INTRODUCTION
Pharmaceutical excipients are usually derived from natural (animal, vegetable, and mineral) and synthetic origin (Giorgio and Patrizia, 2003). The additives developed from natural sources are still the best for the production of pharmaceuticals. This is because of their reduced toxicity, low cost, local availability, soothing action, non-irritant nature, relative abundance, biocompatibility, improved patient tolerance, and public acceptance compared to synthetic excipients (Anroop et al., 2005; Bharadia et al., 2004; Kaushik et al., 2016; Kulkarni et al., 2002; Pawar and D’mello, 2004; Varshosaz et al., 2006). Direct compression is a method of tablet production among other methods such as wet granulation and dry granulation; it simply involves the direct compression of the blend of powdered materials into tablets without modification of the physical characteristic of the materials. This method is becoming very popular because of it being economical, cheap, and an efficient technological process. Direct compression involves few unit operation processes and manufacturing steps, reduced processing time and costs, and less number of equipment (Hindiyeh et al., 2018; Singh et al., 2014). Hydrophilic polymers are popular and suitable for delaying drug release, and interest continues in the use of polymers in controlled drug delivery systems (Gade and Murthy 2011; Genc et al., 1999; Jan et al., 2012; Khan and Jiabi 1998; Manjula et al., 2014; Muhammad et al., 2014; Reddy and Archana 2018).
Hydrophilic polymers in matrix systems do not disintegrate; in the presence of an aqueous medium, hydration develops immediately with the production of a highly viscous gelatinous boundary, which serves as a barrier that controls the release of drug from the matrix system (Talukder et al., 1996). These systems are called swellable controlled release systems. The hydrophilic polymeric matrix system is made up of the hydrophilic polymer, the drug, and other adjuvants, which are evenly distributed within the matrix. These systems generally depended on wetting and hydration of the polymer, and dissolution to achieve modulated drug release (Williams et al., 2002).
Tramadol hydrochloride is a centrally acting opioid analgesic. It is used in the management of pains. The peak concentration of sustained release tramadol preparation is reached after 4.9 hours with an oral bioavailability of 87%—%. It has an elimination half-life of 6 hours (Grond and Sablotzki, 2004) and thus requires dosing every 6 hours in order to maintain optimal blood concentration for the relief of pain as reported by Raber et al. (1999).
Cissus gum obtained from Cissus populnea (Guill and Per), a climbing plant, has been explored in other studies as a binder, controlled release agent, suspending agent, and emulsifying agent (Abioye et al., 2000; 2001; Adeleye et al., 2011; Eichie and Amalime 2007; Emeje et al., 2009; Ibrahim et al., 2002). In order to expand the usefulness of cissus gum, an attempt was made to evaluate its properties in direct compression. The aim of the present study, therefore, was to evaluate the compactibility, mechanical, and release properties of tramadol hydrochloride tablets prepared by direct compression using cissus gum, as a directly compressible excipient. This was done in comparison with xanthan gum, a naturally sourced standard adhesive as a directly compressible excipient.
MATERIALS AND METHODS
Materials
Tramadol hydrochloride used in the study was a gift from Uripharm Specialties Ltd (Lagos, Nigeria). Cissus gum was extracted in the Laboratory of the Department of Pharmaceutics and Industrial Pharmacy, University of Ibadan, Ibadan, Nigeria. Xanthan gum was obtained from Jungbenzlauer Ges.M.B.H. Handelsgericht Wien, Germany. Lactose was also obtained from DMV Veghel, Netherlands. Other solvents and chemicals were of analytical grade.
Methods
Preparation of Cissus gum
The gum was obtained from the stem of Cissus populnea according to the method adapted by Adeleye et al. (2015a). The sliced stem was macerated in distilled water for 24 hours, followed by filtration of the viscous solution with a muslin bag. The viscous solution was then treated with acetone to precipitate the gum. The precipitate was dried at 50°C for 24 hours in an oven and pulverized using a laboratory blender (Model 857 Williamette Industries, Bowing Green Kentucky USA).
