
INTRODUCTION
Cancer is a foxy disease that can start and progress without any warning. This, combined with the difficulty of its treatment, makes cancer a serious global health problem that can strike anyone at any time (Miller et al., 2016). Cancer is a condition in which the cellular monitoring of apoptosis is lost leading to uncontrolled proliferation of cells. This results in the formation of neoplastic tumor that starts benign until its progression to the malignancy as it metastasizes to other tissues or organs (Qiao et al., 2016).
The use of natural products in the treatment of various human diseases including cancer became an interesting area of research in the last decades (Rayan et al., 2017). Phytochemicals with antitumor activity can exert their effect through different mechanisms such as induction of apoptosis, inhibition of topoisomerase I or II, and modification of cellular metabolism (Demain and Vaishnav, 2011).
Phenolic compounds are a group of key secondary metabolites found mainly in the plant kingdom and they have a wide spectrum of physiological activities (Lin et al., 2016). One of the most interesting classes of such compounds is a hydroxycinnamic acids family, to which sinapic, p-coumaric, ferulic, vanillic, and caffeic acids belong (Goufo and Trindade, 2014). Sinapic acid (Fig. 1) is frequently found in human diet because of its high prevalence in many vegetables (e.g., white onion), fruits (e.g., lemon), cereal grains (e.g., rice), and herbs (e.g., borage). Like other members of its family, sinapic acid is naturally found in the free form or as a glycoside (Nićiforović and AbramoviÄ, 2014).
Many reports exploring in vivo and in vitro pharmacological properties of sinapic acid indicated that it has several effects including anti-inflammatory (Zhang et al., 2017), antioxidant (Chen, 2016), antibacterial (Kim et al., 2018), analgesic (Bhanuvalli et al., 2018), anxiolytic (Yoon et al., 2007), and antidiabetic (Cherng et al., 2013) effects. Until now, there is a limited data concerning the antitumor activity of sinapic acid. Based on literature, sinapic acid showed dose- and time-dependent cytotoxic effect against the following cancer cell lines: MDA MB 468, HBL 100 and T47D (breast), SW 480 and HT-29 (colon), HEp-2 (larynx), and HeLa (cervix) (Hameed et al., 2016; Kampa et al., 2004; Hudson et al., 2000; Janakiraman et al., 2014; Rosa et al., 2016).
The aim of this work is to synthesize sinapic acid and nine of its novel analogues starting from gastrodigenin (4-hydroxybenzyl alcohol) through simple multi-step synthetic routes and to study their preliminary cytotoxic effect on four cancer cell lines, which are: Michigan Cancer Foundation-7 (MCF-7) (breast), Adrenomyeloneuropathy 3 (AMN3) (murine mammary adenocarcinoma), Skala Koma Glasgow (SKG) (esophageal), and HeLa (cervix) utilizing 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) viability assay.
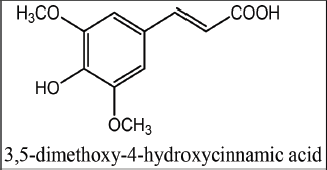 | Figure 1. Chemical structure of sinapic acid. [Click here to view] |
MATERIALS AND METHODS
Chemicals and solvents used in the synthesis were purchased from Tokyo Chemical Industry and Sigma-Aldrich while MTT stain (42000092-1) was obtained from Bio-World. Melting points were determined on an electrochemical CIA 9300 instrument and they were uncorrected. Analytical TLC was performed on silica gel Si 60 F254 plates produced by Merck, and the mobile phase comprised of CHCl3: acetone (4:1). IR spectra of the synthesized products were detected on Bruker-Alpha ATR while their UV spectra were determined on Varian UV/Visible spectrophotometer. 1H-NMR and 13C-NMR spectra were scanned on Bruker 500 MHz AVANCE III HD NMR Spectrometer using CDCl3 as a solvent and Tetramethylsilane (TMS) as an internal standard. Chemical shifts are recorded in parts per million (ppm), while coupling constants (J) are reported in hertz. The abbreviations used to describe the signals are: s (singlet), d (doublet), t (triplet), and m (multiplet). Shimadzu Liquid Chromatography Mass Spectrometry (LCMS-2020) Single Quadrupole Liquid Chromatograph Mass Spectrometer was used to measure the mass spectra, which are expressed as a mass-to-charge (m/z) ratio.
Synthesis
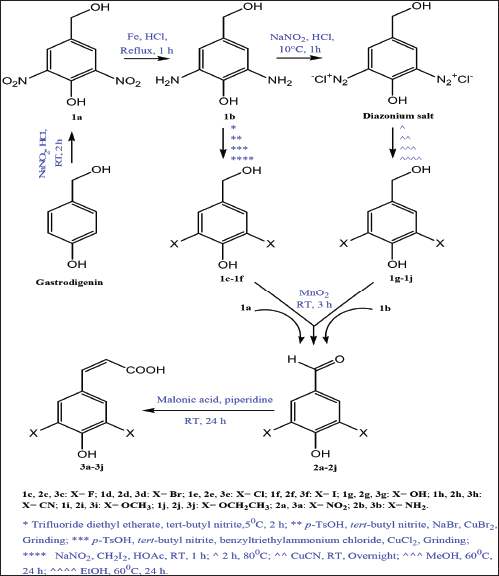
The synthetic pathway for the preparation of sinapic acid and its analogues is displayed in Scheme 1. IR spectral data of the intermediates (1a–1j), intermediates (2a–2j), and of the final products (3a–3j) are listed in Tables 1–3, respectively. The physical properties of the synthesized compounds are shown in Table 4.
Synthesis of 3,5-dinitrogastrodigenin (1a)
To an aqueous solution of gastrodigenin (10 mmol, 1.24 g), a solution of NaNO2 (25 mmol, 2.125 g) in 10-ml H2O was added dropwise. The resultant solution was acidified with 1-N HCl to acheive a pH value of 2 and then stirred for 2 hours at room temperature (RT). The crude product was extracted by CHCl3 (20 ml × 2) and the combined organic layer was dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure. The product was recrystallized from MeOH (Patnaik and Khoury, 2004).
4-Hydroxymethyl-2,6-dinitrophenol (1a) 1H-NMR (500 MHz) δ ppm: 7.42 (s, 2H, Ar H), 6.12 (s, 1H, Ar-OH), 4.60 (s, 2H, Ar-CH2), and 2.24 (s, 1H, CH2OH); 13C- NMR (125 MHz) δ ppm: 150 (Ar C-OH), 144 (Ar C-NO2), 141 (Ar C-CH2), 120 (Ar C), and 68 (Ar C-CH2); LC/MS (ESI) m/z: 215 [M + H]+.
