
INTRODUCTION
Over the last two decades, funding from international donors, increased political will, and international trade flexibilities have contributed towards strengthened HIV and AIDS programs and improved access to antiretroviral (ARV) medicines in sub-Saharan Africa. Regrettably, market proliferation with ARVs has met with an influx of poor quality generics, which threaten to undermine country program gains accrued (WHO, 2007). South Africa’s (SA) HIV epidemic is amongst the largest in the world (UNAIDS, 2012) and by 2015, the ARV program provided treatment for nearly 3.4 million people (mainly) in the public sector (UNAIDS, 2016). Moreover, the country’s adoption of a universal test and treat and pre-exposure prophylaxis policy in 2016 has resulted in further expansion of the program (DOH, 2016).
In 2013, SA recommended a generic fixed-dose combination of emtricitabine (FTC) (200 mg), tenofovir (TDF) (300 mg), and efavirenz (EFV) (600 mg) as the first-line treatment (once daily dose) for HIV in adults (DOH, 2013) to reduce the pill and supply burden. As of 2017, four of these generic combinations were supplied on public tender by local, licensed pharmaceutical companies. While the generic finished pharmaceutical products (FPP) and the innovator, Atripla®, are registered by the South African Health Products Regulatory Authority (SAHPRA) based on bioequivalence and in vitro quality data, there is uncertainty about the level of surveillance of these products after their market authorization, apart from port clearance controls and routine inspections of companies against Good Manufacturing Practice and Good Distribution Practices (GDP) standards. The demand for new ARV fixed-dose combinations (FDCs) has seen many national regulatory authorities adopt an accelerated approval process at a pace that exceeds the development of pharmacopoeial monographs for these combination medicines. As such, approval is based on available compendial specifications for the individual ingredients or existing combinations containing two of the three ingredients.
Post-market quality control involves routine quality testing of products at various levels of the supply chain, as well as voluntary reporting and testing of suspected poor quality medicines by healthcare workers and trained personnel, respectively. This quality assurance undertaking for thousands of products on the market requires intensive financial and physical investment, which poses a challenge for South Africa’s stringent, yet resource constrained, regulatory authority, where surveillance of quality, though legislated, is generally reactionary to reported quality complaints (Lehmann et al., 2018a; Maigetter et al., 2015; Patel et al., 2012). In response to reports of quality defects, SAHPRA submits suspect samples to independent WHO-accredited laboratories for identity and content assays and if the substandard quality is confirmed, an investigation (including supplier audit) and possible medicine recall ensues.
Generic companies are the main suppliers of first-line ARV FDCs to the global market (Chien, 2007). A generic formulation typically includes pre-formulation studies to characterize aqueous solubility and particle size distribution of the active pharmaceutical ingredient (API), as well as API to API and API-excipient compatibility (Aulton and Taylor, 2017). The inherent complexities of mixing two or three APIs in an FDC increase the risk of manufacturing challenges, instability, and consequent bioequivalence issues (EMA, 2007; WHO, 2003). To the extent described in the summary of product characteristics of the originator FPP, the generic manufacturer typically uses similar excipients in the generic FPP. However, excipient grades and amounts, as well as the method of manufacture may differ and could potentially lead to poor quality FPPs.
Poor quality medicines can be classified as falsified (deliberate mislabeling with respect to source, ingredients, or quantities), substandard (authorized for use but fails to meet specifications or quality standards), and unregistered (not authorized for use by National Regulatory Authority) (WHO, 2017). The distinction is important in developing strategic interventions to circumvent their supply and consumption (Newton et al., 2010). There are several reports of falsified ARV combinations on the market of several African countries assisted by USAID (Primo-Carpenter and McGinnis, 2008). Fewer accounts of substandard ARVs are documented; in a WHO survey to assess the quality of ARVs either as single or multiple compound preparations in various African countries, the percentage of “out of specification” medicine was very low (1.8%). Interestingly, three ARV FDC products failed either dissolution, disintegration, or content assays, in spite of being WHO-prequalified1 products (WHO, 2007). This underscores the need for routine post-market surveillance of products irrespective of their authorization for in-country use. Contrastingly, a post-market quality study conducted on ARV combination products sampled at various levels of the supply chain in Cameroon confirmed the quality of these WHO-prequalified products by in vitro testing and those sourced from local drug stores in Nigeria also met desired specifications (Djobet et al., 2017; Joshi et al., 2010).
