
INTRODUCTION
Lung cancer is widely recognized as the most prevalent form of cancer across the globe and ranks as the second-highest cause of cancer-related fatalities [1]. Non-small cell lung cancer (NSCLC) comprises roughly 80%–85% of all lung cancer cases and is frequently detected at an advanced stage [2]. Angiogenesis, invasion/metastasis, motility/migration, and growth/survival of cancer cells are all affected by the MET (mesenchymal-epithelial transition factor) receptor tyrosine kinase (RTK) and its ligand, hepatocyte growth factor (HGF). In the therapy for NSCLC, MET-RTK and HGF have emerged as significant targets. METexon14 skipping mutations are found in approximately 3%–4% of NSCLC patients, making it the fourth most common oncogenic driver in this context [3]. METexon14 skipping is a newly identified alteration that can be targeted and is a major driver of long-term activation of the MET receptor in 3%–4% of NSCLCs. Currently, MET-RTK inhibitors such as tepatinib, capmatinib, savolitinib, and crizotinib are used to treat METexon14 skipping NSCLC. The Type I inhibitors of MET-RTK strongly and specifically inhibit the MET receptor by competitively binding to ATP while also interacting with the MET activation loop. This inhibition results in increased specificity for MET and fewer off-target interactions [4,5].
In March 2020, Tepotinib (TPT) became the first MET-RTK approved in Japan for the treatment of NSCLC. In February 2021, it received fast approval from the US FDA under the brand name “Tepmetko” for managing METexon14 skipping mutations and metastatic NSCLC in adult patients [6–10]. As the first orally available MET-targeted RTK, TPT has the advantage of once-daily dosing, which could reduce the number of pills required for chemotherapy regimens. It also received approval from the European Union in February 2022. The IUPAC name of TPT is “3-[1-[[3-[5-[(1-methylpiperidin-4-yl)methoxy]pyrimidin-2-yl]phenyl]methyl]-6-oxopyridazin-3-yl]benzonitrile” (see Fig. 1).
In the pharmaceutical industry, it is crucial to have an efficient analytical method for analyzing individual drugs or drug combinations. Upon reviewing the literature, it was found that only a few reported methods exist for estimating TPT using RP-HPLC [11–14]. However, these methods lack sensitivity and accuracy when quantifying TPT at low concentrations. Additionally, these methods are expensive, time-consuming, and not suitable for routine analysis. Therefore, we have chosen to utilize a cost-effective photodiode array (PDA)-coupled HPLC technique, which is widely available in laboratories with limited financial resources and requires routine monitoring of TPT in its formulation. Considering these factors, the objective of this study is to develop and validate an RP-HPLC method for quantifying TPT in tablet formulation following ICH guidelines under Q specification [15].
MATERIALS AND METHODS
Chemicals and reagents
In this study, high-grade HPLC reagents and solvents were utilized. The solvents used were Acetonitrile (CH3CN), potassium dihydrogen orthophosphate (KH2PO4), methanol (CH3OH), and orthophosphoric acid (H3PO4), which were supplied by SD Fine Chem Ltd., located in Mumbai, India. The API for TPT was obtained from Ascentyo BioSciences in Hyderabad, India.
Instruments
The study was conducted using the Shimadzu HPLC system (Model No. LC-20AD) coupled with a PDA detector (Model No. SPD-M30A). Data acquisition was done using Empower software version 2. Chromatography was performed using a Phenomenex Luna C18 column (dimensions: 250 mm × 4.6 mm, 5 µm). Weighing was done using an analytical balance from Mettler Toledo.
Chromatographic conditions
The chromatographic conditions for the analysis were as follows: a mobile phase consisting of a potassium dihydrogen orthophosphate and acetonitrile in the ratio of 15:85 v/v was used in isocratic mode. The analysis was conducted at an ambient temperature with a flow rate of 1.0 ml/minute for the mobile phase. Each run involved injecting 10 µl of the sample into the HPLC. The PDA detector was set to a detection wavelength of 284 nm to detect TPT in the effluents from the column (see Fig. 2).
Buffer preparation
To prepare the diluent, 1.36 g of KH2PO4 were dissolved in 1,000 ml of water. Formic acid was used to adjust the pH to 2.5. The solution was then filtered using a nylon membrane filter (0.45 μm) and degassed before being used as the diluent in the HPLC analysis.