Synthesis of 3,5-diaminogastrodigenin (1b)
A mixture of 1a (10 mmol, 2.14 g) in 25-ml EtOH and iron powder (37.5 mmol, 2.1 g) was heated to 60°C using a water bath; to this, concentrated HCl (10 ml) was added dropwise over 30 minutes with stirring. The reaction mixture was refluxed for 1 hours, poured into a cold H2O (250 ml); the resultant mixture was neutralized with 1-N NaOH and filtered. The crude product was extracted by ethyl acetate (25 ml × 2) and the combined organic layer was dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure. The product was recrystallized from a mixture of EtOH and H2O (Wakamatsu et al., 2014).
4-Hydroxymethyl-2,6-diaminophenol (1b) 1H-NMR (500 MHz) δ ppm: 7.36 (s, 2H, Ar H), 6.68 (s, 1H, Ar-OH), 4.58 (s, 2H, Ar-CH2), 4.16 (s, 4H, Ar-NH2), and 2.28 (s, 1H, CH2OH); 13C-NMR (125 MHz) δ ppm: 157 (Ar C-OH), 146 (Ar C-NH2), 140 (Ar C-CH2), 117 (Ar C), and 68 (Ar C-CH2); LC/MS (ESI) m/z: 155 [M + H]+.
Synthesis of the diazonium salt solution
A solution of 1b (10 mmol, 1.54 g) in a mixture of concentrated HCl (10 ml) and H2O (8 ml) was placed in a salt-ice bath. As the solution temperature dropped to 0°C, a cold solution of NaNO2 (24 mmol, 1.66 g) in 10-ml H2O was added dropwise with stirring and the reaction temperature was kept below 10°C. The resultant solution was stirred for 1 hour at that temperature and then used immediately in the next step of the synthetic route (Wang et al., 2017).
Synthesis of 3,5-difluorogastrodigenin (1c)
To a cold solution of 1b (10 mmol, 1.54 g) in 50-ml CH2Cl2, boron trifluoride diethyl etherate (30 mmol, 5 ml) was added and the reaction temperature was kept below 5°C. To this, tert-butyl nitrite (24 mmol, 2.2 ml) was slowly added from a graduated pipette with stirring in an ice bath. The reaction mixture was stirred for 2 hours at that temperature and the formed solid was filtered, washed with H2O and then with hexane. The crude product was re-dissolved in 60-ml CH2Cl2, refluxed for 24 hours, and filtered. The filtrate was evaporated under reduced pressure to afford the titled compound which was purified by recrystallization from a mixture of CHCl3: ether (1:2) (Garel and Saint-Jalmes, 2006).
4-Hydroxymethyl-2,6-difluorophenol (1c) 1H-NMR (500 MHz) δ ppm: 7.56 (s, 2H, Ar H), 6.66 (s, 1H, Ar-OH), 4.58 (s, 2H, Ar-CH2), and 2.34 (s, 1H, CH2OH); 13C-NMR (125 MHz) δ ppm: 164 (Ar C-F), 154 (Ar C-OH), 140 (Ar C-CH2), 118 (Ar C), and 66 (Ar C-CH2); LC/MS (ESI) m/z: 161 [M + H]+.
 | Scheme 1. General synthetic pathway of sinapic acid and its analogues. [Click here to view] |
Synthesis of 3,5-dibromogastrodigenin (1d)
To an agate mortar, compound 1b (10 mmol, 1.54 g), anhydrous p-TsOH (24 mmol, 4.12 g), tert-butyl nitrite (24 mmol, 2.2 ml), NaBr (24 mmol, 2.48 g), a catalytic amount of CuBr2, and a few drops of H2O were added and grinded. As the evolution of N2 finished, H2O (25 ml) was added and the crude product was extracted with CH2Cl2 (20 ml × 2). The combined organic layer was dried over anhydrous MgSO4, evaporated under reduced pressure, and the titled product was recrystallized from CHCl3 (Moon et al., 2010).
4-Hydroxymethyl-2,6-dibromophenol (1d) 1H-NMR (500 MHz) δ ppm: 7.70 (s, 2H, Ar H), 6.43 (s, 1H, Ar-OH), 4.51 (s, 2H, Ar-CH2), and 2.46 (s, 1H, CH2OH); 13C-NMR (125 MHz) δ ppm: 160 (Ar C-OH), 137 (Ar C-CH2), 126 (Ar C-Br), 117 (Ar C), and 61 (Ar C-CH2); LC/MS (ESI) m/z: 283 [M + H]+.
Synthesis of 3,5-dichlorogastrodigenin (1e)
The same method used for the synthesis of 1d was applied except that NaBr was replaced with benzyltriethylammonium chloride, CuBr2 was replaced with CuCl2, and the product recrystallized from a mixture of CHCl3: EtOH (2:1).
4-Hydroxymethyl-2,6-dichlorophenol (1e) 1H-NMR (500 MHz) δ ppm: 7.24 (s, 2H, Ar H), 6.34 (s, 1H, Ar-OH), 4.47 (s, 2H, Ar-CH2), and 2.41 (s, 1H, CH2OH); 13C-NMR (125 MHz) δ ppm: 153 (Ar C-OH), 134 (Ar C-CH2), 120 (Ar C-Cl), 115 (Ar C), and 60 (Ar C-CH2); LC/MS (ESI) m/z: 194 [M + H]+.
Synthesis of 3,5-diiodogastrodigenin (1f)
A mixture of 1b (10 mmol), NaNO2 (50 mmol, 3.45 g), CH2I2 (40 mmol, 35.6 ml) in a solvent system consisted of CH2Cl2 (10 ml) and H2O (10 ml) was stirred for 15 minutes. To this, HOAc (40 mmol, 2.52 ml) was slowly added in one portion, and then stirred for 1 hour at RT. The resulted mixture was concentrated under reduced pressure and then hexane (20 ml) was added to the residue. The precipitate was filtered, washed with hexane and then with water. The product was recrystallized from a mixture of EtOH and ether (Leas et al., 2017).
4-Hydroxymethyl-2,6-diiodophenol (1f) 1H-NMR (500 MHz) δ ppm: 7.56 (s, 2H, Ar H), 6.39 (s, 1H, Ar-OH), 4.83 (s, 2H, Ar-CH2), and 2.42 (s, 1H, CH2OH); 13C-NMR (125 MHz) δ ppm: 176 (Ar C-OH), 139 (Ar C-CH2), 124 (Ar C), 87 (Ar C-I), and 66 (Ar C-CH2); LC/MS (ESI) m/z: 377 [M + H]+.