This quantitative cross-sectional study compared the quality profile of four generics combinations containing EFV (600 mg), FTC (200 mg), and TDF (300 mg) in the tablet dosage form against the innovator product (Atripla®) and according to the only available monograph for this FDC published in the International Pharmacopoeia (IP) (WHO, 2016a). To date, no studies have assessed the post-market quality of generic FDCs containing these three ingredients and this paper sheds light on the quality of products at the distribution level of a relatively well-regulated market.
MATERIALS AND METHODS
Materials
Acetonitrile [99.9% high-performance liquid chromatography (HPLC) gradient grade], methanol (99.8% HPLC grade), sodium dodecyl sulfate, sodium dihydrogen orthophosphate monohydrate, and potassium phosphate dibasic were purchased from Merck, Germany and South Africa. Fumaric acid was obtained from Sigma Aldrich, SA. Primary reference standards (RS) for EFV (99.8% w/w), FTC (99.7% w/w), and TDF (98.8% w/w) were sourced from the European Directorate for the Quality of Medicines and Healthcare, France and stored at 5°C ± 3°C. High purity de-ionized water was obtained from the Milli-RO 4 water purification system, USA.
Sampling
Four generics on the public sector tender for the period 2015–2018 were sourced from a provincial depot (with recently updated Medicines Registration Authority licensure, confirming compliance with GDP) in one of South Africa’s nine geographical provinces. The originator FDC was purchased from a local private sector community pharmacy. The generic and originator samples, all film-coated tablets, were stored in their original container closure system under controlled climatic conditions (temperature not exceeding 30°C) for the duration of experimentation (2015–2016). All samples were batch specific and remained within expiry by the end of the last experiments. The names of the tender companies and the trade names of FDCs are not disclosed so as to protect the anonymity of the companies. The FDCs are hereafter referred to as originator (O), generic 1 (G1), generic 2 (G2), generic 3 (G3), and generic 4 (G4).
Uniformity of weight
Twenty tablets (n = 20) were randomly selected from each container of the different FPPs and weighed individually using an electronic analytical balance and the average, standard deviation (SD), and the % relative standard deviation (RSD) of the 20 tablets were calculated. Tablets passed the weight uniformity tests if not more than two weights of the 20 individual tablets had more than 10% RSD (WHO, 2016b).
HPLC assay
The HPLC assay test conditions stipulated, as per the monograph (WHO, 2016a) were: reversed phase (RP) HPLC using an analytical HPLC column (25 cm × 4.6 mm) packed with chemically-bonded silica (5 μm), i.e. C18, as the stationary phase maintained at a temperature of 35°C; potassium dihydrogen phosphate (5%) in high purity deionized water and 70%:25% acetonitrile:potassium dihydrogen phosphate constituted mobile phases A and B, respectively, at a flow rate of 1 ml/minute and with UV detection of the analytes at 280 nm. In line with these requirements, the local system best suited was an Agilent 1200 series modular HPLC system equipped with a quaternary pump, diode-array detector, and a thermostatted column compartment housing an Ascentis® C18 column 5 μm particle size (25 cm × 4.6 mm) with the column temperature, mobile phases flow rate, and UV wavelength maintained as per the monograph. Transferring the monograph assay method onto the available HPLC system proved unsuccessful, in that peak resolution between the compounds of interest could not be resolved according to International Conference on Harmonization (ICH) guidelines (ICH, 2005).