Preparation of standard stock solution
In a volumetric flask (VF) with a capacity of 100 ml, 10 mg of TPT powder was added. The flask was then filled with the diluent and sonicated for 20 minutes. The volume was adjusted to 100 ml using the diluent to obtain a primary standard stock solution having a concentration of 100 µg/ml. The solution of primary standard stock was stored at 2 ºC–8ºC until further use. Next, 0.8 ml of the solution was transferred to a 10 ml volumetric flask and filled with 10 ml of diluent to achieve a concentration of 8 µg/ml of TPT.
Selection of the detection wavelength
HPLC analysis was performed using an 8 µg/ml solution of TPT. This was accomplished by diluting the primary standard stock solution with a mobile phase containing potassium dihydrogen orthophosphate buffer (pH 2.5) and acetonitrile in a 15:85 (v/v) ratio. The produced solution was then scanned in a UV spectrophotometer between 190 and 400 nm, using the mobile phase as a blank reference. The purpose of this scanning process was to identify the wavelength at which the TPT compound absorbs light the most, which would be optimal for its detection in subsequent HPLC analyses.
Preparation of sample solution from dosage form
Twenty film-coated tablets of Tepmetko® (225 mg dose) were weighed, and their average weight was determined. The tablets were then crushed into powder form, and a 10.0 mg equivalent of the powder was transferred into a VF (100 ml). The flask was filled with the diluent and subjected to sonication for 20 minutes. Next, 0.8 ml of the resulting solution was transferred into a VF (10 ml) and diluted with 10 ml of diluent to achieve a concentration of 8 µg/ml of TPT.
Analytical method development
The method development process for HPLC analysis of TPT involved optimizing various parameters while keeping specific constants. Mobile phase composition (solvent systems and ratios), column selection, and flow rates were systematically adjusted to achieve desirable chromatographic separation. However, to ensure consistency and facilitate validation, several parameters remained unchanged: detector type, injection volume (10 µl), oven temperature (25ºC ± 2ºC), and elution mode. For each set of chromatographic conditions, a spectrum was recorded at the chosen detection wavelength. Ultimately, the selection of the appropriate wavelength depended on the peak purity achieved in the resulting chromatogram. During the method›s development, other criteria that were taken into account were peak height, column pressure, accuracy, resolution, analysis time, and solvent efficiency per run.
Validation
After obtaining suitable chromatographic conditions, the method was validated as per the ICH Q2 guidelines [15]. Furthermore, the stability of reagents and solvents were investigated.
Evaluation of system suitability
System suitability tests were conducted to ensure the reliability of the HPLC system. Six injections at a concentration of 8 µg/ml were made to measure column efficiency, plate count, and tailing factor. The results met the predetermined criteria, confirming the consistency of the system and its adherence to specified limits.
Specificity and selectivity
The method’s specificity was confirmed by analyzing three samples: a blank solution, a placebo matrix-extracted solution, and a standard solution containing TPT using a PDA detector.
Linearity
The linearity of TPT was evaluated by preparing dilutions ranging from 2–12 µg/ml from the standard stock solution. Peak area responses were measured for each concentration in the HPLC analysis.
Precision
Precision was assessed by conducting six repeated injections of the standard solution, which resulted in a low % RSD of 0.9, indicating high precision and consistent results. Similarly, six injections of the test solution showed a % RSD of 1.0, within the acceptable limit for precision (2.0%), demonstrating the reliability of the method in sample analysis.
Intermediate precision (IP)
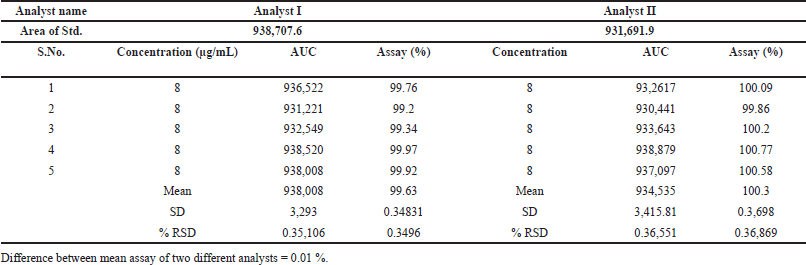
Intermediate precision was evaluated by having two analysts use separate HPLC instruments in different labs on different days to analyze the standard solution. Despite time-based variations, both obtained nearly identical assay results, with a negligible difference of 0.01% and an RSD below the acceptable limit of 2.0% on both days.