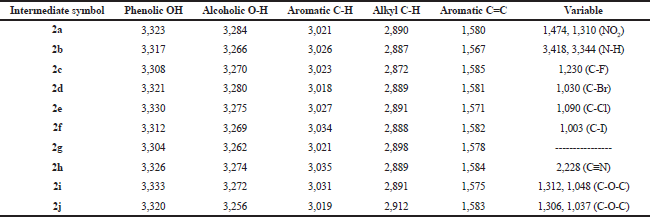 | Table 1. IR spectral data (ν, str., cm−1) of the intermediates (1a–1j). [Click here to view] |
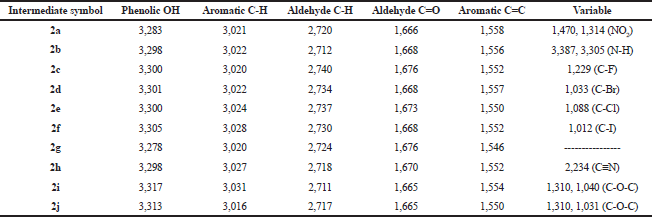 | Table 2. IR spectral data (ν, str., cm−1) of the intermediates (2a–2j). [Click here to view] |
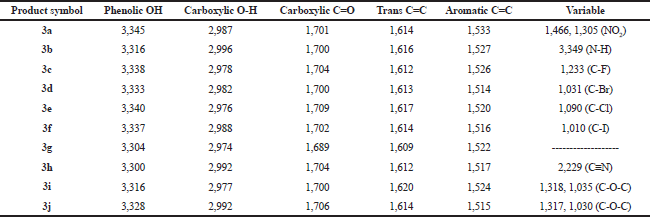 | Table 3. IR spectral data (ν, str., cm−1) of the final products (3a–3j). [Click here to view] |
Synthesis of 3,5-dihydroxygastrodigenin (1g)
The temperature of the previously prepared diazonium salt solution was gradually raised to RT and then the solution was heated for 2 hours at 80°C. The reaction mixture was left to cool and its pH was adjusted to 5 using 10% Na2CO3 solution. The crude yield was extracted with ethyl acetate (25 ml × 2) and the combined organic layer was dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure. The product was recrystallized from MeOH (Kazem-Rostami, 2017).
5-Hydroxymethyl-benzene-1,2,3-triol (1g) 1H-NMR (500 MHz) δ ppm: 7.14 (s, 2H, Ar H), 6.06 (s, 1H, Ar-OH), 5.38 (s, 2H, Ar-OH), 4.59 (s, 2H, Ar-CH2), and 2.15 (s, 1H, CH2OH); 13C-NMR (125 MHz) δ ppm: 152, 134 (Ar C-OH), 144 (Ar C-CH2), 111 (Ar C), and 72 (Ar C-CH2); LC/MS (ESI) m/z: 157 [M + H]+.
Synthesis of gastrodigenin3,5-dicarbonitrile (1h)
To a freshly prepared aqueous solution of cuprous cyanide (20 mmol), cold diazonium salt solution (10 mmol) was added stepwise in an ice bath. The resulted solution was stirred at 20°C for 40 minutes, at 70°C for 40 minutes and overnight at RT. The solid was filtered under vacuum and the product was recrystallized from a mixture of EtOH and CHCl3 (2:3) (Nielsen et al., 2004).
 | Table 4. Physical properties of the intermediates (1a–1j and 2a–2j) and of the final products (3a–3j). [Click here to view] |
2-Hydroxy-5-hydroxymethyl-isophthalonitrile (1h) 1H-NMR (500 MHz) δ ppm: 7.63 (s, 2H, Ar H), 6.45 (s, 1H, Ar-OH), 4.88 (s, 2H, Ar-CH2), and 2.14 (s, 1H, CH2OH); 13C-NMR (125 MHz) δ ppm: 166 (Ar C-OH), 148 (Ar C-CH2), 124 (Ar-CN), 118 (Ar C), 102 (Ar C-CN), and 72 (Ar C-CH2); LC/MS (ESI) m/z: 175 [M + H]+.
Synthesis of 3,5-dimethoxygas trodigenin (1i)
To a cold solution of diazonium salt (10 mmol), absolute MeOH (50 ml) was added and then stirred for 24 hours at 60°C. The crude yield was extracted with ethyl acetate (20 ml × 2), dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. The product was recrystallized from a mixture of CHCl3 and ether (3:1) (Shriver et al., 2009).
4-Hydroxymethyl-2,6-dimethoxyphenol (1i) 1H-NMR (500 MHz) δ ppm: 7.32 (s, 2H, Ar H), 6.40 (s, 1H, Ar-OH), 5.06 (s, 2H, Ar-CH2), 3.95 (s, 6H, OCH3), and 2.26 (s, 1H, CH2OH); 13C-NMR (125 MHz) δ ppm: 162 (Ar C-OH), 150 (Ar C-OCH3), 132 (Ar C-CH2), 122 (Ar C), 72 (Ar C-CH2), and 58 (Ar-OCH3); LC/MS (ESI) m/z: 185 [M + H]+.
Synthesis of 3,5-diethoxygastrodigenin (1j)
The same method used for the synthesis of 1i was applied except that absolute MeOH was replaced with absolute EtOH and the product was recrystallized from ether.
4-Hydroxymethyl-2,6-diethoxyphenol (1j) 1H-NMR (500 MHz) δ ppm: 7.08 (s, 2H, Ar H), 6.24 (s, 1H, Ar-OH), 5.12 (s, 2H, Ar-CH2), 4.30 (m, 4H, J = 6.8 Hz, OCH2), 2.26 (s, 1H, CH2OH), and 1.56 (t, 6H, J = 6.8 Hz, CH3); 13C-NMR (125 MHz) δ ppm: 160 (Ar C-OH), 150 (Ar C-OCH2), 132 (Ar C-CH2), 110 (Ar C), 72 (Ar C-CH2), 64 (Ar-OCH2), and 18 (CH3); LC/MS (ESI) m/z: 213 [M + H]+.
General method for synthesis of syringaldehyde and its analogues (2a–2j)
A suspension prepared from 10 mmol of each of intermediates 1a–1j and MnO2 (4.35 g, 50 mmol) in 50-ml CHCl3 was stirred for 3 hours at RT, filtered through a plug of purified glass wool, and washed with warm CHCl3 (2 × 10 ml). The filtrate was concentrated to dryness under reduced pressure, re-dissolved in 30 ml acetone, and filtered. The solvent was evaporated and the final product was then recrystallized from EtOH (Uchiyama et al., 2000).
4-Hydroxy-3,5-dinitrobenzaldehyde (2a) 1H-NMR (500 MHz) δ ppm: 10.14 (s, 1H, Ar-CHO), 8.92 (s, 2H, Ar H), and 6.08 (s, 1H, Ar-OH); 13C-NMR (125 MHz) δ ppm: 193 (Ar-CHO), 152 (Ar C-OH), 143 (Ar C-NO2), 133 (Ar C-CHO), and 126 (Ar C); LC/MS (ESI) m/z: 213 [M + H]+.
4-Hydroxy-3,5-diaminobenzaldehyde (2b) 1H-NMR (500 MHz) δ ppm: 9.92 (s, 1H, Ar-CHO), 7.58 (s, 2H, Ar H), 6.68 (s, 1H, Ar-OH), and 4.12 (s, 4H, Ar-NH2); 13C-NMR (125 MHz) δ ppm: 190 (Ar-CHO), 164 (Ar C-OH), 146 (Ar C-NH2), 132 (Ar C-CHO), and 120 (Ar C); LC/MS (ESI) m/z: 153 [M + H]+.