Development of an alternative “in-house” RP-HPLC assay proceeded using the Perkin Elmer Flexar HPLC system with slight modifications to the method described by Raju and Begum (2008). The modular HPLC system comprised an autosampler equipped with a 100 μl injection loop, a binary LC pump, and a photodiode-array detector. The Discovery® HS C18 column (15 cm × 4.6 mm internal diameter × 5 μm particle size) was intuitively determined as the column of choice for optimum retention and resolution. Column temperature was controlled at ambient laboratory conditions of 20°C. The mobile phases consisting of 0.02 M sodium dihydrogen orthophosphate monohydrate buffer (Mobile Phase A) adjusted to pH 3.6 and (85:15) methanol:water (Mobile phase B) pumped at a flow rate of 1.0 ml/minute. A gradient was applied as shown in Table 1. The detection wavelength was determined at 260 nm by investigation of the UV spectra. The developed method was validated for system suitability, robustness, limit of detection, limit of quantitation, specificity, linearity, precision, and accuracy in accordance with ICH guidelines.
Preparation of standard solutions for HPLC assay validation
The validation targeted the quantitation of samples subjected to the dissolution test. Individual stock solutions for each RS were accurately weighed and diluted to the 100 ml mark with methanol (containing 0.4% w/v SDS) into different volumetric flasks, followed by vortex for 2 minutes to effect dissolution and thereafter filtered through a 0.22 μm nylon syringe filter. A standard combination stock solution of 10 ml containing a combination of 5 mg of EFZ RS, 2.5 mg of TDF RS, and 1.6 mg of FTC RS was then prepared. Working standards were prepared from the standard combination stock solution by dilution of stock aliquots with methanol to six different concentrations for each API (0.08, 0.1, 0.11, 0.12, 0.13, and 0.14 mg/ml for EFV; 0.04 mg, 0.05 mg/ml, 0.055 mg/ml, 0.06 mg/ml, 0.065 mg/ml, and 0.07 mg/ml for TDF; and 0.026, 0.033, 0.036, 0.04, 0.043, and 0.046 mg/ml for FTC).
Assay test
The assay test proceeded by weighing 20 tablets for each FPP, powdering by mortar and pestle and accurately weighing and dispersing a quantity of each of the powders containing 10 mg TDF into 100 ml of methanol, followed by sonication and filtering of the resulting solution through a 0.22 μm Nylon syringe filter. Six replicates for each FDC, having a final solution concentration of 0.0667 mg/ml of FTC, 0.1 mg/ml of TDF, and 0.2 mg/ml of EFV, were prepared. This was completed by an injection of 10 μl of each replicate into the HPLC and resulting peak areas determined by integration. The average and the SD were calculated. All the FPP FDCs were stored at 20°C until the time of the analysis, none of the products exceeded its expiry date before the end of the experiment.
Dissolution test
Dissolution tests were carried out as described in the IP 6th Edition (WHO, 2016a) to compare the quality of each generic FDC to the originator counterpart. The test was conducted using the USP Type II apparatus (paddle) at a speed of 100 rpm, at a temperature of 37°C ± 0.5°C. The medium comprised 1,000 ml of 2% v/v sodium dodecyl sulfate (SDS) and samples were collected at 30 minutes. Six tablets from each FPP were used. After 30 minutes, samples of 5 ml each were automatically withdrawn, filtered using 0.22 μm Nylon syringe filters, and thereafter diluted with methanol to obtain a final concentration of 0.4% w/v SDS. This sample was injected (10 μl) onto the HPLC for quantification of FTC, TDF, and EFV in accordance with the in-house RP-HPLC conditions described above. According to the IP monograph for this FDC, not less than 80% (Q) of the labelled amount of each active ingredient should be released in 30 minutes.
Disintegration test
The test was done using a disintegration apparatus (Electrolab® disintegration tester ED 2AL) as per the IP general method (WHO, 2016c). The media were heated to 37°C and the timer was set at 30 minutes. Six tablets (n = 6) were randomly selected from each sample bottle and placed into each of the six tubes of the basket. The disks were added to the top of the tablets. The apparatus was operated using distilled water as the media. After 30 minutes, the basket was lifted to observe the tablet disintegration.
