
INTRODUCTION
As per the U.S. Food and Drug Administration (USFDA), 16 (32%) of the top 50 drugs by manufacturer revenue in the year 2023 (Through Q3 2023) were drug-device combination products (DDCPs). This highlights the growing preference for DDCPs over conventional drug products. A combination product is a product composed of two or more regulated components, such as drug/device, biologic/device, or drug/device/biologic components, that are physically, chemically, or otherwise combined or mixed and produced as a single entity or packaged together in a single package or cross-labeled [1,2].
The various types of pharmaceutical dosage forms (Fig. 1), such as prefilled syringes, prefilled pens, autoinjectors, ophthalmic containers, inhalation pumps, and metered dose inhalers, are among the DDCPs.
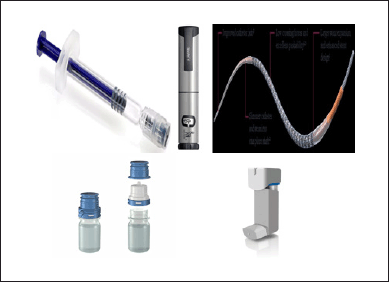 | Figure 1. Examples of commercially available pharmaceutical dosage forms of DDCPs [3–7]. [Click here to view] |
This article reviews the history and evolution of DDCPs and discusses key regulatory pathways to help manufacturers choose appropriate business models and strategies for development and regulatory filing.
Combining device engineering and pharmacotherapy: A shift from traditional pharmaceutical dose forms to drug delivery and control systems
Robin S. Porter received the hypodermic syringe patent in 1914. Historically, hypodermic syringes and glass vials were used for administering drugs through subcutaneous and intramuscular methods. The intersection of patient convenience with advancements in design, materials, and manufacturing technologies was recognized by a variety of European companies in the early 1980s. Beyond their sheer functionality, pen injectors and systems have revolutionized drug delivery. It is now necessary to comprehend two additional concepts in order to achieve success: form and form. The key dimensions that provide precision need to be determined and managed for a good match. At the same time, rigorous ergonomic designs that promote usability and address the form factor are necessary for the entire user experience. By improving compliance and quality of life, the quest for convenience in the self-administration of medications results in better health outcomes and lower expenses. In the 1980s, Novo, Nordisk, and Hoechst introduced multidose 1.5 and 3.0 ml glass insulin cartridges for prefilled and reusable pen injectors, advancing system designs. Novo created the first multidose pen injector, the 1.5 ml NovoPen I, in 1985. In 1987, Hoescht introduced the OptiPen I, ranking second. The Novolet, the first prefilled insulin pen, was developed as a consequence of the 1988 merger between Novo and Nordisk. The early 1990s saw Lilly’s 1.5 ml insulin cartridge available in Germany and the EU alongside Haselmeier’s Diapen, as well as globally through Becton Dickinson’s reusable BD Classic and Owen Mumford’s Autopen. Then, came 3.0 ml cartridges and pens. All three components—pen, cartridge, and needle—were exclusively manufactured by Novo [8,9].
Safeguarding drug products with device engineering innovations: Intellectual property protection
All the novel designs of combination products are protected with exclusive patent rights. Intellectual property (IP) rights block companies from making acceptable mimics of innovators. Uncertainty in patent claims makes it difficult for generic manufacturers to mimic the patented design. Trade dress can prevent companies from using the same device design. Generic manufacturers can challenge patents as invalid, unenforceable, or not infringed when filing the respective abbreviated new drug applications (ANDAs) under the paragraph IV pathway. For the 53 branded inhalers approved from 1986–2020, for seven products, generic manufacturers, filed paragraph IV ANDA applications. The manufacturers sued for infringement in 26 of 68 patents listed at the time of filing or after filing. Sixteen (62%) of these 26 patents pertain to devices. This clarified most innovator companies’ strategies to protect products with device patents as shown in Tables 1 and 2. From the facts and figures, it is also concluded that 28 of these 53 products have device patents that are last to expire from the product patent landscape, which further emphasizes the argument [10–12].
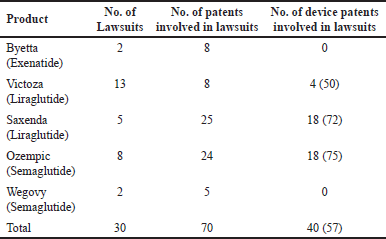 | Table 1. Device-related patent strategies for combination products. [Click here to view] |
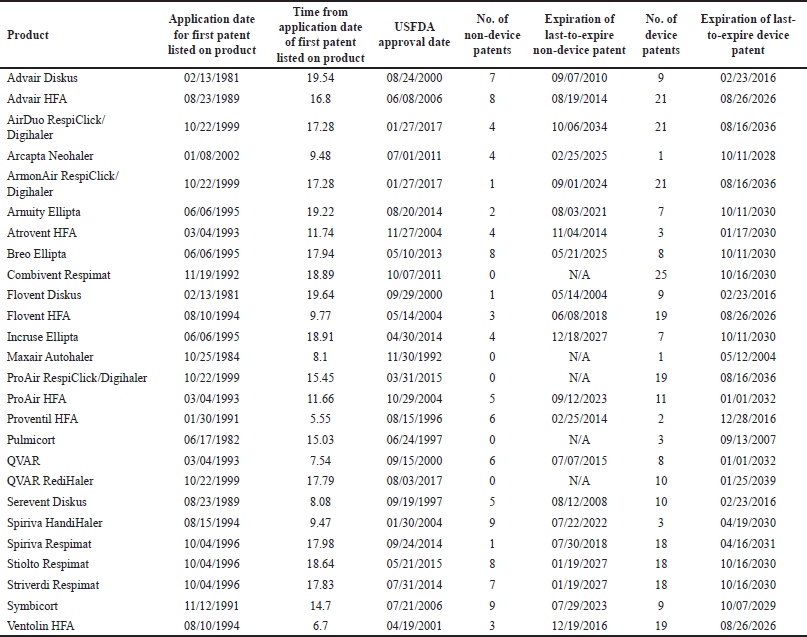 | Table 2. Using device patents as a strategy to extend market exclusivity. [Click here to view] |
Over the last 30 years, innovators of inhaler-based drug products have listed numerous patents with claims on devices but not drugs, and these device patents have been proven to be the strategy to secure market exclusivity for the long-term and inhibit early competition from generic product filers. In the United States, only three products in the group encountered generic competition through 2021: Advair Diskus (with the first generic launched in the first quarter of 2019), ProAir HFA (where the first generic emerged in the first quarter of 2020), and Proventil HFA (the first generic released in the second quarter of 2020). Only the ProAir HFA experienced early generic competition prior to the expiry of its patents. This item was granted 27.2 years of anticipated patent protection set to expire in 2032, comprising 18.9 years from non-device patents and an extra 8.3 years from device patents solely (none of which referenced active ingredients) [13].
Types and classes of devices and their applicability in pharmaceutical dosage forms
Based on safety and criticality, the devices are categorized below by the USFDA and the European Medicines Agency (EMA). When these different classes of devices are combined with drug products, regulatory authorities further verify their clinical safety and associated risk through their respective regulatory bodies based on the primary mode of action (PMOA)/principal action classification of devices by the USFDA and EMA showed in Figure 2.