Accuracy or recovery studies
The accuracy of the HPLC method was verified through a triplicate recovery study. TPT at concentrations of 4, 8, and 12 µg/ml was injected into pre-analyzed samples. The mean recovery percentage was computed from these studies to validate accuracy.
Robustness
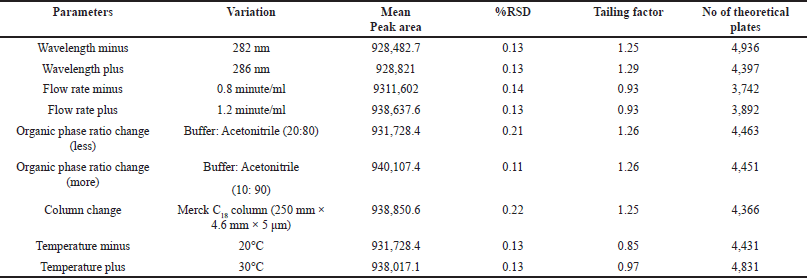
The robustness of the HPLC method was assessed by deliberately adjusting the flow rate and wavelength. These variations did not significantly alter the chromatogram, tailing factor, or plate count, indicating that the method remains robust and unaffected by fluctuations in flow rate and wavelength, ensuring accuracy and precision.
Solution stability
Sample and standard solution stability assessments were conducted to ensure their integrity throughout the analysis. To assess sample stability, triplicate samples were kept at ambient temperature and refrigerated (2°C–8°C) for 1 and 2 days, respectively. The percent relative difference between these stored samples and a freshly prepared sample was calculated. This confirmed that the samples remained stable during storage. Similarly, standard solution stability was tested. Duplicate standard solutions were stored at room temperature and under refrigeration for 1 and 2 days. The percent assay and %RD were calculated by comparing the stored solutions with freshly prepared standards. This verified the stability of the standard solutions throughout the analysis period.
To ensure mobile phase stability, its appearance, system suitability parameters, and repeatability were evaluated after 1 and 2 days of storage at room temperature. This assessment confirmed the suitability of the mobile phase for consistent analysis throughout the planned duration.
Application of the developed method
The validated HPLC method was effectively employed to determine and quantify the amount of TPT in marketed TPT tablets. As described in section 2.7, sample solutions were prepared following the established protocol. Each sample was then injected into the HPLC system three times (triplicate injections) to ensure data accuracy and reliability.
RESULT AND DISCUSSION
Method development and optimization
This research aimed to develop and validate a reliable method using PDA-coupled RP-HPLC to quantify the amount of TPT in film-coated tablets. We combined theoretical knowledge with practical experimentation to successfully achieve this goal. The theoretical understanding of TPT’s physico-chemical properties, derived from available literature, guided the selection of suitable chromatographic conditions. The preliminary approach was developed using complete TPT information such as molecular mass, solubility, partition coefficient, chemical structure and functionality, and UV absorption spectra. Solubility is critical in determining the best mobile phase compositions and columns for HPLC method development. TPT exhibits limited solubility in water but readily dissolves in organic solvents such as dimethyl sulfoxide and acetonitrile. This characteristic makes acetonitrile a suitable choice for preparing stock solutions of TPT. The compound has a molar mass of 492.58 g per mole and an Alog p value of 4.01.
TPT displayed notable ultraviolet (UV) absorbance at 284 nanometers (nm) in a potassium dihydrogen orthophosphate: acetonitrile solution (15:85 v/v), as shown in Figure 2. This conveniently coincided with the optimal wavelength for quantifying TPT (Fig. 2). Leveraging this initial observation, the HPLC method was carefully developed and optimized by modifying various parameters, as detailed in the methods section.
 | Figure 1. Chemical structure of TPT. [Click here to view] |
For an ideal analytical method, the detection wavelength should strike a balance between achieving the lowest Limit of Detection (LOD) and maximizing peak area. Fortunately, in this case, both criteria were met at 284 nm. This wavelength offered not only the minimum detectable concentration of TPT but also resulted in the largest peak area, ensuring accurate quantitation. Furthermore, 284 nm was free from potential interference from excipients co-eluting with TPT at its retention time of 6.97 minutes. The purity of the obtained peak at this wavelength further solidified the choice of 284 nm as the optimal detection wavelength.