Statistical analysis
Graph Pad Prism 6 (GraphPad Software, Inc., San Diego, CA) was used to analyze the data. Results are presented as mean, SD, and percentage SD (%SD). Statistical analysis using One-way ANOVA, Tukey’s multiple comparisons test was carried out to compare the release (dissolution testing) and the content (assay) of EFV, FTC, and TDF between the originator and the generics and between the generics themselves with alpha set at 0.05.
RESULTS
Uniformity of weight
All generic FDCs and the originator (O) samples (G1, G2, G3, and G4) passed uniformity of weight tests with all samples below the 5% RSD of the average tablet weight (Table 2). These findings reflect acceptable intra-batch consistency.
Assay method verification and method development
We found that the IP assay method as described in the 6th Edition provided unsatisfactory resolution of the peaks arising from FTC, TDF, and EFV when either pure reference standards were injected simultaneously or when the tablet sample was injected. Resolution between the peaks was less than 1.5 despite attempts to slightly modify the monograph method by, for example, changing the sample diluent (increasing the percentage of methanol from 50% to 80%). Figure 1 is a representative chromatogram of the FTC, TDF, and EFV reference standards when injected as a mixture.
The gradient RP-HPLC method which was developed provided satisfactory separation of the peaks arising from FTC, TDF, and EFV in both standard injections (Fig. 2) and when the tablet sample was injected (Fig. 3). We found that the method showed robustness with respect to changes in pH (from 3.5 to 3.69) with a slight change in the retention times, while the decrease of the flow rate (from 1 to 0.8 ml/minute) increased the retention times of all the APIs, especially for EFV. The retention time increased from 9.53 to 10.56 minutes for FTC, 12.56 to 13.88 minutes for TDF, and from 14.25 minutes to more than 20 minutes for EFV. A noteworthy observation made during the method development stage was the phenomenon around the fluctuating retention behavior of EFV when storing solutions at low temperature in a fridge or for a lengthy time in the autosampler carousel at room temperature. Subsequent experimental investigation found that by allowing cooled solutions to initially acclimatize to ambient room temperature, then followed by a high vortex for at least 2 minutes immediately prior to the HPLC analysis, restored the retention behavior of EFV. For time-exposed solutions, only the 2-minute high vortex sufficed. This phenomenon, attributed to a solubility issue of EFV in the prescribed diluent, did not require further investigation as the additional preparation step adequately resolved the retention issue and maintained estimation values. All system suitability parameters were within acceptable limits.
 | Table 1: Mobile phases gradient elution conditions of the “in-house” HPLC method for the simultaneous quantification of EFV, FTC, and TDF (Raju and Begum, 2008). [Click here to view] |
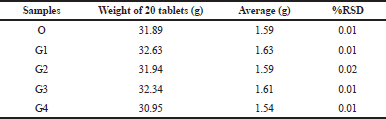 | Table 2: Uniformity of weight for each of the FDC samples. [Click here to view] |
 | Figure 1. Representative chromatogram for FTC, TDF, and EFV eluted under IP method conditions. [Click here to view] |
The specificity, linearity, precision, and accuracy of the assay (in-house method) were acceptable (Table 3). The three APIs were identified and quantified (content assay) in all the FPPs using the validated RP-HPLC method. The retention times (Figs. 2 and 3) and UV spectra (Figs. 4 and 5) for FTC, TDF, and EFV in the sample FDC tablets were comparable to the reference standards, confirming the identity of the three active ingredients in the generics and originator products.
 | Figure 2. Representative chromatogram of FTC (9.53 minutes), TDF (12.61 minutes), and EFV (14.25 minutes) reference standards eluted under the “in-house” method conditions. [Click here to view] |
 | Figure 3. Representative chromatogram of FTC (9.56 minutes), TDF (12.65 minutes), and EFV (14.37 minutes) identified in the originator tablet formulation. [Click here to view] |
Content of FTC, TDF, and EFV in the FDC products
In all samples, the percentage content of EFV, TDF, and FTC was consistent with label claims and within the IP range of 90%–110% (Fig. 6). Although all content was within limits, statistically significant differences in content were observed between the originator and the generics, as well as among the generics themselves (refer to Table 4).