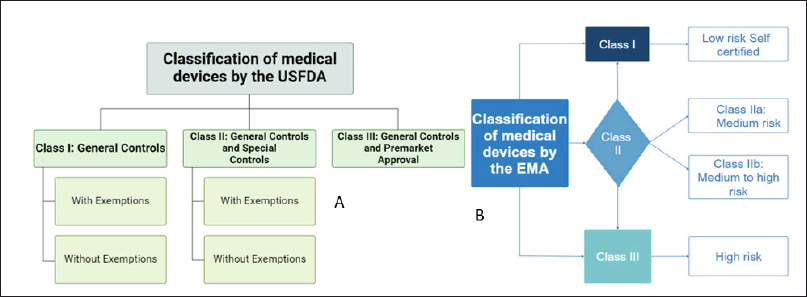 | Figure 2. Classification of devices by the (A) USFDA and (B) EMA. [Click here to view] |
REGULATORY CONSIDERATIONS FOR DDCPS BY THE USFDA
As detailed in the definition of DDCPs, the drug and device components are regulated by different regulatory bodies of the USFDA. The drugs are regulated by the Center for Drug Evaluation and Research (CDER) and are regulated by the Center for Biologics Evaluation and Research (CBER), whereas the devices are regulated by the Center for Devices and Radiological Health (CDRH). Since the components of DDCPs fall under different USFDA regulatory bodies, the selection of the primary regulatory pathway depends on the PMOA, which determines the main therapeutic function of the product. For example, when an insulin-prefilled syringe is filled with an agency, the PMOA of the insulin prefilled syringe, that is, to treat diabetes, is the function of the drug product, and the delivery of the drug product is supported by a prefilled syringe. Hence, the CDER that regulates drug products will review the application. On the other hand, when a drug-eluting stent is filed with the agency, the PMOA of the drug-eluting stent, that is, to dilate the blood vessel and prevent blockage, is the function of the stent, and the drug being eluted can be an auxiliary therapy. Hence, the CRDH regulates the devices to review the application. When DDCPs are filed for marketing authorization, the applicant’s responsibility is to ensure that both drug- and device-related regulations are complied. The core requirement of a combination product should be to ensure that the product is safe and effective, including delivery to the intended site of action. The fundamental requirements outlined in regulations ensure the implementation of systems that oversee the correct design, monitoring, and control of manufacturing processes and facilities. This involves establishing a rigorous quality management system (QMS), utilizing suitable high-quality raw materials, creating strong manufacturing and control procedures based on solid design principles, and identifying and investigating instances of product quality deviations. Furthermore, these regulations mandate the continuous evaluation of systems and the adoption of corrective measures as needed. The manufacturer of the combination product shall evaluate the PMOA and decide the pathway to be followed for approaching regulators. When the PMOA is chosen as the drug, the drug-related QMS regulations with respect to current good manufacturing practices (CGMPs), 21 CFR 210, and CFR 211 should be followed. In addition, manufacturers should demonstrate compliance with applicable device regulations. Similarly, when the applicant decides on the PMOA device, the device-related QMS regulations with respect to the CGMP, 21 CFR 4, and CFR 820 should be followed. In addition, manufacturers should also demonstrate compliance with applicable drug regulations. In the case of cross-labeled combination products, the organization should establish a policy governing assurance over ensuring compliance with respect to drug regulation for drug product manufacturing and device regulations for device components. For single-entity combination products and co-packaged combination products, part 4 outlines two methods for demonstrating compliance with CGMP requirements. The first method requires manufacturers to comply with all CGMP regulations applicable to each constituent part of the combination product. The second method allows for a streamlined approach, where manufacturers of DDCPs that include both a drug and a device can choose to comply with either the drug CGMPs (21 CFR parts 210 and 211) or the device quality system regulation (QSR) (21 CFR part 820) while also meeting specific provisions from the other set of CGMP requirements [14–16]. The USFDA, in its guidance for CGMP requirements for combination products, clarified options that can be adopted by combination product manufacturers to ensure CGMP compliance as shown in Tables 3 and 4.
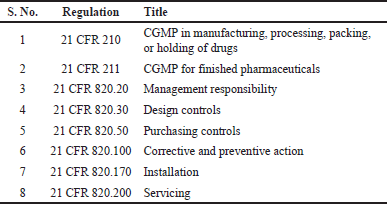 | Table 3. Applicable regulations when PMOA is a drug. [Click here to view] |
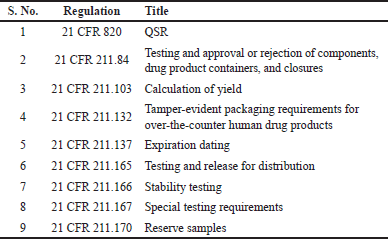 | Table 4. Applicable regulations when the PMOA is a device. [Click here to view] |
Regulatory considerations for DDCPs by the EMA
Like the USFDA , the EMA also has different regulatory bodies overseeing the approval process of drugs and devices. In Europe, drugs are regulated as per “Directive 2001/83/EC,” and devices are regulated by “REGULATION (EU) 2017/745.” During premarket assessment, the application will be reviewed under Directive 2001/83/EC for drug-related aspects, while the device component must comply with the general safety and performance requirements (GSPRs) of REGULATION (EU) 2017/745. Designated notified bodies (NBs) are appointed to review device applications. These firms are also responsible for reviewing combination product applications for compliance with REGULATION (EU) 2017/745. The assessment of NBs will be critically evaluated by authorities responsible for NBs as part of a risk-based approach and sampling of relevant documentation.
This risk-based review considers various factors, such as “action location,” where the device performs its function within or on the human body; “introduction or application site,” where the site on or in the body where the device is introduced or applied; and “systemic absorption,” where the substances that make up the device, or their metabolic products, are absorbed systemically (i.e., throughout the body).
Like the USFDA’s PMOA approach, the EMA determines the regulatory pathway via the concepts of the “principal” and “ancillary” modes of action. When a DDCP, either a single entity or a co-packaged product, is submitted for premarket authorization and if its PMOA/principal action is from a medical product rather than the device, the assessment of such an application shall be performed against Directive 2001/83/EC or Regulation (EC) No. 726/2004 of the European Parliament and the Council. In such cases, the relevant GSPRs set out in Annexure I of Regulation (EU) 2017/745 apply as far as the safety and performance of the device part are concerned [2]. In cases where the PMOA/principal action is from the device rather than the medical product, the designated NBs that are conducting the assessment of the device shall consult the designated member states or EMA for the medicinal product-related assessment performed according to Directive 2001/83/EC for the evaluation of absorption, distribution, metabolism, excretion, local tolerance, toxicity, interaction with other devices, medicinal products or other substances and potential for adverse reactions.
During the process of NB assessment for class II (class IIa and IIb), the designated regulatory organization will evaluate the combination product manufacturer’s technical documentation, clinical data, and QMS demonstrating the risk assessment and mitigation strategies, and compliance against applicable standards to confirm that the combination product is suitable for intended use. In the case of class III devices, the same regulatory organization viz., designated NB, will verify the outcome of clinical safety assessment and provides a legal approval with CE certificate.
Depending on the class of device (Class I, II, III), the combination product manufacturers should adequately justify the adoption of applicable regulations, and the approach being followed should be filed with the USFDA and EMA as part of the Common Technical Document (CTD) under the manufacturing section of the premarket submission. When the DDCP manufacturers/applicants of the marketing authorization application adopt a streamlined approach for design, development and manufacturing ensure that the device component is reviewed by relevant regulators and that appropriate approval is sought, as detailed in Table 6.
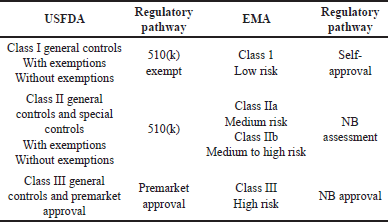 | Table 6. Regulatory approval for different classes of devices. [Click here to view] |
Additionally, a new drug application/ANDA should be submitted for drug product safety, efficacy, and quality evaluation. Regulators have set clear policies and procedures for the development of new drugs and incentivize innovators with market exclusivities for new molecular entities and new therapeutic indications. However, the trends of extended market exclusivity strategies from innovators over the last 30 years make it clear that although device patents do not claim a link with active ingredients, they are used to extend the market exclusivity associated with new drug approvals. With recent USFDA guidelines on CGMPs for DDCP and draft guidelines on comparative analysis and comparative use of human factor (HF) studies and EMA medical device regulations, generic DDCP manufacturers clearly compare their generic designs with innovators to prove that there are no other differences despite providing device performance for primary functions, making it difficult for generic manufacturers to enter the market. Thus, it is imperative for generic filers to find alternate strategies to design devices that are not critically different from innovator design and, at the same time, do not infringe on device patents to derive a path for successful filing and entering the market.
OVERVIEW OF USFDA-APPROVED DEVICES THAT ARE USED IN PHARMACEUTICAL DOSAGE FORMS AS DDCPS
Among the many devices that are regulated by the USFDA, the device types that are widely used in pharmaceutical dosage forms and when combined with drugs are treated as DDCPs are listed in Table 5. As detailed in Figures 3 and 4, based on the PMOA, the review authority should be determined.
