
INTRODUCTION
Apoptosis or a programmed cell death plays a key role in controlling cell development and tissue homeostasis. Its dysregulation is well known as a hallmark of cancer. Apoptosis that occurs through the mitochondrial pathway was regulated by B cell lymphoma-2 (Bcl-2) family proteins by engineering the integrity of the outer mitochondrial membrane. Bcl-2 proteins consist of antiapoptotic proteins and proapoptotic proteins. In a cancer cell, apoptosis was averted through dysregulation antiapoptotic proteins, including Bcl-2 resulting in their aberrant expression which alters the balance between proapoptotic and antiapoptotic members of the Bcl-2 family leading to preserved cancer cell survival (Ashkenazi et al., 2017). Therefore, targeting antiapoptotic Bcl-2 was considered a rational strategy to restore apoptosis function for cancer treatment.
Several small molecules with structural diversity are reported as Bcl-2 inhibitors such as Navitoclax, Venetoclax, and Obatoclax (Davids and Letai, 2013; Lin et al., 2016; Salem et al., 2016; Trudel et al., 2007; Vervloessem et al., 2018). Navitoclax has been approved by Food and Drug Administration (FDA) for curing chronic lymphocytic leukemia, the most frequent leukemia found in adults, and Obatoclax for curing different types of cancer. Despite their advantages, several miserable side effects were observed associated with the clinical use of those agents. The thrombocytopenia was found to be a major dose-limiting toxicity for Navitoclax, particularly in a single- agent setting (Ashkenazi et al., 2017). Meanwhile, the side effects of Obatoclax are largely neurological, including fatigue, euphoria, somnolence, ataxia, diarrhea, nausea, thrombocytopenia, anemia, and pneumonia (Wu et al., 2017). Those facts prompted tremendous efforts worldwide for finding more potent Bcl-2 inhibitors which have fewer side effects.
Porphyrins are well known for their crucial function such as transporting oxygen (haem) and mediating photosynthesis process (chlorophyll) (Ptaszyńska et al., 2018). Porphyrin has been widely used in therapeutic applications. Variations of porphyrin structure have been well reported both through periphery substitution and central nitrogen metalation in order to tune their therapeutic values (Carneiro et al., 2018). Porphyrin with positive charge was also reported to increase interaction with a molecule bearing negatively charged groups such as DNA and RNA in cancer cells. Porphyrin bearing pyridyl and pyrazolium groups attached to the meso position have attracted much interest for their therapeutic values. It was reported that the porphyrin bearing either pyridyl or pyrazolium as periphery substituents increases interaction with cancer cells (Carneiro et al., 2018).
Here, we report the Bcl-2 binding of porphyrin conjugated with anthraquinone group by using the computational approach. Rapid advances in computational approach in drug discovery have been well documented. It saves time and cost in the drug discovery process. In this study, variation on the structure of porphyrin was made by attaching either pyridyl or pyrazolium groups in the meso position. It is found that porphyrin with a mono substituent, H2PyP-AQ, has a comparable affinity with the cognate ligand of Bcl-2.
COMPUTATIONAL METHODS
Macromolecule and ligand preparation
The structure of Bcl-2 was taken from the Protein Data Bank, PDB id 4LVT, resolution 2.05 Å (Souers et al., 2013). Six porphyrin derivatives were built using Gaussian 09 (Frisch et al., 2009) following our previous procedure (Arba et al., 2017). The porphyrin built are mono-H2PyP-AQ, bis-H2PyP-AQ, tris-H2PyP-AQ, mono-H2PzP-AQ, bis-H2PzP-AQ, and tris-H2PzP-AQ. Figure 1 shows the 2D structure of porphyrin derivatives. The optimized ligand was then used for further docking study.