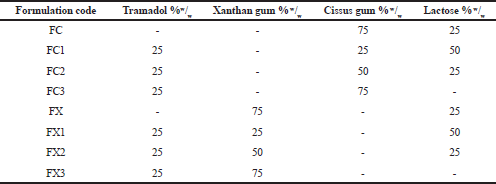 | Table 1. Tramadol hydrochloride matrix tablet formulations. [Click here to view] |
Preparation of powder blend
The formulations in Table 1 were prepared by mixing tramadol hydrochloride, the polymer—Cissus gum or Xanthan gum—and lactose (where required) uniformly. The mixing was done in a tumbling mixer for 10 minutes to form a homogenous blend.
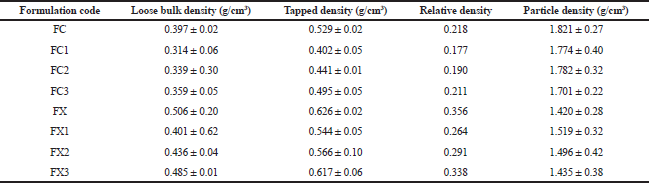
Bulk, tapped, and relative density measurements
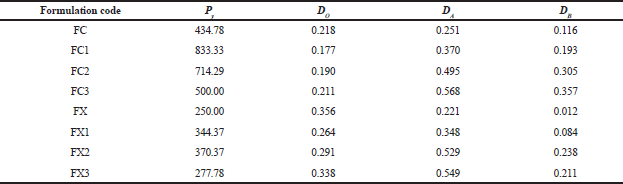
The bulk and tapped densities of each of the formulations were determined by pouring 25.0 g of each of the formulations gradually into a 100 ml graduated glass measuring cylinder with a diameter of 12.6 mm through a funnel at an angle of 45°. The height reached by the powder was measured and the volume and density were calculated appropriately (Mohammadi and Harnby, 1997).
The bulk density (loose), P was calculated using Eq. (1):
P = m/v (1)
Where, m, in grams, is the weight of formulation in the cylinder and v, in cm3, is the volume occupied by the formulation. Determinations were done in triplicate.
Tapped density was determined by tapping 25 g of formulation in the graduated measuring cylinder manually on a wooden surface at height of 7 inches. One hundred taps were applied at a standard rate of 38 taps per minute (Reus-Medina et al., 2004).
Relative density Do of each formulation was obtained from the ratio of the loose density to its particle density (Heckel, 1961).
Particle density measurement
The particle density of each formulation was determined by the pycnometer method using liquid immersion technique with xylene as the displacement liquid (Odeniyi et al., 2011). The pycnometer bottle (50 ml) was weighed empty with the stopper (W). It was filled with xylene to the brim and excess was wiped off with an absorbent paper, and the weight with the stopper was noted as (W1). The difference between W and W1 was recorded as W2. Two gram of each formulation was weighed (W3) and transferred into the pycnometer bottle and filled with xylene to the brim. The excess solvent was wiped off and the bottle weighed again with the stopper (W4). The particle density, Pt (g/cm3), was calculated using Eq. (2):
Pt = W2·W3/50 (W3 – W4 + W + W2) (2)
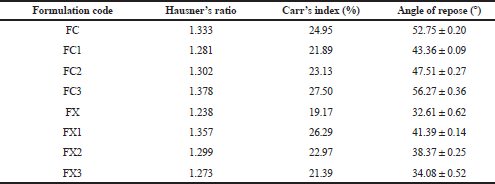
Angle of repose
This was determined by allowing a specific mass of material to flow freely through a funnel from a particular height to form a conical heap on a white paper placed on a horizontal surface. The diameter of the cone was measured and the angle of repose was determined by Eq. (3) (Potu et al., 2011).
tan Ѳ = h/r (3)
Where Ѳ is the angle of repose (the angle made by the heap with the base), h is the height of the heap of powder, and r is the radius of the cone. Determinations were made in triplicate.
Hausner’s ratio and Carr’s index
The Hausner’s ratio and Carr’s compressibility index were calculated using Eqs. (4) and (5), respectively (Carr, 1965; Hausner, 1967).