The statistical analysis indicated a significant difference (p < 0.05) in FTC content between the originator and G1 and among other pairs of generics. A significant difference was observed in TDF content between the originator and G2 and between two other generic pairs. In relation to EFV, significant differences in content were observed between the originator and all four generics (G1–G4). However, it is important to note that these statistically significant differences in content may not translate to any differences in clinical efficacy since all products were within the pharmacopoeial allowable limits of 90%–110% at shelf-life.
Dissolution of FTC, TDF, and EFV from the FDC products
Three of the FDC products released at least 80% (Q) of the active ingredients within 30 minutes. However, we observed that G2 failed the pharmacopoeial dissolution test (refer to Fig. 7). At stage 1, 73.99% ± 10.78% of EFV was released and only two of the six tablets had a release of 85.07% and 81.08%. This product also failed stage 2 limits as the average amount of EFV released from 12 tablets was 62.23% ± 20.43%, and more than one tablet released less than 60% [limits state that at stage 2 average of the 12 (S1 + S2) units should be ≥ Q and no unit should be less than Q minus 15%]. The Q-release values of 10 out of the 12 tablets ranged from 22.04% to 78.73%. The differences in Q-release of EFV between G2 and other generics and against the originator also yielded statistically significant differences (see Table 5).
 | Table 3: Summary of validation results for the in-house RP-HPLC method for simultaneous quantification of FTC, TDF, and EFV at 260 nm. [Click here to view] |
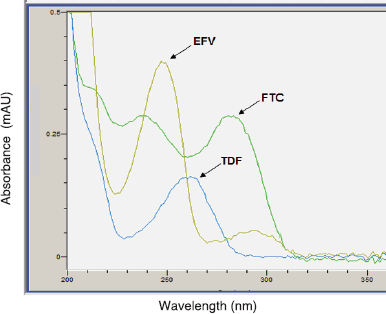 | Figure 4. Typical UV spectra for the reference standards (FTC, TDF, and EFV). [Click here to view] |
 | Figure 5. Typical UV spectra for the FPP (tablets). [Click here to view] |
 | Figure 6. Percentage content (w/w) of FTC, TDF, and EFV in the originator (O) and generic FDCs (G1, G2, G3, and G4). All products were found to contain FTC, TDF, and EFV within the pharmacopoeial limit of 90%–110% w/w (limit indicated by the red rectangle). [Click here to view] |
 | Figure 7. Q-release values of FDCs after 30 minutes. Results shown are the mean and SD of n = 6 tablets. [Click here to view] |
 | Table 4: Comparative content (% w/w) of active ingredients between generics and originator and among the generics. [Click here to view] |
 | Table 5: Average Q-release of EFV in originator and generic samples. [Click here to view] |
Disintegration test
The six selected tablets of each product disintegrated completely within 30 minutes and comply with the WHO IP specifications for film-coated tablets.
DISCUSSION
Apart from a few studies aimed at developing and validating HPLC methods for the FDC containing EFV, TFV, and FTC, none have attempted to assess and compare the quality profile of these medicines subsequent to market authorization or approved WHO status (prequalification) (Raju and Begum, 2008; Raju et al., 2008; Ramaswamy and Dhas, 2014). Our study developed and validated an RP-HPLC method and subsequently compared the quality profiles of different generic versions of this first-line ARV combination. We found that the identification and assay methods described in the IP sixth edition (replicated in the seventh edition) were unsuitable in this study for the simultaneous detection and quantification of EFV, TDF, and FTC either as standards or within the FDCs. With no other monograph available for this combination ARV, apart from monographs for individual ingredients and combinations of two ingredients (e.g. FDC + TDF)—which possibly form the basis for specifications in the registration process in South Africa—it is imperative that the IP method is reproducible and thus suitable for post-market quality assessments in various countries. We recommend further studies to evaluate the suitability of this method.