 | Figure 3. Determination of the lead center for review of the application. [Click here to view] |
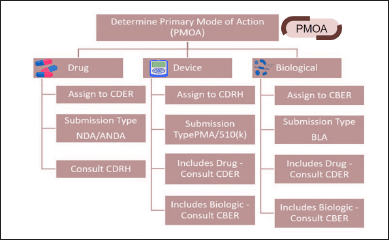 | Figure 4. Consultation between departments based on PMOA. [Click here to view] |
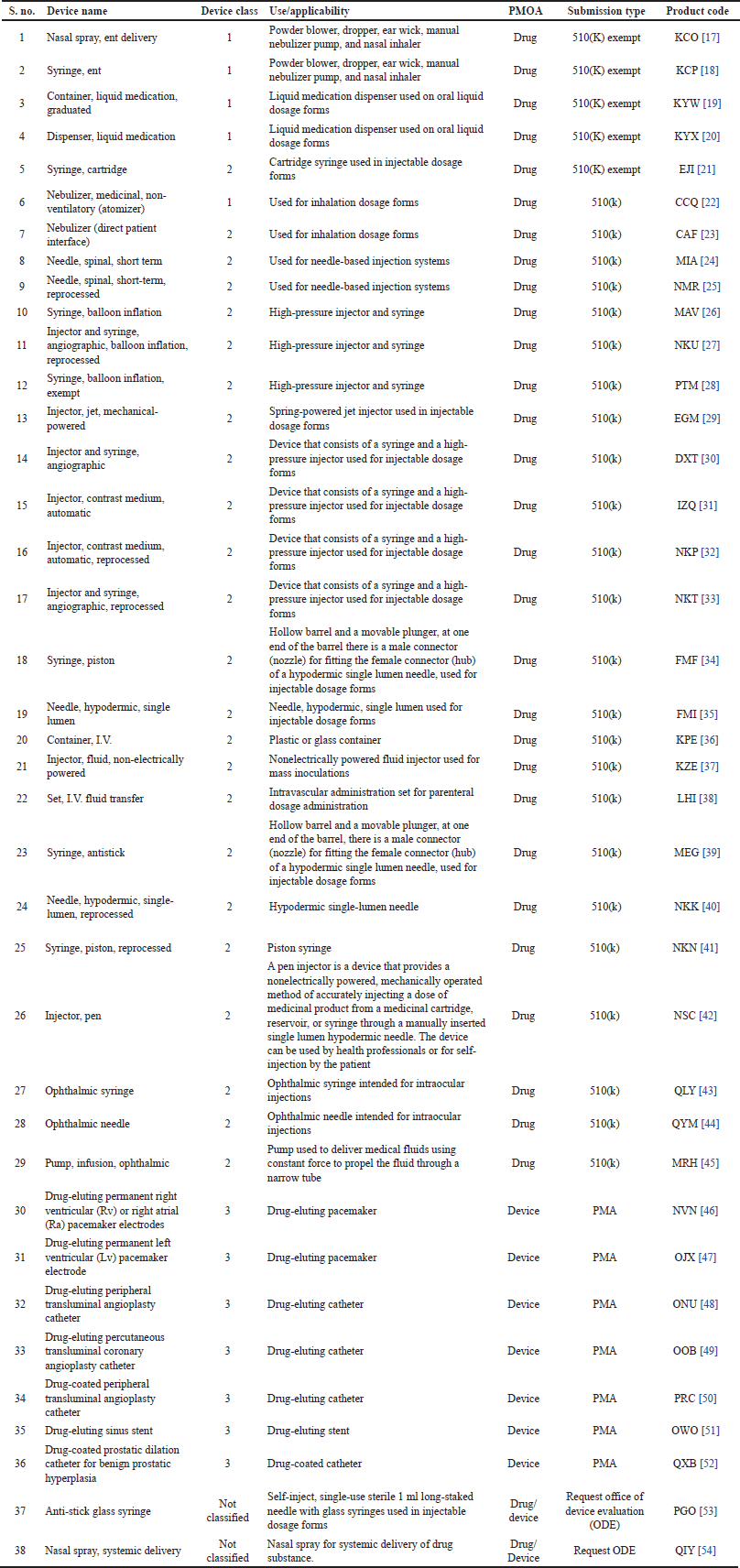 | Table 5. USFDA-approved devices used in pharmaceutical dosage forms as DDCPs. [Click here to view] |
DESIGN AND DEVELOPMENT OF DDCPS
Regardless of whether the PMOA is drug- or device-based, the goal of DDCP manufacturers is to prevent medication errors. These errors may arise from design flaws, manufacturing defects, or usability issues when a product is used by healthcare professionals or patients for self-administration. Therefore, incorporating robust design principles, risk assessments, and usability studies throughout the development process is critical. Hence, it is imperative for the manufacturers of DDCPs to consider the design-, manufacturing-, and usability-related errors that hinder successful medication administration. This can be achieved by comprehending the user’s requirements and correlating these requirements to build a robust design of DDCPs through risk assessment.
The user requirements can be segregated into three types: design requirements, process requirements, and application or usability requirements. The design requirements include the number of components that can come together to form the device and the ancillary components that are required to assemble with the actual device to enable dosage administration, the material for constructing all the device components, and the dimensional compatibility of all the components with each other to finally assemble the device. The process requirements include the number of steps involved in manufacturing the drug product, the device components, and its assembly. Each process or unit operation enlists the process steps involved, and a control strategy is established for identifying critical process parameters. The application requirements can be derived starting from how the user can access the commercially available pack and the steps involved in setting up the dose and administering the required dosage. The requirements for combination products should include considerations such as performance characteristics observed in predicated devices or from the literature searches, regulatory requirements, and operational requirements with respect to manufacturing and user safety requirements and should be identified and traced throughout the development process.
Design inputs should be derived early in the product development process to ensure that the development efforts are consistent with the intended use. Design outputs must be developed based on those inputs. The design outputs are the work produced at the end of design efforts, such as specifications, engineering drawings, compliance certifications to various regulatory requirements and safety-related certifications, assembling instructions, and usage instructions. During the design development process, it is important to identify the primary functions of the respective combination product, which, when not intended, will cause potential harm to the patient or will not accurately deliver the medicinal product. The consistency of the identified primary functions should be verified during design verification as part of combination product release testing, stability testing, stress conditional testing, and at the end of shelf-life testing.
Design verification data generated by device manufacturers as part of a streamlined approach should be considered as part of the combination product design verification. Wherever the functional tests are dependent on drug product compatibility with devices, they should be verified on combination product batches. However, documented evidence must be collected from device manufacturers to ensure that the primary functions are tested and that quality is ensured by device manufacturers. As part of design verification, DDCPs should be preconditioned for stress studies such as transportation simulations, and any failure that occurs during primary function testing post-exposure indicates that the secondary and tertiary packing does not provide sufficient protection to the DDCPs, and upon redesigning the secondary and tertiary packing, the study should be repeated. For example, in the case of a typical prefilled pen, the design requirements can be as follows: the fill volume of the cartridge that contains the drug product; the material used to construct the primary container that comes into contact with the drug product, such as a rubber stopper or needle; the dimensional design requirements of the cartridge height, diameter, rubber stopper, and cartridge housing; the thread dimension of the cartridge housing to be compatible with a suitable needle; and the ability to fix features of the cartridge housing to the pen engine body. Furthermore, the mechanical interactions of components within the pen engine assembly require intervention from the mechanical engineering domain. The process requirements can be enlisted as the fill volume required to administer the labeled dose, which can be derived from the summation of the residual volume and deliverable volume of a cartridge. The volume is a function of the process parameters of the filling machine, such as the stopper insertion depth, type of insertion, either by force or by vacuum, and mode of filling, followed by stopper insertion or stopper insertion followed by filling, depending on the type of filling machine. Similarly, the unit operation and critical process parameters for each of the unit operations of the device assembly should be evaluated as part of the device manufacturing and assembling process. The manufacturing process of devices can include molding the plastic or glass granules into the required design through heating, holding, and cooling. The assembly process of the device can include various stations working in sequence for placing the device components, applying adhesive, and assembling the device components either through mechanical force or heat.
The user requirements can be derived as ease of opening the marketing pack; accessing its contents, such as prefilled pens, needles, patient information leaflets, and instructions for use; easily understandable steps and depictions of instructions for use; fixing the opening cap of the prefilled pen by the targeted age group of users; the dimensional compatibility of the needle with the prefilled pen; setting up the required dose in the case of multiple doses; and the forces required to actuate the pen and inject the dose by the targeted age group of users. Adequate risk assessment of each of these aspects of design, process, and usability is necessary to ensure that when all the components of the drug and device come together as a combination product, the dosage administered should be accurate.
The quality is built into design and processing to ensure that the final DDCPs confirm the intended and committed specifications. The QMS should be designed to ensure that the appropriate material is selected through qualified vendors and that incoming material should have inspection control to ensure that the right material is selected for manufacturing the DDCPs. Appropriate process control should be designed to ensure that the processes are operating within the design space of parameters and, when tested as a pool of samples, provide a representative overview of the population of batches, that is, manufactured. Acceptance activities should be adequately derived to control the critical quality attributes of DDCPs. Any nonconforming product should be investigated to identify the root cause and appropriate corrective, and preventive actions should be implemented. In the case of drug products, safety can be ensured by conducting successful clinical trials. In the case of generic drug products, bioavailability, and bioequivalence in line with the innovator product can ensure the safety of the drug product. The efficacy of drug products can be measured by evaluating the quality of the active ingredient available in the drug product through an assay and ensuring biological performance through simulated testing, such as the pharmacokinetic profile of in vitro dissolution, and the quality of the drug product can be confirmed through testing for degradation products and uniformity within batches and between batches for conformance to the specification and process control.