4-Hydroxy-3,5-difluorobenzaldehyde (2c) 1H-NMR (500 MHz) δ ppm: 10.04 (s, 1H, Ar-CHO), 7.78 (s, 2H, Ar H), and 6.60 (s, 1H, Ar-OH); 13C-NMR (125 MHz) δ ppm: 187 (Ar-CHO), 158 (Ar C-F), 150 (Ar C-OH), 138 (Ar C-CHO), and 122 (Ar C); LC/MS (ESI) m/z: 159 [M + H]+.
4-Hydroxy-3,5-dibromobenzaldehyde (2d) 1H-NMR (500 MHz) δ ppm: 9.82 (s, 1H, Ar-CHO), 7.74 (s, 2H, Ar H), and 6.41 (s, 1H, Ar-OH); 13C-NMR (125 MHz) δ ppm: 192 (Ar-CHO), 165 (Ar C-OH), 137 (Ar C-CHO), 127 (Ar C-Br), and 119 (Ar C); LC/MS (ESI) m/z: 281 [M + H]+.
4-Hydroxy-3,5-dichlorobenzaldehyde (2e) 1H-NMR (500 MHz) δ ppm: 10.15 (s, 1H, Ar-CHO), 7.77 (s, 2H, Ar H), and 6.30 (s, 1H, Ar-OH); 13C-NMR (125 MHz) δ ppm: 197 (Ar-CHO), 160 (Ar C-OH), 133 (Ar C-CHO), 121 (Ar C-Cl), and 118 (Ar C); LC/MS (ESI) m/z: 192 [M + H]+.
4-Hydroxy-3,5-diiodobenzaldehyde (2f) 1H-NMR (500 MHz) δ ppm: 9.95 (s, 1H, Ar-CHO), 7.78 (s, 2H, Ar H), and 6.37 (s, 1H, Ar-OH); 13C-NMR (125 MHz) δ ppm: 191 (Ar-CHO),182 (Ar C-OH), 133 (Ar C-CHO), 127 (Ar C), and 89 (Ar C-I); LC/MS (ESI) m/z: 375 [M + H]+.
3,4,5-Trihydroxybenzaldehyde (2g) 1H-NMR (500 MHz) δ ppm: 10.18 (s, 1H, Ar-CHO), 7.54 (s, 2H, Ar H), 6.38 (s, 1H, Ar-OH), and 5.62 (s, 2H, Ar-OH); 13C-NMR (125 MHz) δ ppm: 198 (Ar-CHO), 155, 140 (Ar C-OH), 138 (Ar C-CHO), and 116 (Ar C); LC/MS (ESI) m/z: 155 [M + H]+.
5-Formyl-2-hydroxy-isophthalonitrile (2h) 1H-NMR (500 MHz) δ ppm: 10.12 (s, 1H, Ar-CHO), 7.98 (s, 2H, Ar H), and 6.22 (s, 1H, Ar-OH); 13C-NMR (125 MHz) δ ppm: 193 (Ar-CHO), 177 (Ar C-OH), 141 (Ar C-CHO), 124 (Ar-CN), 121 (Ar C), and 101 (Ar C-CN); LC/MS (ESI) m/z: 173 [M + H]+.
4-Hydroxy-3,5-dimethoxybenzaldehyde (2i) 1H-NMR (500 MHz) δ ppm: 9.92 (s, 1H, Ar-CHO), 7.78 (s, 2H, Ar H), 6.38 (s, 1H, Ar-OH), and 3.98 (s, 6H, OCH3); 13C-NMR (125 MHz) δ ppm: 190 (Ar-CHO), 158 (Ar C-OH), 150 (Ar C-OCH3), 134 (Ar C-CHO), 126 (Ar C), and 58 (Ar-OCH3); LC/MS (ESI) m/z: 183 [M + H]+.
4-Hydroxy-3,5-diethoxybenzaldehyde (2j) 1H-NMR (500 MHz) δ ppm: 9.88 (s, 1H, Ar-CHO), 7.80 (s, 2H, Ar H), 6.22 (s, 1H, Ar-OH), 4.25 (m, 4H, J = 6.8 Hz, OCH2), and 1.60 (t, 6H, J = 6.8 Hz, CH3); 13C-NMR (125 MHz) δ ppm: 190 (Ar-CHO), 158 (Ar C-OH), 145 (Ar C-OCH2), 127 (Ar C-CHO), 113 (Ar C), 65 (Ar-OCH2), and 16 (CH3); LC/MS (ESI) m/z: 211 [M + H]+.
General method for synthesis of sinapic acid and its analogues (3a–3j)
To a solution prepared from 10 mmol of each of intermediates 2a–2j in 10-ml pyridine, mixture of malonic acid (20 mmol, 2.08 g) and piperidine (2.2 mmol, 0.2 ml) in 10-ml pyridine was added. The reaction mixture was stirred for 24 hours at RT and to this, concentrated HCl (6.6 ml) and then H2O (100 ml) were added sequentially. The crude product was filtered and then recrystallized from CHCl3 (Zhang et al., 2017).
3,5-Dinitro-4-hydroxycinnamic acid (3a) 1H-NMR (500 MHz) δ ppm: 12.69 (s, 1H, COOH), 8.48 (s, 2H, Ar H), 7.58 (d, 1H, J = 16.6 Hz, Ar-CH=CH), 6.62 (d, 1H, J = 16.6 Hz, Ar-CH=CH), and 6.12 (s, 1H, Ar-OH); 13C-NMR (125 MHz) δ ppm: 174 (COOH), 148 (Ar C-OH), 140 (Ar C-CH=), 138 (Ar C-CH=), 133 (Ar C-NO2), 120 (Ar C), and 108 (Ar-CH=CH); LC/MS (ESI) m/z: 255 [M + H]+.
3,5-Diamino-4-hydroxycinnamic acid (3b) 1H-NMR (500 MHz) δ ppm: 12.36 (s, 1H, COOH), 7.72 (d, 1H, J = 16.2 Hz, Ar-CH=CH), 7.08 (s, 2H, Ar H), 6.54 (d, 1H, J = 16.2 Hz, Ar-CH=CH), 6.16 (s, 1H, Ar-OH), and 4.00 (s, 4H, Ar-NH2); 13C-NMR (125 MHz) δ ppm: 170 (COOH), 151 (Ar C-OH), 140 (Ar C-NH2), 137 (Ar C-CH=), 131 (Ar C-CH=), 123 (Ar C), and 101 (Ar-CH=CH); LC/MS (ESI) m/z: 195 [M + H]+.