Molecular docking and MD simulation
Molecular docking was performed by using AutoDock Vina (Trott and Olson, 2010). Protein and ligand were prepared using AutoDockTools and saved as “PDBQT” files. A config file was prepared containing the binding site box which was in the center of a cognate ligand of Bcl-2 with the dimension of 46 × 46 × 46 Å on x y z directions. The docked conformation with high binding energy was chosen for further MD simulation which was performed using AMBER16 software (Case et al., 2005; Salomon-Ferrer et al., 2013). For MD simulation, each protein and ligand was parameterized with ff14SB (Maier et al., 2015) and GAFF (Wang et al., 2004) force fields as well as AM1-BCC (Jakalian et al., 2002). The protein-ligand complex was solvated with the truncated octahedron TIP3P water model with a minimum distance of 10 Å around the system. System electroneutralization was achieved by the addition of Na+ counterions using Leap module of AMBER16.
Before proceeding to MD simulation, each system was minimized using Sander module of AMBER16. Minimization was performed by following our previous procedure (Arba et al., 2017). System was then heated from 0 to 100, 100 to 200, and 200 to 300 K, respectively. Each was done for 50 ps with a timestep of 0.0005 ps and backbone atom restraints for protein (k = 5 kcal mol−1 Å−2). Each system was then equilibrated at 300 K during two 100 ps with restraints (k = 5 and k = 3 kcal mol−1 Å−2), respectively, which was followed by 100 ps constant-temperature, constant-pressure (NPT) ensemble equilibration without restraint. The equilibrated system was then subjected to 50 ns MD production in NPT ensemble by using pmemd.cuda module in AMBER16 (Salomon-Ferrer et al., 2013). All other MD settings were adopted from our previous work (Arba et al., 2017). Analysis was performed by using CPPTRAJ module (Roe and Cheatham III, 2013) of AMBER16, while visualization was achieved with the help of Visual Molecular Dynamics software (Humphrey et al., 1996).
Binding free energy calculations
Our last in silico protocol was the calculation of the binding free energy, which was performed using the Molecular Mechanics-Poisson Boltzmann solvent accessible surface area (MM-PBSA) method (Arba et al., 2018a; 2018b; Kollman et al., 2000). The binding energy was calculated using 200 snapshots taken from 20 to 40 ns simulation trajectories with the python modules of AMBER16 (Miller et al., 2012). The MM-PBSA method for binding energy calculation was based on an assumption that the binding free energy of complex (ΔGbind) is the difference between the free energies of the complex (ΔGcomplex) and the unbound receptor (ΔGrec) and the free ligand (ΔGlig), as shown in the following equations:
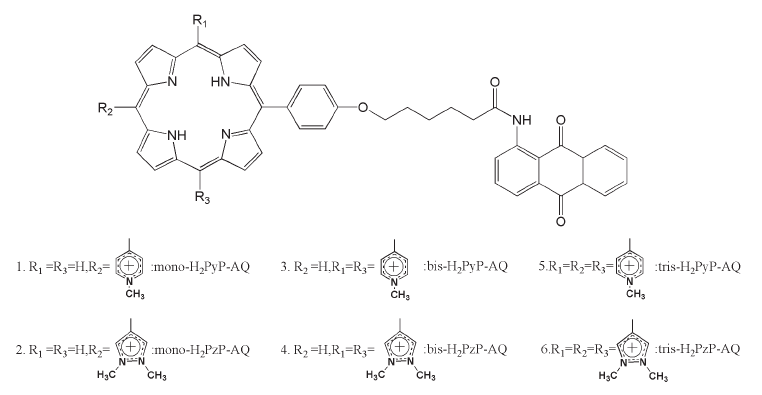 | Figure 1. The 2D structure of porphyrin derivatives. [Click here to view] |
The binding energy (ΔGbind) was a summation of the gas phase binding energy (ΔEMM), solvation free energy (ΔGsol), and the conformational entropy change upon ligand binding at temperature T (TΔS), respectively. Furthermore, bond energy, angle energy, torsion energy, van der Waals energy, and electrostatic energy, all constitute the ΔEMM, while polar contribution to solvation free energy (ΔGPB) was calculated by solving the Poisson-Boltzmann (PB) equation using a grid size of 0.5 Å, and the non-polar contribution (ΔGSA) which was calculated using the solvent accessible surface area (SASA) with solvent-probe radius set to 1.4 Å, both of which constitute ΔGsol.