In the case of a device, efficacy and quality can be ensured by verifying the device design for its intended performance through testing for delivering the required labeled amount of drug per dose setting or actuation, and safety can be evaluated by studying usability-related errors and ensuring adequate controls to mitigate usability errors. Device attributes that are necessary to achieve targeted safety and functionality through demonstrating that the DDCPs consistently deliver the drug product according to the specification and are biocompatible. It is important to evaluate HF studies that are specific to the context of the use of DDCP. The context of use can include various aspects, such as urgency, emergency versus nonemergency, frequency of use, single use versus repeated use, end-users, patients, caregivers, healthcare professionals, the environment of use, clinical hospitals, clinics or non-clinical homes, schools, the patient population, dexterity issues, and incapacitation. The tasks associated with administering should be segregated as critical and noncritical to the administration of medication.
USFDA outlined a development approach to HF studies demarcating the different phase phases viz., formative and validation studies. The formative HF studies to be conducted at the early stage of new combination product development and the learning outcomes of this formative study would serve as feedback studies to identify risk during the early stages of development and mitigate the design-related risk through rework/redesigning the combination product. Once the combination product design is verified for its performance in vitro as per respective standards specific to different device types, the final combination product shall be evaluated through final HF validation studies. In the case of EMA, though the agency does not warrant the requirement of specific HF studies such as USFDA, it requires clinical evaluation and safety assessment reports, which can provide assurance that the product does not pose any use-case-related errors. This is further evaluated and ensured as part of CE certification through clinical usability data and risk management and mitigation strategy files [55–57].
The effectiveness of device usability for generic DDCPs can be demonstrated through comparative use-related HF studies and has become more imperative in the case of generic DDCPs. For each physical, task, or labeling comparison performed during comparative analysis, one should conclude whether there are any differences and if the differences exist, whether they are minor or other external design differences.
The differences are minor if the difference in the user interface of the proposed generic combination product, in comparison to the user interface of the innovator, does not affect an external critical design attribute (Fig. 5). The other external design differences are those if any aspect of the comparative analyses suggests that differences in the design of the user interface of a proposed combination product compared with the innovator may impact an external critical design attribute that involves the administration of the product. When the comparative analysis identifies other design differences, it is necessary to redesign the user interface to minimize the difference from the innovator or predicate device.
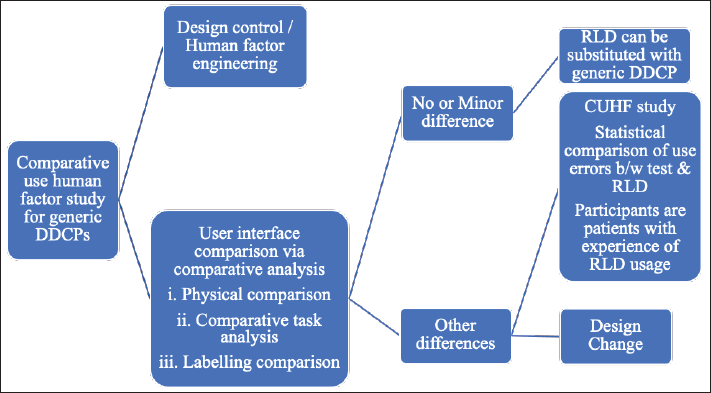 | Figure 5. Comparative use of HF studies for generic combination products. [Click here to view] |
Business model for the design and development of DDCPs
The first and foremost important aspect of the design and development of DDCPs is to identify the various parties that are involved in the project. Often, deciding the various engineering, biomedical, and pharmaceutical aspects of DDCPs is time consuming. Any critical aspects that are not given enough attention can result in costly failures and will be detected only upon their occurrence. Hence, companies can adopt different strategies in deciding their business model [58–60].
In the vertical business model, the end-to-end responsibility is to design, develop, and manufacture both the drug and the device being handled by a single firm. This model requires functional and subject matter expertise in various areas, such as engineering, biomedical, and pharmaceutical development and manufacturing. This can be a costlier approach and time-consuming, as the project plan is difficult to execute in parallel. In the horizontal business model, multiple firms that are experts in individual domains can come together to execute the project in parallel, for example, parallel development of formulation and device design. However, the integration and alignment of individual owners with a common goal can be a potential rate-limiting step. The vertical business model is more suitable for single-entity combination product manufacturers, where the manufacturer can adopt a streamlined approach for complying with either 21 CFR 211 and the applicable section of 21 CFR 820 or vice versa, depending on the PMOA. In contrast, for ease of operability and QMS governance, the horizontal model is best suited for organizations that are involved in the manufacturing of cross-labeled DDCPs, where separate organization entities are responsible for manufacturing drug and device constituents and can ensure compliance with the respective regulations of 21 CFR 211 for drugs and 21 CFR 820 for devices. The approach for the appropriate business model section can be evaluated for its strengths and weaknesses to choose the best-fit model for the organization.
A case-driven analysis and strategic framework for business models in the development of DDCPs
The strategic business plan that supports the development and market launch of DDCPs is just as vital to their commercial success as the technology that goes into them. As regulatory requirements, worldwide supply chains, and cross-functional product design become more complicated, companies need to create business models that fit their own strengths, partnerships, and goals for the product’s lifecycle.
In-house integrated development
Integrated in-house development means that all parts of drug, device, and interface development are done in-house. This gives you the most control over IP, system design, and compliance with regulations. This concept is popular with big pharmaceutical companies that already have experience in device R&D because it lets them combine formulation optimization, software, and hardware all under one roof.
Case study 1: The NovoPen Echo Plus (Novo Nordisk with Medtronic connectivity) is a case study that shows how careful control over the design of the device leads to better alignment between the drug and the device. Novo Nordisk made it all by themselves, with help from Medtronic for the digital part. The connection made it easier to include digital features such as Bluetooth transmission and insulin logging, and it solved interoperability problems early in development. This made the USFDA and European Union Medical Device Regulation (EU MDR) compliance procedure more efficient [61,62].
Case study 2: The Accu-Chek Insight pump system, created fully in-house by Roche Diabetes Care, had built-in glucose monitoring and automatic insulin dosing. Compared to standard care, this single-entity model had therapeutic benefits, such as a 0.8% drop in HbA1c over 6 months, and it allowed for the best integration of software and hardware [63].
Strategic alliances and joint ventures
This model includes co-development partnerships that bring together the resources and skills of drug and device partners. It is especially good for companies that want to find a balance between sharing risks, specializing in external devices, and the speed of innovation.
Case study 3: The Amgen-Medtronic Neuromodulation Platform for Migraine Treatment Amgen and Medtronic worked together to build a neurostimulation platform that leverages erenumab and Medtronic’s neuromodulation technology. The agreement made it possible for the two companies to share the work of developing and regulating, which sped up the process [64].
Case study 4: BD-Medimop smart prefilled syringe for rheumatoid arthritis biologics, BD and Medimop Medical Projects worked together to make prefilled syringes with radio-frequency identification (RFID) technology that make it easier to trace cold-chain shipments. A study done at several centers found that embedded RFID technology made handling drugs easier, which led to a 15% decrease in drug waste [65].
Case study 5: Lilly-Ypsomed Bluetooth-enabled smart pen.
Lilly worked with Ypsomed to make an insulin pen with Bluetooth by using Ypsomed’s knowledge of EU MDR regulations. This made the development process less risky and sped up the process of getting European regulatory approval [66,67].
Advantages:
- Sharing the cost of development and technical risk.
- Having specialist abilities, including knowing how to integrate software and the EU MDR.
- Faster access to the market with clear roles and data sharing.
Limitations:
- Disagreements over IP and governance.
- Goals for making money that do not match up.
- Problems with integration while scaling up.
Contract Development and Manufacturing Organizations’ participation
Businesses can access specialized platforms for design, development, and production without having to spend money on their own infrastructure by collaborating with Contract Development and Manufacturing Organizations. This model is very useful for biological-device interfaces since it lets you easily change the size of the capacity and speed up the time it takes to make changes.
Case study 6: Sanofi-Unilife Wearable Injectors Sanofi hired Unilife to make wearable injectors for biologics. This made it possible to quickly customize the injectors and get them to market faster. This partnership made it possible to meet accelerated development goals for new biologics that need delivery systems that are easy for patients to use [68].
Case study 7: Teva-Catalent elastomeric pump.
Teva was able to make the first batches of the device for humans 3 months ahead of schedule and cut down on the time it took to scale up by 30% by leveraging Catalent’s technology and production platform. The product, which delivered migraine therapy subcutaneously, benefited from Catalent’s knowledge of regulations and their GMP-grade device testing methods.