3,5-Difluoro-4-hydroxycinnamic acid (3c) 1H-NMR (500 MHz) δ ppm: 12.38 (s, 1H, COOH), 7.74 (d, 1H, J = 16.4 Hz, Ar-CH=CH), 7.16 (s, 2H, Ar H), 6.68 (d, 1H, J = 16.2 Hz, Ar-CH=CH), and 6.23 (s, 1H, Ar-OH); 13C-NMR (125 MHz) δ ppm: 172 (COOH), 155 (Ar C-F), 143 (Ar C-OH), 140 (Ar C-CH=), 126 (Ar C-CH=), 120 (Ar C), and 109 (Ar-CH=CH); LC/MS (ESI) m/z: 201 [M + H]+.
3,5-Dibromo-4-hydroxycinnamic acid (3d) 1H-NMR (500 MHz) δ ppm: 12.32 (s, 1H, COOH), 7.70 (d, 1H, J = 16.9 Hz, Ar-CH=CH), 7.18 (s, 2H, Ar H), 6.59 (d, 1H, J = 16.9 Hz, Ar-CH=CH), and 6.38 (s, 1H, Ar-OH); 13C-NMR (125 MHz) δ ppm: 176 (COOH), 166 (Ar C-OH), 150 (Ar C-CH=), 129 (Ar C-Br), 126 (Ar C-CH=), 120 (Ar C), and 110 (Ar-CH=CH); LC/MS (ESI) m/z: 323 [M + H]+.
3,5-Dichloro-4-hydroxycinnamic acid (3e) 1H-NMR (500 MHz) δ ppm: 12.35 (s, 1H, COOH), 7.71 (d, 1H, J = 16.1 Hz, Ar-CH=CH), 7.23 (s, 2H, Ar H), 6.62 (d, 1H, J = 16.1 Hz, Ar-CH=CH), and 6.29 (s, 1H, Ar-OH); 13C-NMR (125 MHz) δ ppm: 170 (COOH), 162 (Ar C-OH), 153 (Ar C-CH=), 135 (Ar C-CH=), 126 (Ar C-Cl), 118 (Ar C), and 108 (Ar-CH=CH); LC/MS (ESI) m/z: 234 [M + H]+.
3,5-Diiodo-4-hydroxycinnamic acid (3f) 1H-NMR (500 MHz) δ ppm: 12.38 (s, 1H, COOH), 7.72 (d, 1H, J = 16.4 Hz, Ar-CH=CH), 7.27 (s, 2H, Ar H), 6.56 (d, 1H, J = 16.4 Hz, Ar-CH=CH), and 6.38 (s, 1H, Ar-OH); 13C-NMR (125 MHz) δ ppm: 175 (Ar C-OH), 170 (COOH), 154 (Ar C-CH=), 132 (Ar C-CH=), 124 (Ar C), 111 (Ar-CH=CH), and 88 (Ar C-I); LC/MS (ESI) m/z: 417 [M + H]+.
3,4,5-Trihydroxycinnamic acid (3g) 1H-NMR (500 MHz) δ ppm: 12.09 (s, 1H, COOH), 7.70 (d, 1H, J = 16.2 Hz, Ar-CH=CH), 7.23 (s, 2H, Ar H), 6.55 (d, 1H, J = 16.2 Hz, Ar-CH=CH), 6.32 (s, 1H, Ar-OH), and 5.61 (s, 2H, Ar-OH); 13C-NMR (125 MHz) δ ppm: 168 (COOH), 159, 147 (Ar C-OH), 150 (Ar C-CH=), 133 (Ar C-CH=), 127 (Ar C), and 112 (Ar-CH=CH); LC/MS (ESI) m/z: 197 [M + H]+.
3,5-Dicyano-4-hydroxycinnamic acid (3h) 1H-NMR (500 MHz) δ ppm: 12.63 (s, 1H, COOH), 7.83 (s, 2H, Ar H), 7.72 (d, 1H, J = 16.8 Hz, Ar-CH=CH), 6.65 (d, 1H, J = 16.8 Hz, Ar-CH=CH), and 6.24 (s, 1H, Ar-OH); 13C-NMR (125 MHz) δ ppm: 172 (COOH), 167 (Ar C-OH), 150 (Ar C-CH=), 140 (Ar C), 131 (Ar C-CH=), 123 (Ar-CH=CH), 118 (Ar-CN), and 102 (Ar C-CN); LC/MS (ESI) m/z: 215 [M + H]+.
3,5-Dimethoxy-4-hydroxycinnamic acid (Sinapic acid, 3i) 1H-NMR (500 MHz) δ ppm: 12.46 (s, 1H, COOH), 7.69 (d, 1H, J = 16.4 Hz, Ar-CH=CH), 7.21 (s, 2H, Ar H), 6.66 (d, 1H, J = 16.4 Hz, Ar-CH=CH), 6.38 (s, 1H, Ar-OH), and 3.94 (s, 6H, OCH3); 13C-NMR (125 MHz) δ ppm: 170 (COOH), 162 (Ar C-OH), 153 (Ar C-OCH3), 147 (Ar C-CH=), 136 (Ar C-CH=), 121 (Ar C), 115 (Ar-CH=CH), and 58 (Ar-OCH3); LC/MS (ESI) m/z: 225 [M + H]+.
3,5-Diethoxy-4-hydroxycinnamic acid (3j) 1H-NMR (500 MHz) δ ppm: 12.46 (s, 1H, COOH), 7.74 (d, 1H, J = 16.3 Hz, Ar-CH=CH), 7.23 (s, 2H, Ar H), 6.74 (d, 1H, J = 16.3 Hz, Ar-CH=CH), 6.20 (s, 1H, Ar-OH), 4.26 (m, 4H, J = 7.0 Hz, OCH2), and 1.43 (t, 6H, J = 7.0 Hz, CH3); 13C-NMR (125 MHz) δ ppm: 170 (COOH), 155 (Ar C-OH), 149 (Ar C-OCH2), 142 (Ar C-CH=), 138 (Ar C-CH=), 109 (Ar C), 69 (Ar-OCH2), and 16 (CH3); LC/MS (ESI) m/z: 253 [M + H]+.
Preliminary cytotoxicity study
Cytotoxicity study was conducted according to the method reported by Mustafa et al. (2018), which involved the use of the MTT assay to test the ability of the synthesized compounds to induce cytotoxicity utilizing 5-fluorouracil and Dimethyl sulfoxide (DMSO) as positive and negative controls, respectively (Mustafa et al., 2018). This test was applied on the following cancer cell lines: MCF-7 (breast), AMN3 (murine mammary adenocarcinoma), SKG (esophageal), and HeLa (cervix).
RESULTS AND DISCUSSION
Synthesis
The synthesis of sinapic acid and its analogues starting from gastrodigenin was carried out through six successive synthetic steps as shown in Scheme 1. The nitration of gastrodigenin afforded compound 1a, which was subject to Bechamp reduction reaction to form compound 1b. The amino groups of 1b were diazonated, and the resultant diazonium salt compound was transformed via Sandmeyer reaction to give compounds 1c–1h. Aryl ethers containing compounds 1i and 1j were prepared via nucleophilic aromatic substitution reaction of the diazonium salt with MeOH and EtOH, respectively.