Hausner ratio = Tapped density/ Bulk density (4)
Carr’s Index = Tapped density – Bulk density/Tapped density 100 (5)
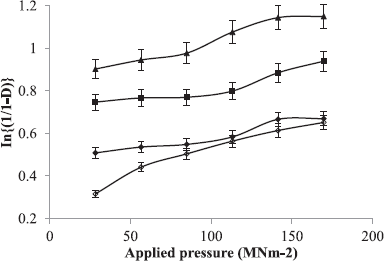
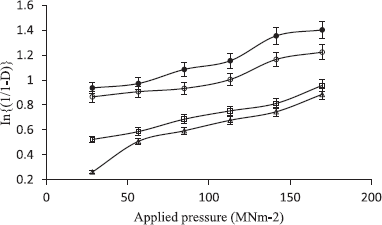
Tablet compression
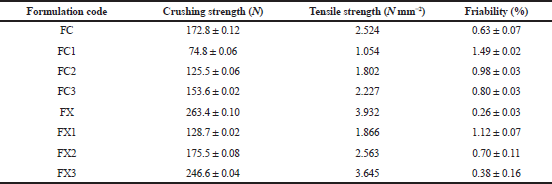
The formulations containing the powder mixture were compressed for 30 seconds into 400 mg tablet compacts at six different compression loads using a hydraulic hand press (Model C, Carver Jnc., Menomonee Falls, W1) with a 10.5 mm die and flat-faced punches lubricated with a 2% dispersion of magnesium stearate in acetone before each compression. The tablets were ejected and stored in airtight containers over silica gel for 24 hours to allow for elastic recovery and hardening.
Tablets compressed with a compression load of 113.16 M Nm−2 were selected for the evaluation of mechanical and release properties of the tablets.
Tablet tensile strength
The force (N) required to break each tablet was determined by the procedure of Fell and Newton (1970) using a tablet hardness tester (DKB instrument, Mumbai. Model EH 01). Tablets were placed between the spindle and the anvil of the hardness tester, and the pressure was applied until the tablet split diametrically. Determinations were made in triplicate for each batch of the tablet tested.
The tensile strength, T, in N mm-2, of the tablets was calculated from Eq. (6) (Fell and Newton, 1970; Hiestand et al., 1977):
T = 2·F/ π·d·t (6)
Where F is the force required to cause breakage, π is 3.14, d is the tablet diameter, and t is the tablet thickness.
Tablet friability
The percentage friability of the tablet was determined using the Veego tablet friability apparatus (Veego Scientific Devices, Mumbai, India). The weights of 10 tablets were taken collectively and the tablets were placed in the friabilator, which was then operated for 4 minutes at 25 rpm. The tablets were collected, dusted, and weighed again (Adeleye et al., 2015b). The percentage weight loss was calculated as the percent friability. Determinations were made in triplicate.
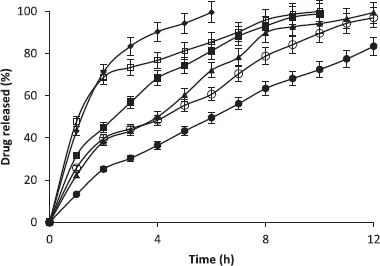
Tablet dissolution test
The release rate of tramadol hydrochloride from the tablets was examined by rotating basket USP Dissolution Apparatus (Model NE4-COPD, Copley scientific, Nottingham, UK) operated at 50 rpm with 900 ml of 0.1 mol.l−1 hydrochloric acid at 37.0°C ± 0.5°C as the dissolution medium. Five milliliter samples were withdrawn and immediately replaced with 5 ml of fresh 0.1 mol.l−1 hydrochloric acid at 1-hour interval for 12 hours maintained at the same temperature. The amount of tramadol released was analyzed using a spectrophotometer (Jenway UV-780 print UV-Spec) at a wavelength of 271 nm.
Statistical analysis
Statistical analysis was performed with GraphPad Prism 5. Some data were presented as mean ± standard deviation (XÌ… ± SD). Student’s t-test was used to identify differences between the parameters evaluated. Differences were considered to be statistically significant at a p value of <0.05.