Case study 8: Genentech-SHL cancer autoinjector.
Genentech hired SHL Medical to help them combine their cancer biologic with an easy-to-use autoinjector. It got USFDA approval in 26 months, which is around 30% faster than Genentech’s own projects of the same magnitude [69].
Advantages:
- Less money is spent upfront.
- Infrastructure that is already in place speeds up development.
- Support for regulatory papers and manufacturers that can grow.
Limitations:
- Problems with IP protection and oversight.
- Relying on quality methods from outside.
- There is a risk in putting off solving problems during design verification.
Platforms as a source of innovation
Platform models use delivery systems that have been validated and are modular, so they may be used with diverse medication portfolios. These systems allow quick development cycles, regulatory bridging strategies, and a lower strain for revalidating devices.
Case study 9: Amgen Repatha® with West Pharmaceutical SmartDose®.
Amgen used West’s SmartDose® technology to give Repatha® under the skin. Using a platform device made it easy to add more monoclonal antibody products and cut down on the time it took to get regulatory approval [70,71].
Case study 10: Philips Caire® companion inhaler platform.
This platform makes it easier to give corticosteroids and bronchodilators, and it is licensed to a number of pharmaceutical partners. A registry study that found a 20% drop in COPD flare-ups backed up the need for modular, reusable designs even more [72].
Case study 11: Teva calcitonin gene-related peptide (CGRP) migraine delivery system
Teva sped up the launch of its CGRP-targeting biologic by 18 months and made home delivery viable through modular autoinjector technology by using a pre-validated device platform [73–76].
Advantages:
- Less tasks related to making devices and getting them approved by the government.
- Flexibility between goods and portfolios.
- Simplified submission with data bridging.
Limitations:
- Not all formulas will work with them.
- You need to revalidate your certification if there are big changes.
- Not much freedom to change the design.
Buying and licensing
Businesses can bypass early development and get rapid access to unique technologies or delivery systems by buying or licensing inventions from other sources.
Case study 12: GSK buys verb surgical.
Glaxo Smithkline Pharmaceuticals (GSK) immediately entered the market for robotic-assisted pharmaceutical delivery by buying Verb Surgical, which was a cooperation between J&J and Verily. The acquisition put GSK at the top of the list of companies that offer digital surgical remedies [77].
Case study 13: BD infusion device expansion: BD improved DDCP supply chain control and reduced operational redundancy by selectively acquiring infusion device assets to increase their biologics delivery portfolio [78].
Advantages:
- Instant access to cutting-edge technology.
- Increasing the portfolio with little internal research and development work.
- Get to the market quickly to stand out from the crowd.
Limitations:
- Risks that come with integration after a merger and high prices of buying companies.
- There may be a difference between the history of regulations and QSs.
- Problems with old documents and integrating into a new culture.
A global look at the complicated rules and long waits for approvals of DDCPs
Even while DDCPs could lead to new ideas and have therapeutic efficacy, they may have to wait a long time to be approved. These delays are caused by a lack of clear rules in different places, conflicting rules around the world, and different standards for documentation, classification, and HF evaluation.
United States: Centralized coordination even though oversight is not connected
The USFDA’s Office of Combination Products (OCP) is in charge of the U.S. regulatory system. It works with CDER (drugs), CDRH (devices), and CBER (biologics). This centralized coordination aims to make combination product evaluations easier and clarify regulatory authority by using tools, such as the 21 CFR Part 4 cGMP rule, the pre-request for designation (Pre-RFD) system, and formal inter-center consult channels. However, real-world experience has shown that there are still certain long-lasting problems, even with these structural changes:
Case study 14: Betaseron® autoinjector (Bayer).
This device was delayed for 14 months after it was submitted in July 2016 till its approval in May 2017, largely because the USFDA needed further proof of HFs. The delay was blamed on not knowing how to do usability studies and what to expect after submission [79].
Case study 15: Symjepi™ epinephrine injector (Adamis Pharmaceuticals Corp.).
The company submitted the product through the 505(b)(2) regulatory process and worked with the USFDA early on, but the product was delayed by 15 months from March 2016 to May 2017 because of more PMOA considerations and late-stage summative usability testing requirements [80].
These examples show how hard it is for regulators to keep track of everything when there are overlapping jurisdictional evaluations and different expectations, especially for DDCPs that have connectivity features, embedded software, or digital health functions.
European Union: Delays and dual authority caused by Article 117
The EU made big changes to how DDCPs are regulated when they put Regulation (EU) 2017/745 (MDR) into effect. Most importantly, Article 117 said that drug-led combination therapies with a device part must have an opinion from an NB on whether the device is compliant. This sets up a dual authority approach that requires the EMA and a selected NB to work together.
Even though its objective was to improve device supervision, this structure has caused problems with regulation:
Case study 16: Bioton S.A. insulin injector.
The producer had to prove that the integrated device met the standards after the fact, even though it had already been used before the MDR classification. This created a lot of delays in getting an NB opinion [81].
This example show how the complexity of MDR, especially the differences between EMA and NB procedures, can make it harder to launch products at the same time, cause longer delays, and make dossiers more redundant.
Japan: No integrated DDCP framework or parallel routes
There is not just one way to apply for DDCP; instead, Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) has different rules for drugs and devices. This regulatory structure requires parallel evaluations, which usually means more testing, separate dossier requirements, and long approval times.
Case study 17: Neulasta® OnproTM Kit.
The OnproTM kit was held up in Japan for 18 months, even though it was approved in the United States within the normal review times. PMDA called for more post-marketing usability studies than the USFDA did since the two agencies had different expectations for post-approval evidence [82].
Case study 18: Lilly connected smart pen.
The Japanese release of Lilly’s smart insulin pen was delayed by 14 months because the PMDA required revisions to the technical dossier that were particular to Japan and longer surveillance mandates [83].
Case study 19: Dexcom G6 CGM system with insulin pump integration.
The PMDA’s extra review of wireless interoperability and human aspects paperwork that the USFDA did not ask for delayed the approval of this system by 18 months [84].
These examples highlight how Japan’s lack of a single review process leads to broken submissions, unclear rules, and missed chances to make money.
The main reasons for delays among regions
Even though regional authorities have made considerable progress in recognizing DDCPs as separate product classes, the lack of common definitions, submission methods, and review standards still makes it hard for people all around the world to access them. The examples above show how important it is to have regulatory frameworks that are more reliable, consistent, and cooperative, and that take into consideration the hybrid character of DDCPs and how their technology interfaces are always developing.
Regulatory review of DDCPs in different places
As DDCPs include more digital components, complicated delivery methods, and patient-centered features, it has become harder to get quick approvals around the world. National markets, especially the US, EU, and Japan, have come up with rules to control DDCPs. However, these rules are still very different when it comes to how products are classified, how far the rules apply, and what is expected of technical dossiers.
A timeline-based look at how well different regulations work
More and more evidence shows that DDCPs in the EU and Japan take 12–24 months longer to get cleared than those in the United States. This is largely due to submission processes that do not line up, too many HF studies, and PMOA designations that do not agree. The main reason for these delays was that Japan’s PMDA and EMA/NBs required separate evaluations that were the same, especially for HF validation.
Different rules for HFs
One of the most common reasons for delays in DDCPs across jurisdictions is the need for HF assessment, which includes designing and using methodologies to check usability. The USFDA prefers that the final-use environment be integrated into both study design and root cause analysis. It also requires that usability be assessed in a summative way according to IEC 62366. Even though EMA and NBs follow similar rules, they often ask for more risk documentation under EU MDR Annex I. This is because sponsors have to show that their devices meet the GSPRs. The PMDA often needs separate HF investigations that are specific to Japan, even when there is evidence from throughout the world. Sponsors must provide regionally contextualized user interface data to duplicate test infrastructure and extend timelines. Because there is no mutual recognition or universal approval criteria, protocols are broken up, and expensive revalidation attempts are needed.
Problems with classifying the PMOA
The PMOA of a DDCP is very important since it sets the lead regulatory center and helps with the submission process, such as the required paperwork and technical requirements. The USFDA employs the PMOA to decide which combination of goods go to CDER, CDRH, or CBER. This is supported by the Pre-RFD and OCP processes, which give early jurisdictional clarification. The EU can award PMOAs to drug-led products that have device parts under Article 117 of the MDR. This requires an NB opinion, but there is no easy way to settle disputes. Japan, on the other hand, does not have a unified DDCP structure. The PMDA evaluations are split between the Medical Device Evaluation Division and the Pharmaceutical Review Division, and there is no Pre-RFD equivalent to clear up any confusion about PMOA. As with other delayed launches such as OnproTM, Companion Inhaler, and Dexcom G6, this means that the same device is not regulated the same way in different places.