The selective oxidation of primary hydroxyl group found in compounds 1a–1j using MnO2 afforded compounds 2a–2j, which were coupled with malonic acid in the presence of organic base to form compounds 3a–3j in a good yield.
Compounds 1a–1e, 1h, 1j, 2b, 2h, 2j, 3a–3h, and 3j are novel. Compounds 1f, 1g, 1i, 2c, and 2e–2g are available commercially, but there are no methods in the literature for their synthesis. There are methods for the preparation of compounds 2a, 2d, and 2i, but those used in this work are different. Finally, only compound 3i has an established synthetic method which was utilized in this work (Zhang et al., 2017).
In vitro cytotoxicity study
Sinapic acid and its synthesized analogues were scanned for their preliminary cytotoxic activity utilizing MTT test against four cancer cell lines, which are MCF-7 (breast), AMN3 (murine mammary adenocarcinoma), SKG (esophageal), and HeLa (cervix). Eight serial concentrations (3.125, 6.25, 12.5, 25, 50, 100, 200, and 400 μg/ml) of the tested compounds, with DMSO as a negative control and 5-fluorouracil as a positive control were applied in this assay. The results presented in Table 5 indicate that sinapic acid analogues 3a, 3c, 3d, 3e, and 3h have IC50 values lower than that of 5-fluorouracil against the tested cancer cell lines. Also, the results revealed that sinapic acid itself and the remaining synthesized analogues have IC50 values higher than that of positive control. Accordingly, compounds 3a, 3c, 3d, 3e, and 3h can be considered as potential cytotoxic agents.
CONCLUSION
Sinapic acid and nine of its analogues were synthesized starting from gastrodigenin through multi-step synthetic route. The chemical structures of the intermediates and of the final products were characterized by analyzing their IR, 1H-NMR, 13C-NMR, and MS-ESI spectra. The preliminary cytotoxic effect of sinapic acid and its synthesized analogues was tested on MCF-7, AMN3, SKG, and HeLa cancer cell lines using MTT viability assay. The results indicated that sinapic acid analogues 3a, 3c, 3d, 3e, and 3h have IC50 values lower than that of 5-fluorouracil on the tested cancer cell lines, while sinapic acid itself and the other synthesized analogues did not have such cytotoxic effect. With an exception of iodide may be due to its high atomic mass, it is concluded that the presence of electron-withdrawing groups in ortho positions to phenolic hydroxyl group of sinapic acid may improve its cytotoxic activity.
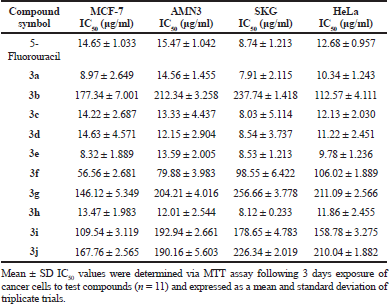 | Table 5. Mean ± SD IC50 values of 5-Fluorouracil as a positive control, sinapic acid and its analogues against MCF-7, AMN3, SKG, and HeLa cancer cell lines. [Click here to view] |
The synthetic steps utilised in this work were easy to apply and successful in yielding the required products with sufficient purity as indicated by their analysis spectra. The simple synthesis did not prevent these compounds from having good cytotoxic activity as shown by the results of the MTT assay. These results revealed that sinapic acid analogues 3a, 3c, 3d, 3e, and 3h have IC50 values lower than that of 5- fluorouracil on the tested cancer cell lines. Therefore, these compounds can be used as a starting point for cytotoxic agent development.
CONFLICT OF INTERESTS
The authors declare that they have no conflicts of interest.
FINANCIAL SUPPORT AND SPONSORSHIP
None.
REFERENCES
Bhanuvalli RS, Lotha R, Sivasubramanian A. Phenyl propanoid rich extract of edible plant Halosarcia indica exert diuretic, analgesic, and anti-inflammatory activity on Wistar albino rats. Nat Prod Res, 2018; 3:1–5.
Chen C. Sinapic acid and its derivatives as medicine in oxidative stress-induced diseases and aging. Oxid Med Cell Longev, 2016; 2016:3571614.
Cherng YG, Tsai CC, Chung HH, Lai YW, Kuo SC, Cheng JT. Antihyperglycemic action of sinapic acid in diabetic rats. J Agric Food Chem, 2013; 61(49):12053–9.
Demain AL, Vaishnav P. Natural products for cancer chemotherapy. Microb Biotechnol, 2011; 4(6):687–99.
Garel L, Saint-Jalmes L. One-pot fluoro-de-diazoniation of anilines in organic medium. Tetrahedron Lett, 2006; 47:5705–8.
Goufo P, Trindade H. Rice antioxidants: phenolic acids, flavonoids, anthocyanins, proanthocyanidins, tocopherols, tocotrienols, γ-oryzanol, and phytic acid. Food Sci Nutr, 2014; 2(2):75–104.
Hameed H, Aydin S, Basaran AA, Basaran N. Assessment of cytotoxic properties of sinapic acid in vitro. Turk J Pharm Sci, 2016; 13:225–32.
Hudson EA, Dinh PA, Kokubun T, Simmonds MS, Gescher A. Characterization of potentially chemopreventive phenols in extracts of brown rice that inhibit the growth of human breast and colon cancer cells. Cancer Epidemiol Biomarkers Prev, 2000; 9(11):1163–70.
Janakiraman K, Kathiresan S, Mariadoss AV. Influence of sinapic acid on induction of apoptosis in human laryngeal carcinoma cell line. Int J Modn Res Revs, 2014; 2(5):165–170. Available via www.journalijmrr.com
Kampa M, Alexaki VI, Notas G, Nifli AP, Nistikaki A, Hatzoglou A, Bakogeorgou E, Kouimtzoglou E, Blekas G, Boskou D, Gravanis A. Antiproliferative and apoptotic effects of selective phenolic acids on T47D human breast cancer cells: potential mechanisms of action. Breast Cancer Res, 2004; 6(2):R63–74.
Kazem-Rostami M. Facile preparation of Λ-shaped building blocks: Hünlich base derivatization. Synlett, 2017; 28:1641–5; doi:10.1055/s-0036-1588180.
Kim G, Dasagrandhi C, Kang EH, Eom SH, Kim YM. In vitro antibacterial and early stage biofilm inhibitory potential of an edible chitosan and its phenolic conjugates against Pseudomonas aeruginosa and Listeria monocytogenes. 3 Biotech, 2018; 8(10):439.
Leas DA, Dong Y, Vennerstrom JL, Stack DE. One-pot, metal-free conversion of anilines to aryl bromides and iodides. Org Lett, 2017; 19(10):2518–21.
Lin D, Xiao M, Zhao J, Li Z, Xing B, Li X, Kong M, Li L, Zhang Q, Liu Y, Chen H. An overview of plant phenolic compounds and their importance in human nutrition and management of Type 2 diabetes. Molecules, 2016; 21(10):E1374.
Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin, 2016; 66(4):271–89.
Moon ME, Choi Y, Lee YM, Vajpayee V, Trusova M, Filimonov VD, Chi KW. An expeditious and environmentally benign preparation of aryl halides from aryl amines by solvent-free grinding. Tetrahedron Lett, 2010; 51:6769–71.
Mustafa YF, Najem MA, Tawffiq ZS. Coumarins from Creston apple seeds: Isolation, chemical modification, and cytotoxicity study. J Appl Pharm Sci, 2018; 8(08):049–56.
Nićiforović N, AbramoviÄ H. Sinapic acid and its derivatives: Natural sources and bioactivity. Compr Rev Food Sci Food Safety, 2014; 13(1):34–51; doi:10.1111/1541-4337.12041.
Nielsen MA, Nielsen MK, Pittelkow T. Scale-up and safety evaluation of a sandmeyer reaction. Org Proc Res Dev, 2004; 8:1059–64. Available via https://pubs.acs.org/doi/pdfplus/10.1021/op0498823.
Patnaik P, Khoury JN. Reaction of phenol with nitrite ion: pathways of formation of nitrophenols in environmental waters. Water Res, 2004; 38(1):206–10.
Qiao A, Gu F, Guo X, Zhang X, Fu L. Breast cancer-associated fibroblasts: their roles in tumor initiation, progression and clinical applications. Front Med, 2016; 10(1):33–40.
Rayan A, Raiyn J, Falah M. Nature is the best source of anticancer drugs: Indexing natural products for their anticancer bioactivity. PLoS One, 2017; 12(11):e0187925.
Rosa LS, Silva NJA, Soares NCP, Monteiro MC, Teodoro AJ. Anticancer properties of phenolic acids in colon cancer: a review. J Nutr Food Sci, 2016; 6:468; doi:10.4172/2155-9600.1000468.
Shriver JA, Flaherty DP, Herr CC. Aryl ethers from arenediazonium tetrafluoroborate salts: from neat reactions to solvent-mediated effects. J Iowa Acad Sci, 2009: 116(1–4):Article 6. Available via https://scholarworks.uni.edu/jias/vol116/iss1/6.