Although three out of four products passed all quality tests, there are concerns about the EFV release in one of the generic combinations currently on the South African market. A failed dissolution test could impact the efficacy of a medicine, potentially leading to therapeutic failure, development of drug resistance and toxic or adverse reactions (WHO, 2007). During the dissolution test, we observed that G2 appeared insoluble in the dissolution media. This could explain the failure in the dissolution of EFV, which is a more hydrophobic and poorly soluble API in comparison to TDF and FTC. We subsequently investigated this observation further by performing a disintegration test on all the FDCs. We performed this test in water and water containing 1% sodium dodecyl sulfate. However, in both situations, G2 disintegrated completely within 30 minutes. This led us to speculate that the failure in the dissolution of G2 could be related to the presence of an excipient that could be retarding the release of EFV. Upon further inspection of the inactive and film-coating ingredients of all the FPPs, an excipient which was unique to G2 was identified as a possible cause of its slower dissolution rates since this excipient happens to be used in extended-release and sustained release tablet formulations. Apart from excipient selection, the unfavorable dissolution of this FDC could also potentially be attributed to the company’s specifications for the particle size distribution of this API or their manufacturing procedures (Al Ameri et al., 2012; Genazzani and Pattarino, 2008). Although we concede that the IP specifications for dissolution of this FDC may differ from the manufacturer’s specifications prescribed in the dossier for market approval by SAHPRA, these compendial standards remain a universally recognized tool for post-marketing surveillance (Nkansah et al., 2017).
Although content assay findings complied with specifications, some inter-product variability exists, which could affect interchangeability of first-line generic ARV regimens in patients, who themselves display inter-individual differences (Genazzani and Pattarino, 2008). When the quality of generic medicines is brought into question, prescribers, dispensers, and patients are reluctant to use them and the knock-on effect is a loss of confidence in the health system which could seriously undermine health and economic gains accrued from health programs and national policies (e.g., pro-generics policies) (Al-Tamimi et al., 2013; Sharrad et al., 2009).
The rapid globalization of the pharmaceutical industry has increased international trade of raw materials and/or finished pharmaceutical products with many more intermediary distributors and role players operating under different national regulatory authorities but in the absence of global coordination. Even stringent regulatory authorities and countries relying on WHO pre-qualified medicine have experienced problems with substandard medicines entering their markets, in spite of efforts to rigorously review dossier applications, audit manufacturers, and distribution centers, as well as clearing every batch of imported medicine according to the manufacturer’s certificate of release (WHO, 2017). Most countries model an inverse relationship between regulatory function and medicine life-cycle with more stringent upstream controls (e.g., registration, inspection of manufacturers) and relatively weaker regulation towards the lower end of the supply chain (e.g., reassessment of quality) (WHO, 2017). If a regulatory agency’s upstream investments in preventing sub-standard medicines are not monitored downstream for the desired outputs (e.g., quality medicine) and outcomes (e.g., undetectable viral loads), then the monitoring and evaluation loop is incomplete and performance cannot be adequately gauged. This study, together with a recent assessment on post-market quality of antibiotics and analgesics on the private sector South African market (Lehmann et al., 2018b) provides evidence that stringency in medicine registration and manufacturer auditing processes does not guarantee on-going integrity of pharmaceutical products and in the absence of routine surveillance, these may go undetected. South Africa, like other lower and middle-income countries, lacks the regulatory capacity to fully enforce existing quality monitoring legislation and strategies to combine surveillance efforts with non-profit, regional, and global organizations should be more actively pursued. Furthermore, a risk-based approach to post-marketing surveillance which prioritizes the on-going reassessment of “high-risk” products (Nkansah et al., 2017) like ARV FDCs that have complex production methods, stability concerns, and forms the therapeutic basis of an extensive health program should be considered to optimize the use of restricted resources.
CONCLUSION
We confirmed the quality of three generic versions of Atripla® tablets and found one to be substandard. This study underscores the need for routine post-market surveillance of combination ARV regimens in South Africa to support and strengthen its large-scale ARV program. We recommend an independent verification of our findings by a WHO prequalified or SAHPRA-recognized quality control laboratory.