Dossier architecture that is not well-organized and document redundancy
There are further problems because there is not a common worldwide CTD for DDCPs. ICH M4 structures can usually be used for drugs, but device parts and interface elements sometimes need their own annexes for each region, such as
- USFDA device master files.
- Opinion packages (EU MDR).
- GSPRs (EU) and QMS documents.
- Dossiers for devices that work on their own (PMDA).
This variety makes it take longer to put together, adds more work for authors, and creates unnecessary paperwork, especially for sponsors that work in all three main marketplaces.
What sponsors should think about while making decisions
These changes in jurisdiction have a big effect on DDCP lifecycle planning:
- A delayed launch period for first-in-class technology makes it harder to defend market share and make money.
- For fragmented submission planning, you need dedicated cross-functional regulatory intelligence teams and specialist ways to gather evidence.
- Emerging countries often utilize the same strict device assessment methods as the EU or Japan, which makes submissions more complicated.
- Therefore, for agile DDCP development to work, there needs to be regulatory convergence or, at the very least, reliable planning tools that work across jurisdictions.
Even while worldwide authorities agree that DDCPs are important and one-of-a-kind, the lack of harmonization within regulatory frameworks makes it harder for people to access multiple markets. The regulatory authorities need to quickly agree on mutual usability data, standardize PMOA interpretation, and create a global CTD architecture that works for DDCPs. Without these changes, product developers will keep having to wait, do unnecessary testing, and use up a lot of resources on regulatory tasks.
Ways to make approvals for international DDCPs easier
To fix the ongoing regulatory inefficiencies talked about in the previous sections, we need strategic frameworks that can systematically cut down on approval delays, get rid of unnecessary testing, and help DDCPs manage their lifecycles in a flexible way across multiple regions. Early development decisions must follow international rules, especially when it comes to quality control, PMOA classification, technical documentation, and HFs.
Early involvement of regulators and clear jurisdiction
The best way to lower the danger of regulatory timeframes is to work with regulatory authorities from the start of development. Early interaction makes sure that everyone agrees on PMOA, the jurisdictional lead authority, and documentation expectations.
Through the USFDA’s Pre-RFD process, sponsors can officially find out early on what the product’s PMOA and center assignment (CDER, CDRH, or CBER) will be. This system has helped firms speed up integrated DDCP review operations and cut down on submission misclassification. EMA Scientific Advice encourages parallel contacts with regulators and NBs, especially when Article 117 reviews are likely to happen. Following the EMA criteria for borderline classification and device documentation, requirements can considerably cut down on the number of review iteration cycles. As long as sponsors get involved early and send in coordinated development plans, Japan’s PMDA Sakigake Designation gives innovative goods that meet important unmet needs a faster path to market. Even though Sakigake is not DDCP-specific, it encourages people to talk about parallel dossier design and regulatory data needs [85].
Integrated design for compliance across courts
DDCP developers should use a “compliance-by-design” approach based on internationally agreed-upon standards from the start of the project. This strategy does away with the requirement for reengineering that is exclusive to a certain area and makes it possible to set up devices and data packages that can grow. Use standards that are approved all across the world, such as ISO 10993 for biocompatibility, ISO 14971 for risk management, ISO 13485 for device QMSs, and IEC 62366 for usability. To make sure that quality, design controls, and process validation are the same all over the world, use the ICH Q8–Q11 criteria and the International Medical Device Regulators Forum (IMDRF) basic principles. Make sure that cross-jurisdictional HF studies are prepared from the outset to meet the needs of the USFDA, EMA, and PMDA all at once. Include explanations for any user interface peculiarities that are specific to each location.
Implementation strategy: Both the injector and the inhaler systems can employ risk control matrices that can be changed and usability procedures that can be added to. A single master design file with regional annexes helps regulatory flexibility and cuts down on rework.
Sequencing of market entry and strategic regulatory mapping
For the best regulatory plan, the combination product developers need to carefully map out regional timelines, the complexity of the dossier, and the commitments that come after marketing. Sponsors should plan market releases carefully based on how strong the dependence routes are and how likely it is that regulations may change. Because the USFDA Type B/C meeting structure is predictable, the PMOA designation tools are better, and the combination product instructions are clearer, U.S. first releases are often given precedence. After the CE-marking of the gadget and the completion of the NB assessments as required by Article 117, EU launches may come second. Japan launches are appropriate as phase 3 entrants after worldwide P3 data availability, although they normally require longer lead times because of dossier fragmentation and additional local testing.
For example, Amgen’s Neulasta OnproTM kit adopted this idea and won clearance in the United States before working out more complicated PMDA and EMA requirements. This step-by-step method got the most out of each market and sped up global debuts.
Global validation strategy and standardized HF protocols
HFs testing is still one of the most unevenly understood parts of the law in all DDCP jurisdictions. The goal of sponsors should be to combine the HF methodology with internationally accepted validation methods. Use IEC 62366-1 as the worldwide reference framework, but explain why local changes are needed based on things like language and differences in healthcare systems. To make it possible to make unified HF data, set up important user groups, task analyses, and use-related risk categories that can be used in all areas. If there are little modifications in the user interface or labeling between markets, make bridge studies or justification reports to cut down on the number of new HF studies that need to be done. By employing unified HF protocols, sponsors can speed up the review and validation process by sending the same basic usability report to the USFDA, EMA (as part of the NB opinion), and PMDA with only small changes made to it.
QMSs are a mix of different types
DDCP developers have a hard time putting together device and drug GMP standards into one, unified QMS. The answer is to build hybrid QMS architectures that mix ISO-compliant systems with the USFDA’s GMP requirement for combination products.
- Make sure that submissions submitted in the United States follow 21 CFR Part 4, which includes device QSR (21 CFR 820) and medication GMP (21 CFR 210/211).
- To make sure that the QMS helps the device meet the GSPR under MDR, make sure it works with ISO 13485 for submissions from the EU.
- Connect quality events and corrective action and preventive action procedures around the world to make sure that change control, treatment of nonconformances, and traceability are all in place for both drug and device parts.
- Use centralized document management systems with separate access roles for device and pharmaceutical quality assurance to make sure that both USFDA and NB inspections are ready at the same time.
Participation in the regulatory convergence and reliance initiative
One approach to help bring the world’s rules into line with each other more quickly is to take part in or back multilateral efforts like:
- IMDRF work items on combination goods include frameworks for classifying devices and templates for clinical evaluations.
- ICH M4 and Q12 suggestions, which are the basis for managing changes after approval and making sure that the lifecycle is consistent.
- The FDA and medicines and healthcare products regulatory agency (MHRA) are working together on pilots to look into how to share usability test data and device parts.
- The Access Consortium (Canada, Singapore, Australia, and the United Kingdom) offers a collaborative evaluation platform that allows for coordinated regulatory submissions across a number of English-speaking countries.
For example, getting the technical file ready early has led to fewer resubmissions and clearer expectations for sponsors that are part of IMDRF pilot programs.
Using early participation, standardized design limitations, and reliance on modular, cross-compatible data frameworks in development can lead to a globally expedited regulatory pathway for DDCPs. By putting money into regulatory information and using global best practices in core development operations, sponsors may reduce the amount of rework needed in different jurisdictions, speed up approvals, and get the most out of the lifetime of new combination medicines.
CONCLUSION AND FUTURE RECOMMENDATION
The demand for specialized delivery systems such as DDCPs is continuously increasing over conventional drug products due to an increase in lifestyle diseases that require self-administration, devoid of potential for use errors. DDCPs are a revolutionary new way to combine cutting-edge engineering with drug development. DDCPs combine biologics, small chemicals, and medical devices into coherent treatment systems that enhance patient outcomes by making it easier for patients to stick to their medication, delivering it to the right place, and giving them personalized care. With the advent of DDCPs from pioneers in pharmaceutical industries such as Novo, Nordisk, and Hoechst, Lilly, Becton Dickinson, which led the path to the development of combination products, it is imperative to choose suitable business models depending on the domain and expertise of a specific firm and to develop new or non-infringing device designs to address unmet patient needs. Although the regulatory pathways remain specific to the processes of the respective regulatory bodies of the USFDA and EMA, the intent of regulators remains the same to ensure the safety and efficacy of drugs and devices. The discretion of DDCP manufacturers is to choose the best-fit business model via a vertical or hybrid approach and to streamline the technical package for regulatory filing while considering the principal function/PMOA of DDCPs. This clinical promise is limited by sophisticated development needs, problems with cross-functional integration, and regulatory fragmentation, which require equally complicated business and regulatory solutions.