In single-component analysis, high peak purity can often be confidently assumed. This is further bolstered by the meticulous optimization conducted here, ensuring peak symmetry, the absence of interfering peaks, and adherence to established criteria for peak shape and plate number. The exceptional resolution achieved in the results serves as a testament to the methodology’s effectiveness in delivering precise and reliable chromatographic data. Notably, the impressive peak area of 99.88% further strengthens the confidence in assuming peak purity.
At 284 nm, the TPT spectra displayed a distinct threshold and a similarity curve that fell below the threshold. This finding is remarkable since spectra recorded over a single-component peak should presumably be identical [16]. Therefore, this result strongly suggests the presence of only TPT in the sample solutions, free from any significant spectral interference. Consequently, considering both sensitivity and method conditions, 284 nm emerged as the optimal detection wavelength, fulfilling all necessary criteria.
Considering the physicochemical properties of TPT, both C8 and C18 analytical columns were evaluated for their suitability in analysis. The C18 column surpassed the C8 column in terms of important chromatographic characteristics such as resolution (Rs), optimal plate number (N), and tailing factor (Tf). This indicates superior peak separation and sensitivity on the C18 column. Subsequently, to determine the optimal detection wavelength, the instrument’s LOD at 284 nm was measured using the PDA detector.
Since TPT remains neutral during separation, the mobile phase can be a simple mixture of a buffer solution and an organic solvent. Among the various tested mobile phases, a mixture of potassium dihydrogen orthophosphate buffer (pH 2.5) and acetonitrile (15:85 v/v) yielded superior results. This combination produced symmetrical peaks for TPT with excellent resolution while also providing an appropriate retention time. Increasing the acetonitrile concentration significantly impacted peak resolution, widening band spacing. However, decreasing the concentration led to stronger retention but with varying selectivity, band spacing, and resolution. This highlights the importance of careful solvent selection and composition for optimal method development. Ultimately, a 15:85 (v/v) mixture of potassium dihydrogen orthophosphate buffer (pH 2.5) and acetonitrile proved to be the ideal mobile phase composition for quantifying TPT.
Flow rate also played a crucial role in separation, influencing TPT’s retention time. While varying the rate from 0.5 to 1.5 mL/minute, the optimal value for plate number (N) and resolution (Rs) between components was found to be 1.0 ml/minute. Notably, the quantification was performed at room temperature (25ºC ± 2ºC) due to TPT›s neutral characteristics. It›s important to remember that, in reversed-phase chromatography, higher temperatures often decrease selectivity for neutral compounds [16].
Leveraging TPT’s neutral nature for optimal separation, the temperature was maintained constant throughout the experiment. Following thorough analysis, the optimal set of parameters for the analytical method was determined to be a C18 column, a flow rate of 1 ml/minute, a mobile phase consisting of potassium dihydrogen orthophosphate buffer (pH 2.5) and acetonitrile in a ratio of 15:85 v/v, and a detection wavelength of 284 nm. Under these optimized conditions, a symmetrical and well-defined peak of TPT was observed with an average retention time of 6.97 minutes, demonstrating the effectiveness of the chosen parameters.
The analysis showed a percent relative standard deviation (%RSD) of less than 2%, indicating exceptional precision. Additionally, the column pressure remained significantly below the recommended limit. For most procedures, column pressures under 150 bar are ideal, while pressures under 200 bar are safe [16].
System suitability parameters
The precision of the HPLC system was assessed by determining the % RSD (relative standard deviation) from six replicate injections of the standard solution. A requirement was set for the RSD not to exceed 2% to be considered acceptable. Since the RSD of the standard solution fell within this limit, indicating precision within the specified criterion, the system proved reliable for accurate TPT quantification in samples. These findings are detailed in Table 1.
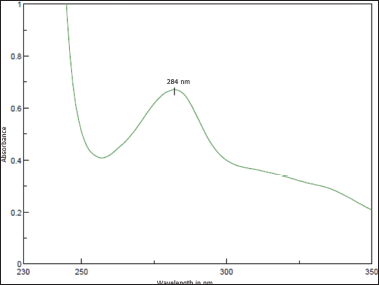 | Figure 2. UV spectrum of TPT. [Click here to view] |
TPT exhibited a retention time (RT) of 6.97 minutes, confirming its effective separation and timely detection. Through meticulous selection of mobile phase composition, optimal wavelength, and parameter fine-tuning, the HPLC method for TPT analysis was tailored to meet the desired criteria: swift analysis and satisfactory resolution. This rigorous optimization ensures reliable and efficient TPT quantification, meeting stringent analytical demands.