ACKNOWLEDGMENTS
The authors would like to thank the Provincial Department of Health for permission to source medicine samples from their ARV depot.
CONFLICT OF INTEREST
There are no conflicts of interest.
REFERENCES
Al Ameri MN, Nayuni N, Kumar KGA, Perret D, Tucker A, Johnston A. The differences between the branded and generic medicines using solid dosage forms: In-vitro dissolution testing. Results Pharma Sci, 2012; 1–8; doi:10.1016/j.rinphs.2011.12.001 CrossRef
Al-Tamimi SK, Hassali MA, Alrasheedy A. Challenges to generic medicines utilization in Yemeni healthcare system. GaBi J, 2013; 2; doi:10.5639/gabij.2013.0202.017 CrossRef
Aulton M, Taylor K (Eds.). Aulton’s pharmaceutics. In: The design and manufacture of medicines. 5th edition, Elsevier, New York, NY, 2017.
Chien C. HIV/AIDS drugs for Sub-Saharan Africa: How do brand and generic supply compare? PLos One, 2007; 2(3):e278; doi:10.1371/journal.pone.0000278 CrossRef
Department of Health (DOH). The South African antiretroviral treatment guideline. Version 14, 2013. Available via http://www.kznhealth.gov.za/medicine/2013_art_guidelines.pdf (Accessed 4 March 2018).
Department of Health (DOH). National Policy on HIV Pre-exposure Prophylaxis (PrEP) and Test and Treat (T&T). Government Printer, South Africa, 2016. Available via http://www.sahivsoc.org/Files/PREP%20and%20TT%20Policy%20-%20Final%20Draft%20-%205%20May%202016%20(HIV%20news).pdf (Accessed 8 August 2018).
Djobet MP, Singhe D, Lohoue J, Kuaban C, Ngogang J, Tambo E. Antiretroviral therapy supply chain quality control and assurance in improving people living with HIV therapeutic outcomes in Cameroon. AIDS Res Ther, 2017; 14:19; doi.org/10.1186/s12981-017-0147-x CrossRef
European Medicines Agency (EMA). Atripla®: EPAR—scientific discussion, 2007. Available via https://www.ema.europa.eu/documents/scientific-discussion/atripla-epar-scientific-discussion_en.pdf (Accessed 5 March 2017).
International Conference on Harmonisation (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use. Validation of analytical procedures: text and methodology Q2 (R1). International Conference on Harmonisation, Harmonised Tripartite Guideline, 2005.
Genazzani A, Pattarino F. Difficulties in production of identical products from a pharmaceutical technology viewpoint. Drugs R D, 2008; 9(2):65–72; doi:10.2165/00126839-200809020-00001 CrossRef
Joshi A, Esseku F, Silva L, Igwilo C, Oqua D, Kunle B, Obodozie O, Inyang U, Adeyeye MC. Postmarketing in vitro/in vivo assessment of fixed dose combination products of first line antiretrovirals. J Pharm Sci, 2010; 99(6):2655–63. Available via https://www.ncbi.nlm.nih.gov/pubmed/20054854 (Accessed 6 April 2017).
Lehmann A, Hofsäss M, Dressman J. Differences in drug quality between South Africa and Germany. J Pharm Pharmacol, 2018a; 70(10):1301–14; doi:10.1111/jphp.12985 CrossRef
Lehmann A, Katerere D, Dressman J. Drug quality in South Africa: a field test. J Pharm Sci, 2018b; 107(10):2720-30; doi:10.1016/j.xphs.2018.06.012
Maigetter K, Pollock AM, Kadam A, Ward K, Weiss MG. Pharmacovigilance in India, Uganda and South Africa with reference to WHO’s minimum requirements. Int J Health Policy Manag, 2015; 4(5):295–305; doi:10.15171/IJHPM.2015.55 CrossRef
Newton PN, Fernández FM, Green MD, Primo-Carpenter J, White NJ. Counterfeit and substandard anti-infectives in developing countries. In: Sosa A, Byarugaba D, Amábile-Cuevas C, Hsueh PR, Kariuki S, Okeke I (eds.). Antimicrobial resistance in developing countries. Springer, New York, NY, 2010. Available via http://emerald.tufts.edu/med/apua/about_us/publications_21_3125925763.pdf (Accessed 3 November 2017). CrossRef
Nkansah P, Smine K, Pribluda V, Phanouvong S, Dunn C, Walfish S, Umaru F, Clark A, Kaddu G, Hajjou M, Nwokike J, Evans L. Guidance for implementing risk-based post-marketing quality surveillance in low- and middle-income countries. U.S. Pharmacopeial Convention, The Promoting the Quality of Medicines Program, Rockville, MD, 2017.