Real-life case studies to teach strategic lessons
Successful DDCPs, such as Advair Diskus®, Neulasta®, and OnproTM, have changed over time, showing how important it is to have scalable delivery forms, early regulatory alignment, and design that works well together. These goods kept their market share and increased their lifecycle value by adding user-friendly features, which were frequently the result of extensive HFs research and proprietary delivery mechanisms. On the other hand, the delayed debuts of SymjepiTM show how much it costs to use reactive design methods, have unclear jurisdiction, and have rules that do not match up. These events show how important it is to start planning for global regulations as soon as feasible and to make sure that testing techniques and paperwork may be used in different parts of the world.
The growing roles of digital, AI, and sustainability in DDCPs
DDCP innovation will be more and more defined by the following in the future
- AI-enabled personalization: Titration algorithms, closed-loop insulin systems, and smart inhaler feedback loops are all making DDCPs more like therapeutic ecosystems that can work on their own. These systems need frameworks for validating data in real time, integrated cybersecurity risk management, and advanced software development lifecycles.
- Creating gadgets with the environment in mind is becoming more and more significant in the field of sustainable technologies. The use of reusable electromechanical autoinjectors, biodegradable subcutaneous injectors, and dry-powder inhalers with low greenhouse gas potential propellants shows a move toward the circular economy. Regulators such as Health Canada and the EMA are beginning to include environmental factors in health technology assessments.
- Validation of real-world performance: The rise of real-world evidence (RWE) frameworks, especially those created by the USFDA, MHRA, and EMA, opens up new opportunities for adaptive approval and post-market label expansion. Smart DDCPs with built-in sensors can provide real-time data to RWE systems. This lets payers evaluate results and do a dynamic risk-benefit analysis.
Using both product and platform economics in hybrid business models
DDCP commercialization is going to be based on hybrid value models that encompass digital health services, data analytics, platform licensing, and pharmaceutical items.
- Cloud connectivity makes subscription-based device ecosystems viable. These models give patients medicine delivery and ongoing services such as dose tracking and reminders to take their medicine.
- Data monetization partnerships: Pharmaceutical companies are working with digital companies more and more to leverage anonymized patient-use data for payer negotiations, adherence analytics, and clinical trial optimization.
- Modular platform extension: Device platforms such as SmartDose® and Caire® companion make it easy to get into related therapeutic areas quickly by allowing design reuse, regulatory bridging, and minimum post-approval validation.
These models call for a growing regulatory framework that may look at the combined pharmaceutical, software, and service parts, in addition to changes inside the company itself.
Global regulatory strategy needs
Sponsors that want to take part in a highly competitive and quickly changing global market need to follow these strategic imperatives while developing and commercializing DDCP:
- Keep the end goal in mind: Make sure that the design inputs fit with long-term market, access, and regulatory goals. Include early HFs, cybersecurity, and supply chain risk management with regulatory foresight.
- Get involved early and often: Before important trials or design lock, use tools such as Pre-RFD, EMA Scientific Advice, and PMDA talks to settle PMOA, documentation, and usability needs.
- Standardize, and then adapt: Make standardized QMSs, HF procedures, and modular submission packages that can be changed to fit the needs of any jurisdiction.
- Get ready for the real world: To stay up with the growing need for real-world data in regulations, put money into digital infrastructure that makes it easier to do post-market surveillance, monitor performance in real time, and create evidence.
Looking ahead
To sum up, the next generation of DDCPs will be more than just drug delivery systems; they will be versatile therapeutic platforms that are part of bigger care ecosystems. The project’s success will depend on how well the sponsors can coordinate the development of technology, compliance with international regulations, and value-based commercialization initiatives. Developers can use the frameworks in this manuscript to get past approval bottlenecks, get into international markets faster, and offer high-impact treatments that raise the bar for care. These frameworks cover business model innovation, regulatory alignment, harmonizing usability protocols, and QS integration.
It is time to act now. Patients, payers, and regulators all want healthcare that is sustainable, connected, and tailored to their needs. DDCP developers must meet these needs with a bold, system-wide change, not just in small steps.
ACKNOWLEDGMENTS
The authors would like to express their sincere gratitude to Manipal College of Pharmaceutical Sciences, Manipal Academy of Higher Education (MAHE), Manipal, for their invaluable support in conducting this work.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the International Committee of Medical Journal Editors (ICMJE) requirements/guidelines.
FINANCIAL SUPPORT
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
The authors report no financial or any other conflicts of interest in this work.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Office of Combination Products, Food and Drug Administration. Current good manufacturing practice requirements for combination products: guidance for industry and FDA staff. U.S. Department of Health and Human Services; 2017 [cited 2024 Aug 7]. Available from: http://www.fda.gov/oc/combination
2. European Union. Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009, and repealing Council Directives 90/385/EEC and 93/42/EEC. Off J Eur Union. 2017 May 5;L 117/1 [cited 2024 Aug 7]. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0745
3. SKU: 47598710, GTIN: 00382904759872, BD: BD-100221, BD Hylok™ SCF 1MLL RF 7025/65 PP SHB. BD [Internet]. [cited 2024 Aug 7]. Available from: https://www.bd.com/en-in/products-and-solutions/products/product-page.47598710
4. Novo Nordisk Global. Pens and needles/NovoPen® 4 [Internet]. [cited 2024 Aug 7]. Available from: https://www.novonordisk.com/our-products/pens-and-needles/novopen-4.html
5. XIENCE Skypoint™ Stent. Abbott [Internet]. [cited 2024 Aug 7]. Available from: https://www.cardiovascular.abbott/us/en/hcp/products/percutaneous-coronary-intervention/xience-family/xience-skypoint.html
6. Ophthalmic Squeeze Dispenser (OSD) Technology Platform. Aptar [Internet]. [cited 2024 Aug 7]. Available from: https://aptar.com/products/pharmaceutical/ophthalmic-squeeze-dispenser-technology-platform/
7. HeroTracker®. Sense – digital solution for respiratory medicine. Aptar [Internet]. [cited 2024 Aug 7]. Available from: https://aptar.com/products/pharmaceutical/herotracker-sense-a-smart-healthcare-solution-for-digital-respiratory-medicine/
8. Jameel F, Skoug JW, Nesbitt RR, editors. Development of biopharmaceutical drug-device products. Berlin, Germany: Springer; 2020. CrossRef
9. Lauritsen KJ, Nguyen T. Combination products regulation at the FDA. Clin Pharmacol Ther. 2009;85(5):468–70. CrossRef
10. Reddy S, Beall RF, Tu SS, Kesselheim AS, Feldman WB. Patent challenges and litigation on inhalers for asthma and COPD: study examines patent challenges and litigation involving inhalers for asthma and COPD. Health Affairs. 2023;42(3):398–406. CrossRef
11. Alhiary R, Kesselheim AS, Gabriele S, Beall RF, Tu SS, Feldman WB. Patents and regulatory exclusivities on GLP-1 receptor agonists. JAMA 2023;330(7):650–7. CrossRef
12. Food and Drug Administration. Approved drug products with therapeutic equivalence evaluations (orange book). 45th ed. Washington, DC: U.S. Department of Health and Human Services; 2025.