RESULTS AND DISCUSSION
To investigate the putative interaction mechanism of porphyrin derivatives with Bcl-2, molecular docking was conducted by using AutoDock Vina. Each porphyrin derivative was docked to the protein allowing the maximum torsion of each compound.
Firstly, the internal validation phase was performed by re-docking 1XJ over the Bcl-2 active site. The root mean square deviation (RMSD) of heavy atoms of re-docked and co-crystallized conformations of 1XJ was utilized to assess the reliability of the docking protocol. The result shows that the RMSD values of the two conformations were 0.77 Å, indicating the validated performance of docking (Goodsell et al., 1996; Ruslin et al., 2017). In the 1XJ docked pose, two hydrogen bonds (hbond) were formed between ligand and Arg104 and Gly142 residues of Bcl-2. Figure 2 depicts the docked and X-ray crystallographic poses of 1XJ and interacting 1XJ over the active site of Bcl-2.
Furthermore, porphyrin binding to the Bcl-2 was predicted using the validated docking protocol. Several interactions between porphyrin derivatives with Bcl-2 occurred such as hydrogen bond (hbond), hydrophobic and electrostatic interactions. Conventional hbond took place with Tyr105 as observed in bis-H2PzP-AQ and tris-H2PzP-AQ. The Glu133, Arg136, and Gly142 were also detected in the conventional hbond in bis-H2PyP-AQ, tris-H2PyP-AQ, and mono-H2PyP-AQ, while Arg143 and Leu198 were, observed in tris-H2PzP-AQ and mono-H2PyP-AQ, respectively. In addition, C−H−O hbonds were observed with Ala146 in bis-H2PyP-AQ and tris-H2PyP-AQ, while Leu134 was detected in bis-H2PyP-AQ binding.
Hydrophobic interactions of porphyrin derivatives consisted of pi–pi stacking, pi–pi T-shaped stacking, and pi-alkyl interactions. Pi–pi stackings were seen between mono-H2PzP-AQ and tris-H2PzP-AQ with Tyr105, while pi–pi T-shaped stackings were observed between mono-H2PyP-AQ, mono-H2PzP-AQ, and bis-H2PyP-AQ with each Tyr105 and Phe101, respectively. Meanwhile, tris-H2PzP-AQ and tris-H2PyP-AQ established two pi–pi T-shaped stackings with both Phe101 and Tyr105. Further, pi-alkyl interactions were found with Ala146 in tris-H2PzP-AQ, tris-H2PyP-AQ, bis-H2PyP-AQ, and mono-H2PzP-AQ, while those with Leu134 were observed with tris-H2PyP-AQ, bis-H2PyP-AQ, and mono-H2PzP-AQ. The ligand mono-H2PzP-AQ and mono-H2PyP-AQ share similar interacting residue with Arg143. Additionally, Ala97, Val145, and Leu198 were bound to mono-H2PyP-AQ, Met112 bound to mono-H2PzP-AQ, and Val130 to bis-H2PyP-AQ.
 | Figure 2. (a) The superimposition of docked and X-ray crystallographic poses of 1XJ; (b) the docked conformation of 1XJ in the binding site of Bcl-2. [Click here to view] |
In addition, pi-cationic interactions were observed between tris-H2PzP-AQ and Arg143, as well as tris-H2PyP-AQ and Arg136, while pi-anion interaction was observed between tris-H2PyP-AQ and Asp108. Figure 3 shows the interaction of each porphyrin derivative with Bcl-2.