Specificity and selectivity
The method demonstrated high specificity and selectivity, allowing for the accurate detection of TPT in the sample. The chromatogram of the TPT reference standard showed a distinct peak, while the blank containing only the diluent showed no response or interfering peaks. This confirms that there are no interferences from other sample components, as shown in Figures 3 and 4 (standard and blank injections, respectively). The chromatogram of the TPT solution displayed a sharp, well-separated, and symmetrical peak at a retention time of 6.97 minutes (Fig. 3). Importantly, there were no interfering peaks from the placebo matrix at this retention time, indicating the excellent specificity of the method. This means that the method can accurately identify and quantify TPT even in the presence of other components from the sample.
Peak purity
The proposed methods’ selectivity was further demonstrated by assessing the referenced drug’s peak purity in a pharmaceutical preparation matrix (placebo) with a PDA detector. PDA’s key feature is the ability to gather spectra over a range of wavelengths for each data point collected throughout a peak, and using software modifications, individual spectra can be compared to assess peak purity [17]. The “purity angle” and “purity threshold” together define the overall peak’s purity. For TPT, the purity angle is 0.106, and purity threshold is 1.013 (see Table 1 and Fig. 6). To meet the peak purity acceptance criterion, the purity angle must be smaller than the purity threshold. Thus, the peak purity of TPT in the sample was passed.
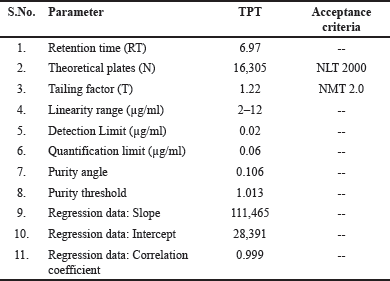 | Table 1. System suitability parameters for TPT. [Click here to view] |
Linearity
A linearity graph was generated for TPT, with concentration in μg/ml on the x-axis and area under the curve (AUC) on the y-axis. Within the range of 2–12 µg/ml, a linear correlation was observed between drug concentrations and peak area responses. The results, depicted in Figure 5 and summarized in Table 2, highlight the importance of linearity in analytical methods. This characteristic ensures precise drug quantification across a wide range of concentrations, based on the measurement of peak areas. The HPLC method exhibits excellent linearity within the specified concentration range, establishing a strong relationship between concentration and peak area response, which facilitates accurate TPT analysis.
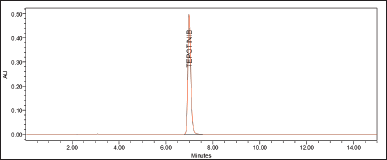 | Figure 3. Optimized chromatogram of the standard. [Click here to view] |
 | Figure 4. Blank chromatogram. [Click here to view] |
 | Figure 5. Linearity plot of TPT. [Click here to view] |
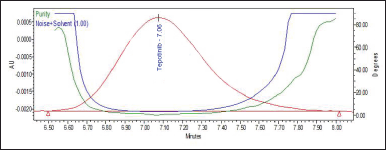 | Figure 6. Peak purity plot of TPT. [Click here to view] |
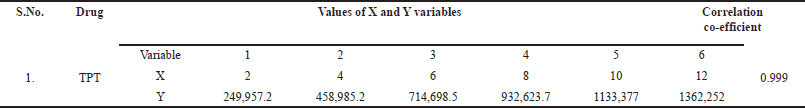 | Table 2. Linearity of TPT. [Click here to view] |
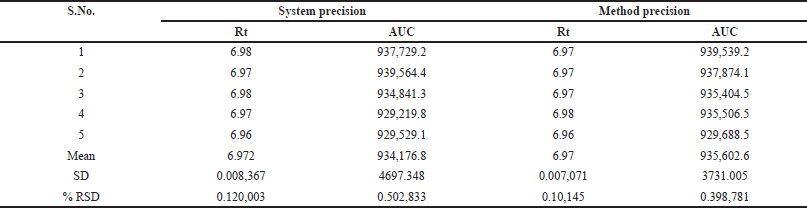 | Table 3. Precision study. [Click here to view] |
 | Table 4. IP or ruggedness study. [Click here to view] |
 | Table 5. Accuracy study. [Click here to view] |
Precision
The precision of the method, including the repeatability of both sample and standard preparations, was found to be satisfactory. Table 3 confirms these findings, demonstrating the reproducibility and reliability of the HPLC method in TPT analysis. This validation supports the routine use of the method for accurate quantification across various types of samples.