Patel A, Gauld R, Norris P, Rades T. Quality of generic medicines in South Africa: perceptions versus reality—a qualitative study. BioMed Central Health Serv Res, 2012; 12(1):297; doi:10.1186/1472-6963-12-297 CrossRef
Primo-Carpenter J, McGinnis M. Matrix of drug quality reports in USAID-assisted countries by the US Pharmacopeia drug quality and information program, 2008. Available via https://pdf.usaid.gov/pdf_docs/Pnadl275.pdf (Accessed 25 September 2018).
Raju NA, Begum S. Simultaneous RP-HPLC method for the estimation of Emtricitabine, Tenofovir disoproxil fumerate and Efavirenz in tablet dosage forms. Res J Pharm Technol, 2008; 1(4):522–5.
Raju NA, Rao JV, Prakash KV, Mukkanti K, Srinivasu K. Simultaneous estimation of Tenofovir disoproxil, Emtricitabine and Efavirenz in tablet dosage form by RP-HPLC. Orient J Chem 2008; 24(2):645–50.
Ramaswamy A, Dhas ASAG. Development and validation of analytical method for quantitation of Emtricitabine, Tenofovir, Efavirenz based on HPLC. Arab J Chem, 2014; 11(2):275–81; doi:10.1016/j.arabjc.2014.08.007 CrossRef
Sharrad AK, Hassali MA, Shafie AA. Generic medicines perceptions of physicians in Basrah, Iraq. Austral Med J, 2009; 1:58–64. Available via https://core.ac.uk/download/pdf/27270684.pdf (Accessed 13 August 2018).
Joint United Nations Programme on HIV/AIDS (UNAIDS). Together we will end AIDS. Geneva, Switzerland, 2012.
Joint United Nations Programme on HIV/AIDS (UNAIDS). Global AIDS update 2016. Geneva, Switzerland, 2016.
World Health Organization (WHO). Fixed-dose combinations for HIV/AIDS, tuberculosis, and malaria—report of a meeting held 16–18 December 2003. Geneva, Switzerland, 2003. Available via http://apps.who.int/iris/handle/10665/68967 (Accessed 7 February 2017).
World Health Organization (WHO). International Pharmacopoeia (IP).– Monographs: Dosage forms: Specific monographs: Efavirenz, emtricitabine and tenofovir tablets (Efavirenzi, emtricitabini et tenofoviri compressi). 6th edition, Geneva, Switzerland, 2016a.
World Health Organization (WHO). International Pharmacopoeia (IP). Methods of analysis: pharmaceutical technical procedures: Uniformity of mass for single-dose preparations 2016. 6th edition, Geneva, Switzerland, 2016b.
World Health Organization (WHO). International Pharmacopoeia (IP). Methods of analysis: Pharmaceutical technical procedures: Disintegration test for tablets and capsules. 6th edition, Geneva, Switzerland, 2016c.
World Health Organization (WHO). Survey of the quality of antiretroviral medicines circulating in selected African countries. Geneva, Switzerland, 2007. Available via http://www.who.int/medicines/publications/ARV_survey.pdf (Accessed 4 March 2017).
World Health Organisation (WHO). WHO Global surveillance and monitoring system for substandard and falsified medical products. World Health Organization, Geneva, Switzerland, 2017. Available via https://www.who.int/medicines/regulation/ssffc/publications/gsms-report-sf/en/ (Accessed 5 August 2018).