Molecular dynamics simulation
Figure 4 depicts RMSD for all complexes. It shows that each complex has reached stability as indicated by stable RMSD values under 3 Å. The RMSD value of 1XJ (red-colored) tends to be slightly higher than those of porphyrin, indicating their better stability.
 | Figure 3. The interaction of (a) mono-H2PzP-AQ, (b) mono-H2PyP-AQ, (c) bis-H2PzP-AQ, (d) bis-H2PyP-AQ, (e) tris-H2PzP-AQ, and (f) tris-H2PyP-AQ, each with Bcl-2. [Click here to view] |
Additionally, fluctuation on amino acid residues was monitored through the root mean square deviation (RMSF) plot. Figure 5 depicts the RMSF plot versus residue number of amino acid. It shows that all complex has a similar fluctuation of amino acids during MD simulation. Generally, the amino acid fluctuation was under 4 Å indicating the stability of the complex. High fluctuation was observed in the Pro201 (Pro139) corresponding to carboxy end of the protein.
MM-PBSA binding free energy
The binding energy was predicted by using MM-PBSA calculation and the result is presented in Table 1, showing clearly that favorable energy contributions originated from van der Waals (ΔEVDW) and nonpolar desolvation energies (ΔEPBSUR), due to hydrophobic character of porphyrin macrocycle. The favorable contribution was also observed from electrostatic energy (ΔEELE), while net unfavorable electrostatic contribution was unfavorable due to positive polar desolvation energy (ΔEPBCAL). The electrostatic energy may be due to several electrostatic interactions which were described previously, including hydrogen bond and pi-cationic interactions. Furthermore, it is observed that porphyrin hybrid having one meso substituent, i.e., mono-H2PzP-AQ, had most negative binding energy (ΔGpred = −51.81 ± 4.75 kcal/mol), only slightly different from that of the cognate ligand with a binding energy of −54.72 ± 3.72 kcal/mol. This indicates the potential of that compound to be further developed in drug discovery.
 | Figure 4. RMSD value of protein heavy atom during MD simulation for (a) 1XJ (red), bis-H2PyP-AQ (green), and bis-H2PzP-AQ (blue); (b) 1XJ (red), mono-H2PyP-AQ (green), and mono-H2PzP-AQ (blue); and (c) 1XJ (red), tris-H2PyP-AQ (green), and tris-H2PzP-AQ (blue). [Click here to view] |
 | Figure 5. RMSF change during 50-ns MD simulation for (a) 1XJ (red), bis-H2PyP-AQ (green), and bis-H2PzP-AQ (blue); (b) 1XJ (red), mono-H2PyP-AQ (green), and mono-H2PzP-AQ (blue); and (c) 1XJ (red), tris-H2PyP-AQ (green), and tris-H2PzP-AQ (blue). [Click here to view] |
 | Table 1. The binding free energies and the corresponding components (kcal/mol). [Click here to view] |
CONCLUSION
In conclusion, porphyrin derivatives were able to interact with Bcl-2. The interaction of porphyrin was mainly supported by electrostatic and van der Waals energies. Bcl-2 binding preferred porphyrin with less number of meso substituents. The designed compounds, i.e., mono-H2PzP-AQ, can potentially be used as a Bcl-2 inhibitor as shown by binding free energy predicted by MM-PBSA, which is comparable with that of cognate ligand, 1XJ.
ACKNOWLEDGMENTS
The authors wish to thank Ministry of Research, Technology and the Higher Education Republic of Indonesia for supporting this research through Hibah Penelitian Dasar Unggulan Perguruan Tinggi (PDUPT) Tahun 2018.
REFERENCES
Arba M, Ihsan S, Ramadhan LOAN, Tjahjono DH. In silico study of porphyrin-anthraquinone hybrids as CDK2 inhibitor. Comput Biol Chem, 2017; 67:9–14.
Arba M, Nur-Hidayat A, Ruslin, Yusuf M, Sumarlin, Hertadi R, Wahyudi ST, Surantaadmaja SI, Tjahjono DH. Molecular modeling on porphyrin derivatives as β5 subunit inhibitor of 20S proteasome. Comput Biol Chem, 2018a; 74:230–8.
