACKNOWLEDGMENTS
The authors would like to thank Dr Reddy’s laboratories Ltd and Andhra University for providing lab facility and required guidance to complete the work.
FINANCIAL SUPPORT
None.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
REFERENCES
Amr MB, Abd El-aziz BA, Abd El-Alim, Ahmed SS. Stability-indicating spectro-photometric methods for determination of Clobetasol propionate in the presence of its alkaline degradation product by derivative spectrophotometric techniques. Drug Test Anal, 2010; 2:130–6.
Angelo T, Cunha-Filho MS, Gelfuso GM, Gratieri T. Chromatographic method for clobetasol propionate determination in hair follicles and in different skin layers. Biomed Chromatogr, 2017; 31(2):1–7. CrossRef
Badilli U, Amasya G, Özkan S, Tarimci N. Simultaneous determination of clobetasol propionate and calcipotriol in a novel fixed dose emulgel formulation by LC-UV. Chromatographia, 2013; 76:133–40. CrossRef
Bassuoni YF, Elzanfaly ES, Essam HM, Zaazaa HE. Development and validation of stability indicating TLC densitometric and spectrophotometric methods for determination of Clobetasol propionate. Bull Fac Pharm Cairo Univ, 2016; 54:165–74. CrossRef
Bhuyian M, Rashid DH, Islam A, Tareque M. Development and validation of method for determination of clobetasol propionate and salicylic acid from pharmaceutical dosage form by HPLC. Br J Pharm Res, 2015; 7:375–85. CrossRef
Fauzee AF, Walker RB. Forced degradation studies of clobetasol 17-propionate in methanol, propylene glycol, as bulk drug and cream formulations by RP-HPLC. J Sep Sci, 2013; 36:849–56. CrossRef
FDA. Available via https://www.fda.gov/downloads/Drugs/Guidances/UCM386366.pdf (Accessed 26 February 2019).
Fontana MC, Bastos MO, Beck R. Development and validation of a fast RP-HPLC method for the determination of clobetasol propionate in topical nanocapsule suspensions. J Chromatogr Sci, 2010; 48:637–40. CrossRef
Gagliardi L, De Orsi D, Manna F, Tonelli D. HPLC determination of clobetasol propionate in cosmetic products. J Liq Chromatogr Relat Technol, 2000; 23:355–62. CrossRef
Gordon ML. The role of clobetasol propionate emollient 0.05% in the treatment of patients with dry, scaly, corticosteroid-responsive dermatoses. J Clin Ther, 1988; 20:26–38. CrossRef
Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet, 2007; 370:263–71. CrossRef
ICH. Available via https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf (Accessed 26 February 2019).
Joseph JK, Stephanie AS, William LJ, Barry EB. Fused-core particle technology in high-performance liquid chromatography: an overview. J Pharm Anal, 2013; 3:303–12. CrossRef
Manoharan G. Development and validation of a stability-indicating RP-HPLC method for the estimation of clobetasol propionate in bulk and ointment dosage form. Eur J Pharm Med Res, 2017; 4:119–26.
Marika K, Katherine F, Jianmin L, Mike CG, Debra F. A sensitive high-throughput HPLC assay for simultaneous determination of everolimus and clobetasol propionate. J Chromatogr Sci, 2008; 46:23–9. CrossRef
Michael ES. UPLC: an introduction and review. J Liq Chromatogr Relat Technol, 2005; 28:1253–63. CrossRef
Mostafa AA, Bebawy LI, Refaat HH. Spectrophotometric determination of clobetasol propionate, halobetasol propionate, quinagolide hydrochloride, through charge transfer complexation. J Pharm Biomed Anal, 2002; 27:889–99. CrossRef
Mrunali RP, Rashmin BP, Jolly RP, Bharat GP. HPTLC method for estimation of Clobetasol propionate in topical gel formulations and in vitro study. Anal Methods, 2010; 2:275–81. CrossRef
Nam YS, Kwon IK, Lee KB. Monitoring of clobetasol propionate and betamethasone dipropionate as undeclared steroids in cosmetic products manufactured in Korea. Forensic Sci Int, 2011; 210:144–8. CrossRef
Neelam D, Sunil K, Sunny R, Jagbir G , Sheefali M, Rekha R. Development and validation of UV spectrophotometric method for quantitative estimation of clobetasol 17-propionate. Asian J Chem Pharm Sci, 2016; 1:36–40 CrossRef
Nidhi P, Pooja P, Dhananjay M. Development and validation for RP-HPLC method simultaneous estimation of nadifloxacin and clobetasol propionate in its pharmaceutical dosage form. Am J PharmTech Res, 2016; 6(5):289–304.
Reepmeyer JC, Revelle LK, Vidavsky I. Detection of clobetasol propionate as an undeclared steroid in zinc pyrithione formulations by high-performance liquid chromatography with rapid-scanning ultraviolet spectroscopy and mass spectrometry. J Chromatogr A, 1998; 828:239–46. CrossRef
Sparidans RW, van Velsen SGA, de Roos MP, Schellens JHM, Bruijnzeel-Koomen CA, Beijnen JH. Liquid chromatography-tandem mass spectrometric assay for clobetasol propionate in human serum from patients with atopic dermatitis. J Chromatogr B, 2010; 878:2150–4. CrossRef
Turabi ZM, Khatatbeh OA. Simultaneous determination of clobetasol (as propionate) and chlorocresol in cream by stability indicating RP-HPLC method. Int J Pharm Sci Drug Res, 2014; 6:140–4. CrossRef
Van Velsen SGA, De Roos MP, Haeck IM, Sparidans RW, Bruijnzeel-Koomen CA. The potency of clobetasol propionate: Serum levels of clobetasol propionate and adrenal function during therapy with 0.05% clobetasol propionate in patients with severe atopic dermatitis. J Dermatol Treat, 2012; 23:16–20. CrossRef
Wenling Y, Xiaomei Y, Fanghua S, Zhigang L, Yongkun L, Liangzhong Y, Ruixun W, Qing L, Kaishun B. Qualitative and quantitative assessment of related substances in the compound ketoconazole and clobetasol propionate cream by HPLC-TOF-MS and HPLC. J Pharm Anal, 2019; 9:156–162.
Whan BK, Dana J, Jensen Y. Diagnosis and management of psoriasis. Can Fam Physician, 2017; 63:278–85.