13. Demkowicz BJ, Tu SS, Kesselheim AS, Carrier MA, Feldman WB. Patenting strategies on inhaler delivery devices. Chest 2023;164(2):450–60. CrossRef
14. U.S. Food and Drug Administration. Title 21, Code of Federal Regulations (CFR): part 210 - Current good manufacturing practice in manufacturing, processing, packing, or holding of drugs; general [Internet]. Revised Apr 1, 2023 [cited 2024 Aug 7]. Available from: https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-210
15. U.S. Food and Drug Administration. Title 21, Code of Federal Regulations (CFR): part 211 - Current good manufacturing practice for finished pharmaceuticals [Internet]. Revised Apr 1, 2023 [cited 2024 Aug 7]. Available from: https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-211
16. U.S. Food and Drug Administration. Title 21, Code of Federal Regulations, Part 820: quality system regulation [Internet]. [cited 2024 Aug 7]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=820https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=1688
17. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: http://accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=1694
18. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=1695
19. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2825
20. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2826
21. U.S. Food and Drug Administration. Code of Federal Regulations, 21 CFR 868.5640 [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=1305
22. U.S. Food and Drug Administration. Code of Federal Regulations, 21 CFR 868.5630 [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=118
23. U.S. Food and Drug Administration. Code of Federal Regulations, 21 CFR 868.5150 [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=82
24. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=145
25. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=163
26. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=920
27. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=953
28. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=1029
29. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=1267
30. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2703
31. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2704
32. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2705
33. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2706
34. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2737
35. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2739
36. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2817
37. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2830
38. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2842
39. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2863
40. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2880
41. U.S. Food and Drug Administration. Code of Federal Regulations, 21 CFR 880.5860 [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2881
42. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2884
43. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2983
44. U.S. Food and Drug Administration. Code of Federal Regulations, 21 CFR 880.5725 [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2997
45. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=1117
46. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=1141
47. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=1154
48. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=1155
49. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=1156
50. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=1170
51. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=1802
52. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=2415
53. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=3069
54. U.S. Food and Drug Administration. Product classification database [Internet]. [cited 2025 Feb 10]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm?id=3073
55. Office of Device Evaluation, Food and Drug Administration. Applying human factors and usability engineering to medical devices: guidance for industry and FDA staff. February 2016 [cited 2025 Apr 24]. Available from: http://www.fda.gov/CombinationProducts/default.htm
56. Center for Devices and Radiological Health, Center for Drug Evaluation Research, Center for Biologics Evaluation and Research, and Office of Combination Products, Food and Drug Administration. Human factors studies and related clinical study considerations in combination product design and development: draft guidance for industry and FDA staff. February 2016 [cited 2025 Apr 24]. Available from: http://www.fda.gov/CombinationProducts/default.htm
57. Committee for Medicinal Products for Human Use (CHMP). Guideline on the quality requirements for drug-device combinations: draft guideline. EMA/CHMP/QWP/BWP/259165/2019. May 2019 [cited 2025 Apr 24]. Available from: https://www.ema.europa.eu/en/search?f%5B0%5D=ema_search_entity_is_document%3ADocument&search_api_fulltext=Draft%20guideline%20on%20the%20quality%20requirements%20for%20drug-device%20combinations%20-%20First%20version
58. Couto DS, Perez-Breva L, Saraiva P, Cooney CL. Lessons from innovation in drug-device combination products. Adv Drug Deliv Rev. 2012;64(1):69–77. CrossRef
59. Choi SH, Wang Y, Conti DS, Raney SG, Delvadia R, Leboeuf AA, et al. Generic drug device combination products: regulatory and scientific considerations. Int J Pharm. 2018;544(2):443–54. CrossRef
60. Burt HM, Hunter WL. Drug-eluting stents: a multidisciplinary success story. Adv Drug Deliv Rev. 2006;58(3):350–7. CrossRef
61. Novo Nordisk Global, Smart Pens, Devices and Apps/NovoPen® 6 and NovoPen Echo® Plus. [cited 2025 May 16]. Available from: https://www.novonordisk.com/our-products/smart-pens/novopen-6.html
62. Medtronic News. Dated Sep 16, 2019. [cited 2025 May 16]. Available from: https://news.medtronic.com/2019-09-16-Medtronic-and-Novo-Nordisk-Enter-Agreement-to-Provide-Integrated-Digital-Solutions-for-People-with-Diabetes
63. Weissmann J, Mueller A, Messinger D, Parkin CG, Amann-Zalan I. Improving the quality of outpatient diabetes care using an information management system: results from the Observational VISION study. J Diabetes Sci Technol. 2015;9(2):244–50. CrossRef
64. Amgen Inc. Amgen annual report. 2021 [cited 2025 May 16]. Available from: https://www.amgen.com/investors
65. Wearable Bolus/High Volume Injectors. ONdrugDelivery September 14, 2015, Issue No 60. East Sussex, UK: Frederick Furness Publishing Ltd. [cited 2025 May 16]. Available from: https://www.ondrugdelivery.com/wp-content/uploads/2015/09/Wearable-Bolus-High-Volume-Injectors-ONdrugDelivery.pdf
66. Lilly Eli. Lilly initiates clinical trial to evaluate the functionality and safety of its automated insulin delivery system. December 2017 [cited 2025 May 16]. Available from: https://investor.lilly.com/news-releases/news-release-details/lilly-initiates-clinical-trial-evaluate-functionality-and-safety
67. YpsoMed. SmartPilot for YpsoMate: reusable add-on transforms YpsoMate into a fully connected device. [cited 2025 May 16]. Available from: yds.ypsomed.com/en/injection-systems/smart-devices/smartpilot.html
68. Fierce Biotec. Unilife inks 15-year deal with Sanofi for wearable injectors. By Joseph Keenan, Oct 7, 2014 8:56 am [cited 2025 May 17]. Available from: https://www.fiercebiotech.com/medical-devices/unilife-inks-15-year-deal-sanofi-for-wearable-injectors.
69. Injectable Drug Delivery. Issue No 120. Frederick Furness Publishing Ltd; May 31st 2021 [cited 2025 May 17]. Available from: https://www.ondrugdelivery.com/wp-content/uploads/2021/05/Injectable-Delivery-ONdrugDelivery-120-May-2021-LR.pdf
70. Amgen press release. FDA approves first and only single monthly injection for A PCSK9 inhibitor. [cited 2025 May 17]. Available from: https://www.amgen.com/newsroom/press-releases/2016/07/fda-approves-first-and-only-single-monthly-injection-for-a-pcsk9-inhibitor
71. Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease. Eur Heart J. 2013;34:3478–90.
72. Respiratory Therapy. AstraZeneca partners to develop smart inhaler adherence program. Therapy Devices; Aug 5, 2015 [cited 2025 May 17]. Available from: https://respiratory-therapy.com/products-treatment/monitoring-treatment/therapy-devices/astrazeneca-partners-develop-smart-inhaler-adherence-program/
73. Steinmetz JD, Seeher KM, Schiess N, Nichols E, Cao B, Servili C, et al. Global, regional, and national burden of disorders affecting the nervous system, 1990–2021: a systematic analysis of the Global Burden of Disease Study 2021. Lancet Neurol. 23(4):344–81. CrossRef
74. Hershey A, Szperka CL, Barbanti P, Pozo-Rosich P, Bittigau P, Barash S, et al. Efficacy and safety of fremanezumab for the preventive treatment of episodic migraine in children and adolescents: a phase 3, randomised, double-blind, placebo-controlled study. Presented at European Headache Congress (EHC); 4-7 December 2024, Rotterdam. ePoster LP036.
75. Steinmetz JD, Seeher KM, Schiess N, Nichols E, Cao B, Servili C, et al. Global, regional, and national burden of disorders affecting the nervous system, 1990–2021: a systematic analysis of the Global Burden of Disease Study 2021. Lancet Neurol. In press.
76. Greene K, Irwin SL, Gelfand AA. Pediatric migraine: an update. Neurol Clin. 2019;37(4):815–33. CrossRef
77. GSK Press release archive. GSK and Verily to establish Galvani Bioelectronics – a new company dedicated to the development of bioelectronic medicines. 01 August 2016 [cited 2025 May 17]. Available from: https://www.gsk.com/en-gb/media/press-releases/gsk-and-verily-to-establish-galvani-bioelectronics-a-new-company-dedicated-to-the-development-of-bioelectronic-medicines/
78. Deena Beasley. Becton Dickinson to buy CareFusion for $12 billion in cash, stock. 2014 [cited 2025 May 17]. Available from: https://www.reuters.com/article/business/becton-dickinson-to-buy-carefusion-for-12-billion-in-cash-stock-idUSKCN0HU0U3/
79. CDER application number: 103471Orig1s5189. BETASERON®. Bayer HealthCare Pharmaceuticals Inc. [cited 2025 May 18]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/103471Orig1s5189.pdf
80. CDER application number: 207534Orig1s000. Symjepi™. Adamis Pharmaceuticals Corp. [cited 2025 May 18]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/207534Orig1s0000MedR.pdf
81. Masierek M, Nabrdalik K, Janota O, Kwiendacz H, Macherski M, Gumprecht J. The review of insulin pens—past, present, and look to the future. Front Endocrinol (Lausanne). 2022 Mar 8;13:827484. CrossRef
82. Nagai N. Evaluation of nanotechnology-based medicines. Tokyo: Pharmaceuticals and Medical Devices Agency (PMDA). [cited 2014 Oct 31]. Available from: https://ss.pmda.go.jp/en_all/search.x?nccharset=6A733F16&nccharset=FEF28A70&q=Neulasta&ie=UTF-8&page=1
83. ElSayed NA, Aleppo G, Aroda VR, Bannuru RR, Brown FM, Bruemmer D, et al. Introduction and methodology: standards of care in diabetes—2023. Diabetes Care 2023 Jan 1;46(Suppl 1):S1–4. CrossRef
84. Pharmaceuticals and Medical Devices Agency (PMDA). List of approved medical devices. April 2004 to September 2024 [cited 2025 May 19]. Available from: https://www.pmda.go.jp/files/000274003.pdf
85. Pharmaceuticals and Medical Devices Agency (PMDA), Ministry of Health, Labour and Welfare of Japan. Strategy of SAKIGAKE. [cited 2025 May 20]. Available from: https://www.pmda.go.jp/english/review-services/reviews/advanced-efforts/0001.html