Arba M, Ruslin, Kalsum WU, Alroem A, Muzakkar MZ, Usman I, Tjahjono DH. QSAR, molecular docking and dynamics studies of quinazoline derivatives as inhibitor of phosphatidylinositol 3-kinase. J Appl Pharm Sci, 2018b; 8(5):001–9.
Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Disc, 2017; 16(4):273–84.
Carneiro J, Gonçalves A, Zhou Z, Griffin KE, Kaufman NEM, Vicente da MGH. Synthesis and in vitro PDT evaluation of new porphyrins containing meso-epoxymethylaryl cationic groups. Laser Surg Med, 2018; 50(5):566–75.
Case DA, Cheatham III TE, Darden T, Gohlke H, Luo R, Merz Jr KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem, 2005; 26(16):1668–88.
Davids MS, Letai A. ABT-199: Taking dead aim at BCL-2. Cancer Cell, 2013; 23(2):139–41.
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision B.01., Gaussian, Inc., Wallingford, CT, 2009.
Goodsell DS, Morris GM, Olson AJ. Automated docking of flexible ligands: applications of AutoDock. J Mol Recognit, 1996; 9(1):1–5.
Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph, 1996; 14(1):33–8.
Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem, 2002; 23(16):1623–41.
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham III TE. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res, 2000; 33(12):889–97.
Lin KH, Winter PS, Xie A, Roth C, Martz CA, Stein EM, Anderson GR, Tingley JP, Wood KC. Targeting MCL-1/BCL-XL forestalls the acquisition of resistance to ABT-199 in acute myeloid leukemia. Sci Rep, 2016; 6(1):27696.
Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: An efficient program for end-state free energy calculations. J Chem Theory Comput, 2012; 8(9):3314–21.
PtaszyÅ„ska AA, Trytek M, Borsuk G, Buczek K, Rybicka-JasiÅ„ska K, Gryko D. Porphyrins inactivate Nosema spp. microsporidia. Sci Rep, 2018; 8(1):1–11.
Roe DR, Cheatham III TE. PTRAJ and CPPTRAJ: software for processing and analysis of molecular synamics trajectory data. J Chem Theory Com, 2013; 9(7):3084–95.
Ruslin, Nirwana, Arba M, Mukhsar, Tjahjono DH. QSAR, molecular docking and dynamics studies of pyrrolo[2,3-b]pyridine derivatives as bruton’s tyrosine kinase inhibitors. J Appl Pharm Sci, 2017; 7(12):001–7.
Salem AH, Agarwal SK, Dunbar M, Enschede SLH, Humerickhouse RA, Wong SL. Pharmacokinetics of venetoclax, a novel BCL-2 inhibitor, in patients with relapsed or refractory chronic lymphocytic leukemia or non-Hodgkin’s lymphoma. J Clin Pharmacol, 2016; 57(4):484–92.
Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh ewald. J Chem Theory Comput, 2013; 9(9):3878–88.
Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput, 2015; 11(8):3696–713.
Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, fairbrother WJ, Huang DCS, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park Ch, Park C-M, Philips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med, 2013; 19(2):202–208.
Trott O, Olson AJ. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem, 2010; 31:455–61.
Trudel S, Zhi HL, Rauw J, Tiedemann RE, Xiao YW, Stewart AK. Preclinical studies of the pan-Bcl inhibitor obatoclax (GX015-070) in multiple myeloma. Blood, 2007; 109(12):5430–8.
Vervloessem T, Kerkhofs M, La Rovere RM, Sneyers F, Parys JB, Bultynck G. Bcl-2 inhibitors as anti-cancer therapeutics: The impact of and on calcium signaling. Cell Calcium, 2018; 70:102–16.
Wang JM, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem, 2004; 25(9):1157–74.
Wu H, Medeiros LJ, Young KH. Apoptosis signaling and BCL-2 pathways provide opportunities for novel targeted therapeutic strategies in hematologic malignances. Blood Rev, 2018; 32(1):8–28.