Intermediate precision (IP)
The consistent performance of the HPLC method across diverse laboratories, instruments, and analysts, even on different days, emphasizes its robustness. Its IP affirms its suitability for routine use, ensuring consistently accurate and reliable results under varying experimental conditions. Table 4 displays the percentage RSD values for IP, and they are below 2.0%, confirming the precision of the method and instrument. This consistency guarantees dependable and minimally variable results, as evidenced by the acceptable recovery rate range of typically 99.63%–100.3%.
 | Table 6. Robustness study. [Click here to view] |
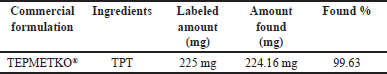 | Table 7. Analysis of marketed formulation. [Click here to view] |
Accuracy
The assay yielded a mean recovery percentage of 100.27%, signifying the accuracy of the HPLC method in quantifying the TPT content within expected values. Results from Table 5, which displays spiked concentrations and mean recovery percentages, validate the reliability of the method for quantitative analysis, showcasing successful TPT recovery from varied spiked samples. This accuracy, as evidenced by the recovery falling within the acceptable range, confirms the satisfactory performance of the method in accurately measuring the TPT content.
Robustness
Table 6 presents the results from robustness studies, detailing the variations tested and their impact on the performance of the method. The confirmed robustness of the HPLC method affirms its suitability for routine application, ensuring consistent and reliable results even under slightly altered operating conditions. Notably, insignificant changes in peak areas and retention times highlight the method’s ability to deliver reliable outcomes across different conditions. In comparison to previously reported analytical methods in the literature, this HPLC method excels with shorter retention times, enhanced theoretical plates (indicating improved resolution), and a mobile phase that promotes better separation of TPT from other constituents. Consequently, its increased efficiency and precision make it more suitable for the routine quantification of TPT across several sample types.
Solution stability
Both the standard and sample solutions remained stable within the acceptance criteria (≤2.0% RD for standards and ≤3.0% RD for samples) after 1 day of storage at both room temperature and refrigerated conditions (2°C–8°C). However, solutions stored for 2 days at either temperature did not meet the criteria, primarily due to changes in the response-to-standard ratio (RST). This indicates that the solutions become unstable after 2 days.
The mobile phase remained clear and free of particles throughout the stability study, consistently meeting the acceptance criteria for system suitability and repeatability. This indicates excellent stability for up to 2 days at room temperature storage, eliminating the need for frequent preparation.
Application of the developed method
TPT in film-coated tablets that are sold commercially was successfully evaluated using the described approach. The assay for TPT in Tepmetko® tablets was determined to be 99.63% ± 0.2% (Table 7). This result aligns favorably with the desired outcome for method development, as a high assay percentage ensures consistent and reliable analysis for other researchers conducting routine analysis of the same compound, potentially across different dosage forms.
CONCLUSION
Following the ICH guidelines, an HPLC method was established and validated. The method utilized a Shimadzu HPLC with PDA detection at a wavelength of 284 nm. The injection volume was 10 µl, and a Phenomenex Luna C18 column (250 × 4.6 mm, 5 μm) was used with isocratic elution. The resulting method is rapid, robust, straightforward, precise, sensitive, and cost-effective. It offers key benefits such as a brief run time (below 7 minutes) and excellent resolution. The % RSD values for all validation parameters met the criteria, confirming the method’s suitability for routine analysis of TPT in laboratories and quality control.
AUTHOR CONTRIBUTIONS
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. All the authors are eligible to be an author as per the international committee of medical journal editors (ICMJE) requirements/guidelines.
FUNDING
There is no funding to report.
CONFLICTS OF INTEREST
The authors report no financial or any other conflicts of interest in this work.
ETHICAL APPROVALS
This study does not involve experiments on animals or human subjects.
DATA AVAILABILITY
All the data is available with the authors and shall be provided upon request.