Uchiyama M, Kimura Y, Ohta A. Stereoselective total syntheses of (±)-arthrinone and related natural compounds. Tetrahedron Lett, 2000; 41:10013–7.
Wakamatsu K, Tanaka H, Tabuchi K, Ojika M, Zucca FA, Zecca L, Ito S. Reduction of the nitro group to amine by hydroiodic acid to synthesize o-aminophenol derivatives as putative degradative markers of neuromelanin. Molecules, 2014; 19(6):8039–50.
Wang H, Xu Q, Shen S, Yu S. Synthesis of quinolines through three-component cascade annulation of aryl diazonium salts, nitriles, and alkynes. J Org Chem, 2017; 82(1):770–5.
Yoon BH, Jung JW, Lee JJ, Cho YW, Jang CG, Jin C, Oh TH, Ryu JH. Anxiolytic-like effects of sinapic acid in mice. Life Sci, 2007; 81(3):234–40.
Zhang Q, Hu JX, Kui X, Liu C, Zhou H, Jiang X, Zeng L. Sinapic acid derivatives as potential anti-Inflammatory agents: Synthesis and biological evaluation. Iran J Pharm Res, 2017; 16(4):1405–14.
SUPPLEMENTARY MATERIALS
 | The IR spectrum of compound 3a [Click here to view] |
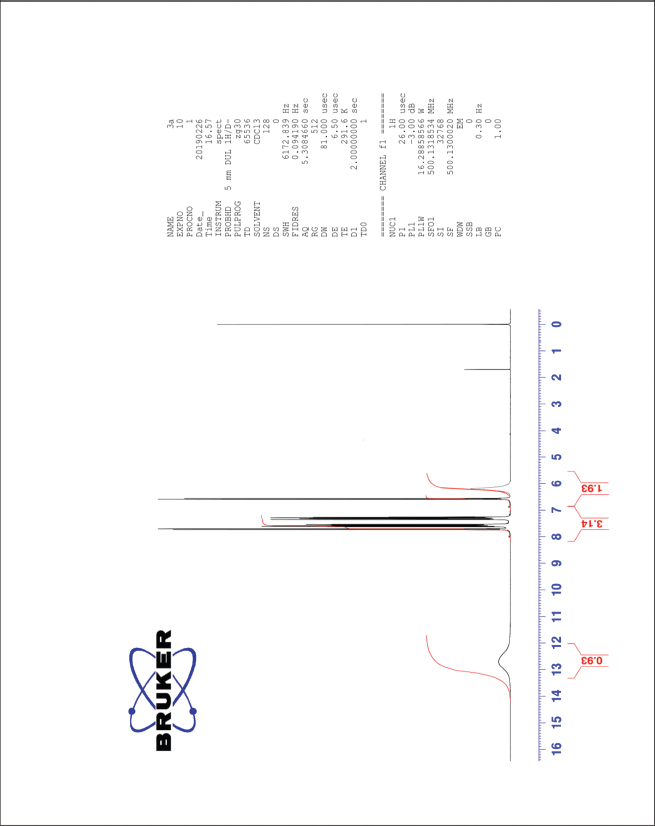 | The 1H-NMR spectrum of compound 3a [Click here to view] |
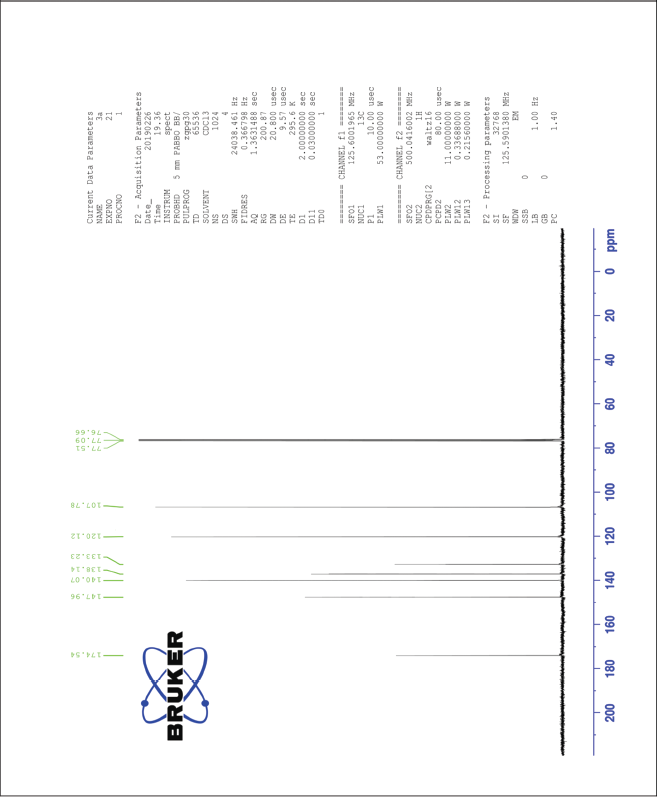 | The 13C-NMR spectrum of compound 3a [Click here to view] |
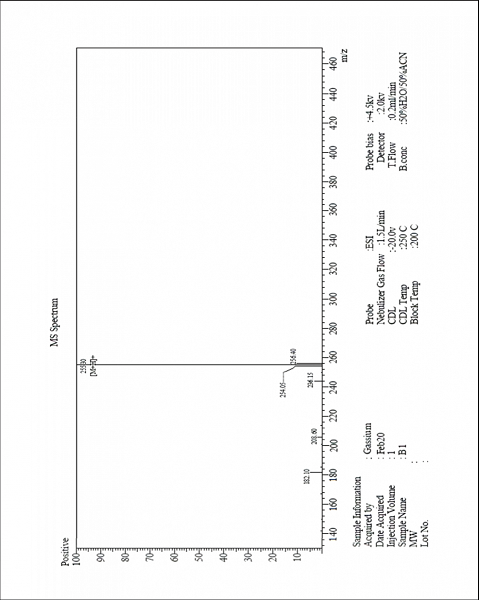 | MS-ESI spectrum (m/z) of the compound 3a operated in a positive mode [Click here to view] |
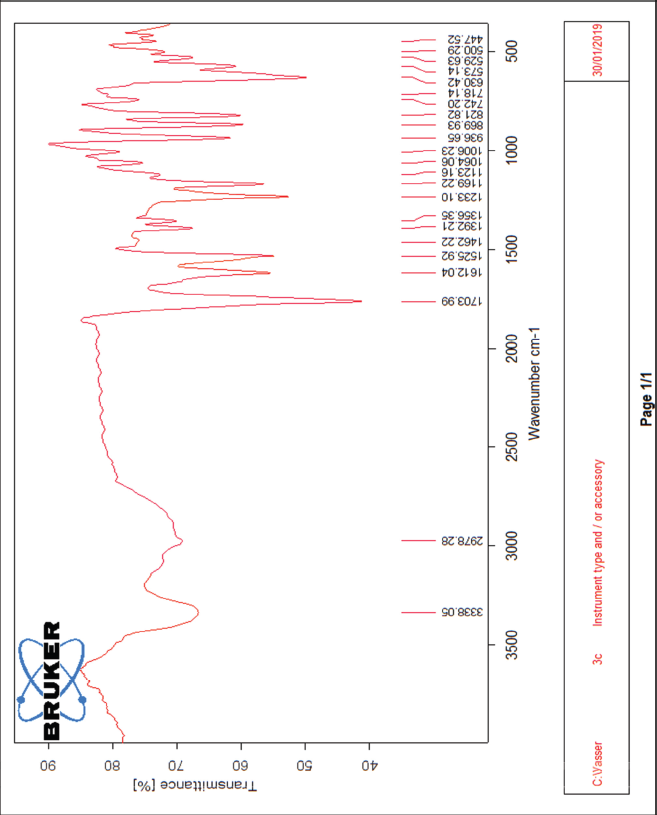 | The IR spectrum of compound 3c [Click here to view] |
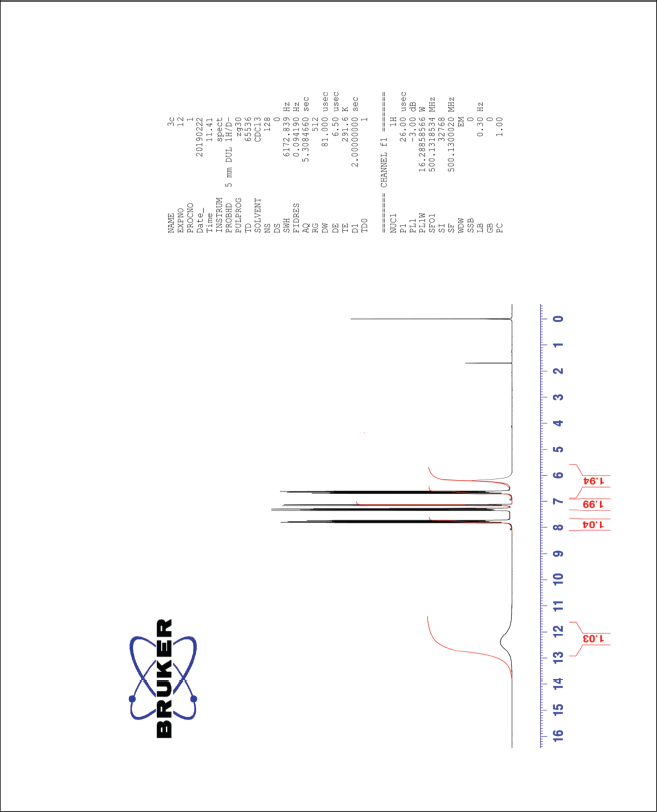 | The 1H-NMR spectrum of compound 3c [Click here to view] |
.png) | The 13C-NMR spectrum of compound 3c [Click here to view] |
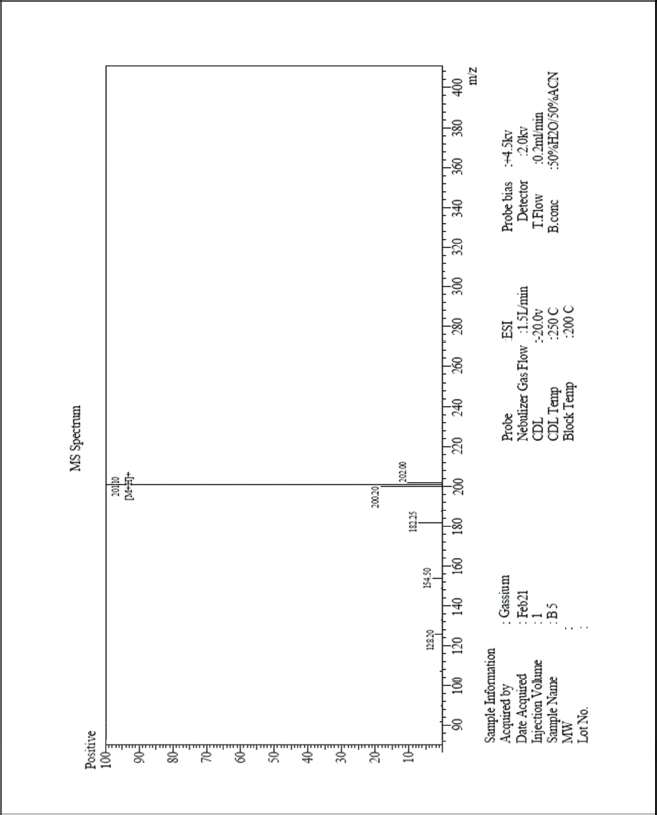 | MS-ESI spectrum (m/z) of the compound 3c operated in a positive mode [Click here to view] |