PUBLISHER’S NOTE
All claims expressed in this article are solely those of the authors and do not necessarily represent those of the publisher, the editors and the reviewers. This journal remains neutral with regard to jurisdictional claims in published institutional affiliation.
USE OF ARTIFICIAL INTELLIGENCE (AI)-ASSISTED TECHNOLOGY
The authors declares that they have not used artificial intelligence (AI)-tools for writing and editing of the manuscript, and no images were manipulated using AI.
REFERENCES
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
2. Bareschino MA, Schettino C, Rossi A, Maione P, Sacco PC, Zeppa R, et al. Treatment of advanced non-small cell lung cancer. J Thorac Dis. 2011;3(2):122–33.
3. Awad MM, Oxnard GR, Jackman DM, Savukoski DO, Hall D, Shivdasani P, et al. MET Exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent met genomic amplification and c-met overexpression. J Clin Oncol. 2016;34(7):721–30. CrossRef
4. Cortot A, Le X, Smit E, Viteri S, Kato T, Sakai H, et al. Safety of MET tyrosine kinase inhibitors in patients with MET Exon 14 skipping non-small cell lung cancer: a clinical review. Clin Lung Cancer. 2022;23(3):195–207. CrossRef
5. Drilon A, Cappuzzo F, Ou SI, Camidge DR. Targeting MET in lung cancer: will expectations finally be met? J Thorac Oncol. 2017;12(1):15–26. CrossRef
6. Sakai H, Morise M, Kato T, Matsumoto S, Sakamoto T, Kumagai T, et al. Tepotinib in patients with NSCLC harbouring MET exon 14 skipping: Japanese subset analysis from the Phase II VISION study. Jpn J Clin Oncol. 2021;51(8):1261–8. CrossRef
7. Paik PK, Felip E, Veillon R, Sakai H, Cortot AB, Garassino MC, et al. Tepotinib in non-small-cell lung Cancer with MET Exon 14 skipping mutations. N Engl J Med. 2020 Sep 3;383(10):931–43. CrossRef
8. Drusbosky LM, Dawar R, Rodriguez E, Ikpeazu CV. Therapeutic strategies in METex14 skipping mutated non-small cell lung cancer. J Hematol Oncol. 2021 Aug 23;14(1):129. CrossRef
9. Yuan P, Xue X, Qiu T, Ying J. MEt alterations detection platforms and clinical implications in solid tumors: a comprehensive review of literature. Ther Adv Med Oncol. 2024 Jan 18;16:17588359231221910. CrossRef
10. Le X, Paz-Ares LG, Van Meerbeeck J, Viteri S, Galvez CC, Smit EF, Garassino M, et al. Tepotinib in patients with non-small cell lung cancer with high-level MET amplification detected by liquid biopsy: VISION Cohort B. Cell Rep Med. 2023 Nov 21;4(11):101280. CrossRef
11. Rao SS, Sahoo S, Kavitha K. Stability indicating method development and validation for the estimation of tepotinib in api and tablet dosage form by rp-hplc. J Innov Dev Pharm Tech Sci. 2023;6(10):14–7.
12. Monika MS, Smitha TK, Shubangi MB, Mallinath K. Development and validation of stability indicating rp-hplc method for the estimation of tepotinib in bulk and formulation. Eur Chem Bull. 2023;12(10):10877–91.
13. Nikhil J, Parthiban C, Sudhakar M, Vijaya SK. Method development and validation for the estimation of tepotinib in pharmaceutical dosage forms by rp-hplc. Int J Pharm Pharm Res. 2022;26 (1):468–77.
14. Hameeda B, Mohammed A, Madiha J. Development and validation of liquid chromatography method for determination of tepotinib in bulk drug and in tablet dosage form. Int J Pharm Sci Rev Res. 2023;80(2):128–13.
15. ICH. Validation of analytical procedure: methodology International Conference on Harmonization. ICH harmonised tripartite guideline—validation of analytical procedures: text and methodology Q2(R1). Geneva, Switzerland: ICH; 2005.
16. Snyder LR, Kirkland JJ, Glajch JL, Practical HPLC method development. Hoboken, NJ: John Wiley & Sons; 2012.
17. Castledine JB, Fell AF. Strategies for peak-purity assessment in liquid chromatography. J Pharm Biomed Anal. 1993 Jan;11(1):1–13. CrossRef